
Abstract
The accumulation of metabolites in the intervertebral disc is considered an important cause of intervertebral disc degeneration (IVDD). Lactic acid, which is a metabolite that is produced by cellular anaerobic glycolysis, has been proven to be closely associated with IVDD. However, little is known about the role of lactic acid in nucleus pulposus cells (NPCs) senescence and oxidative stress. The aim of this study was to investigate the effect of lactic acid on NPCs senescence and oxidative stress as well as the underlying mechanism. A puncture-induced disc degeneration (PIDD) model was established in rats. Metabolomics analysis revealed that lactic acid levels were significantly increased in degenerated intervertebral discs. Elimination of excessive lactic acid using a lactate oxidase (LOx)-overexpressing lentivirus alleviated the progression of IVDD. In vitro experiments showed that high concentrations of lactic acid could induce senescence and oxidative stress in NPCs. High-throughput RNA sequencing results and bioinformatic analysis demonstrated that the induction of NPCs senescence and oxidative stress by lactic acid may be related to the PI3K/Akt signaling pathway. Further study verified that high concentrations of lactic acid could induce NPCs senescence and oxidative stress by interacting with Akt and regulating its downstream Akt/p21/p27/cyclin D1 and Akt/Nrf2/HO-1 pathways. Utilizing molecular docking, site-directed mutation and microscale thermophoresis assays, we found that lactic acid could regulate Akt kinase activity by binding to the Lys39 and Leu52 residues in the PH domain of Akt. These results highlight the involvement of lactic acid in NPCs senescence and oxidative stress, and lactic acid may become a novel potential therapeutic target for the treatment of IVDD.
Similar content being viewed by others


Introduction
Intervertebral disc degeneration (IVDD) is an ageing-related degenerative disease and the leading cause of low back pain and associated disability worldwide, which causes a substantial burden on health care systems [1, 2]. Intervertebral discs lie between two adjacent vertebral bodies, and they are composed of 3 discrete components: the nucleus pulposus (NP), the annulus fibrosus, and the cartilage endplate. Intervertebral discs are the largest avascular tissue in the whole body, and the oxygen and nutrition supply to this tissue mainly depends on permeation between the NP and the cartilage endplate. These anatomical characteristics result in a low-oxygen, low-nutrition microenvironment [3, 4] and the accumulation of many metabolites [5,6,7,8].
Lactic acid is a common metabolic waste that is produced during cellular anaerobic respiration. Due to the hypoxic microenvironment in the intervertebral disc, anaerobic glycolysis is the main mechanism of energy supply for intervertebral disc cells, especially nucleus pulposus cells (NPCs), and it results in the production of a large amount of lactic acid [9, 10]. Under normal conditions, the generation, transport, and clearance of lactic acid occur in a dynamic balance to achieve the maintenance of intracellular pH and homeostasis of the intervertebral disc [11, 12]. However, under pathological conditions, the concentration of lactic acid in the intervertebral disc distinctly rises, especially in the centre of the degenerative disc [13, 14]. The concentration of lactic acid in the degenerate intervertebral disc of patients can reach levels 10 times higher than those in the plasma [15]. The disruption of glycolysis and the imbalance of lactic acid transport and clearance are the main causes of lactic acid accumulation in the intervertebral disc [11, 16,17,18]. The injection of lactic acid into the intervertebral disc of healthy rats elicits degeneration-related symptoms, indicating that lactic acid can directly aggravate IVDD [19, 20].
Although the phenomenon of lactic acid accumulation in the intervertebral disc has been widely reported, the cell biological mechanisms by which lactic acid results in degeneration are still unclear. High concentrations of lactic acid can downregulate the expression of extracellular matrix glycosaminoglycan and downregulate the expression of matrix metalloproteinase 3 [21, 22]. In addition, high concentrations of lactic acid have been reported to regulate the apoptosis and autophagy of NPCs [23]. A recent study also reported that high concentrations of lactic acid can promote NLRP3 inflammasome activation and exacerbate inflammation in NPCs [20]. These phenomena are all relevant manifestations of cellular senescence and oxidative stress, which are the most crucial changes in cell biology that occur during IVDD [24]. However, the effect of lactic acid on NPCs senescence and oxidative stress has not been verified. lactic acid has been proven to be related to senescence in Drosophila [25]. High concentrations of lactic acid have been considered a hallmark of brain senescence [26], and high concentrations of lactic acid contribute to the progression of Alzheimer’s disease, which is a common degenerative brain disease [27, 28]. In addition, lactic acid can induce oxidative stress injury during peripheral axon regeneration and arterial calcification [29, 30]. Therefore, we surmise that lactic acid may promote the senescence and oxidative stress of NPCs to accelerate the progression of IVDD.
To test our hypothesis, primary cultured rat NPCs and a puncture-induced disc degeneration rat model were used to determine the effect of lactic acid on NPCs. RNA-seq and molecular docking were utilized to elucidate the underlying mechanisms. This study is the first to verify the effects of lactic acid on NPCs senescence and oxidative stress. This study may help to better understand the relationship between lactic acid and NPCs senescence and oxidative stress during IVDD and may provide a prospective strategy for IVDD treatment.
Materials and methods
Patient information and NP tissue sample collection
In the present study, 24 human NP tissue samples were collected from patients who underwent surgery between April 2022 and July 2022 at the Department of Orthopedics, Xinqiao Hospital, Army Military Medical University. According to the Pfirrmann grading criteria (Table S1) and MRI examination of patients, Pfirrmann grading was performed for the IVDD degree of patients. Then, 6 patients of every grade between Pfirrmann II and V were selected to perform the subsequent experiments, including 15 males and 9 females aged 18–65 years. Inclusion criteria: Patients with lumbar disc herniation or lumbar spinal stenosis; Body mass index (BMI) between 18 and 24. Exclusion criteria: Patients with diabetes, chronic pulmonary/cardiovascular/hepatic/kideny diseases, metabolic diseases, hematological diseases, lumbar infection, tumor or previous lumbar surgery at the involved level. The details of the patients are shown in Table S2.
Collection of normal disc and puncture-induced disc degeneration (PIDD) samples
The rats in the normal group did not receive any treatment, and the rats in the PIDD group were used to establish a caudal disc degeneration model based on previous reports [31, 32]. Briefly, the rats were anaesthetized with 2% pentobarbital (50 mg/kg), and carprofen analgesia (5 mg/kg) was administered preoperatively. The surgical area was disinfected with iodophor, and a no. 20 G needle was vertically inserted into the 4 consecutive intervertebral discs between coccyx 4 and coccyx 8 with a depth of approximately 5 mm. The needle was rotated 360 degrees and held in place for 30 s. Then, the needle was removed vertically, and the surgical area was disinfected again. Each rat was given analgesics and antibiotics for 3 days for postsurgical care. Four weeks after the operation, the rats were effectively anaesthetized and killed under analgesia, and the caudal vertebrae and intervertebral discs of the rats were collected for subsequent experiments.
Cell isolation, culture, and treatment
Rats were effectively anaesthetized and killed under analgesia, and then, the NP tissue was isolated under aseptic conditions. According to a previous report [33], the NP tissues of rats were mixed and digested with 0.2% type II collagenase (C2-BIOC, Sigma‒Aldrich, USA) for approximately 2 h at 37 °C, followed by centrifugation at 400×g for 5 min at room temperature. The supernatant was discarded, and the sediment was suspended and cultured in DMEM-F12 (BI, Kibbutz Beit-Haemek, Israel) supplemented with 10% fetal bovine serum (C04001500, VivaCell, Shanghai, China) and 1% penicillin/streptomycin (C0222, Beyotime, Shanghai, China). NPCs were placed in a humidified chamber in a 5% CO2–air mixture at 37 °C. The medium was changed every 3–4 days. The cells were subcultured when they reached a confluence of 80–90%. NPCs from the third generation were used throughout the experiments. For lactic acid (L6402, Sigma‒Aldrich) treatment, NPCs were treated with different concentrations of lactic acid (0, 2, 6, 10 mM) for 4 days, and then NPCs were collected for further evaluation and analysis.
Lactic acid content assays
A lactic acid assay kit (BC2235, Solarbio, China) was used to measure the lactic acid content in the NPCs. The extraction solution was added to 1*10^6 NPCs or 50 mg fresh NP tissue, and then homogenate was extracted using an ultrasonic homogenizer (M3000, BioLogics, USA). The homogenate was centrifuged to harvest the supernatant. Standards were prepared by serial dilution of stock standards. The reaction mix was added to each well of the standard and sample. The output was measured on a microplate reader at OD 570 nm. The concentration of lactic acid in the test samples was calculated as follows: Lactic acid concentration = La/Sv*D. La = the amount of lactic acid in the sample well calculated from a standard curve (nmol). Sv = volume of sample added into the well (µL). D = sample dilution fold.
Metabolomics analysis
Metabolomics analysis was performed by MetWare Biotechnology Co., Ltd. (Wuhan, China). Four weeks after needle-puncture surgery, 6 rats were randomly selected from each of the normal group and PIDD group. Fifty milligrams of NP tissue were taken from each rat, and then 500 µL of precooled 70% methanol was added to the samples. Then, a tissue-grinding pestle (MM400, Retsch) was used to grind the tissue samples and the mixture was centrifuged at 12,000 r/min for 10 min at 4 °C, and then 300 µL of supernatant of each sample was collected. Multiple reaction monitoring (MRM) was used for quantitative analysis of metabolites. First, the precursor ions of the target metabolites were screened, and then the prerequisite ions were rescreened after ionization to exclude the interference of nontarget ions. After the mass spectrometry data of different samples were obtained, the chromatographic peaks of all target substances were integrated. Quantitative analysis was carried out by the standard curve of metabolites. The data acquisition instrument system mainly includes ultra performance liquid chromatography (UPLC, ExionLCTM AD) and tandem mass spectrometry (MS/MS, OTRAPB6500+). Liquid chromatographic conditions included the following: column: ACQUITY UPLC BEH Amide column (1.7 μm, 100 mm x 2.1 mm i.d.); mobile phase: A phase, ultrapure water (10 mM acetic acid, 0.3% ammonia); B phase, 90% acetonitrile/water (V/V); flow rate: 0.40 mL/min; column temperature: 40 °C; sample volume: 2 µL; mobile phase gradient: 0-1.2 min A/B is 5:95 (VV), 8 min A/B is 30:70 (V/V), 9.0–11 min A/B is 50:50 (V/V), and 11.1–15 min A/B is 5:95 (V/V). The mass spectrometric conditions mainly included an electrospray ionization (ESI) temperature of 550 °C, a positive ion mode mass voltage of 5500 V, a negative ion mode mass voltage of -4500 V, and a curtain gas (CUR) of 35 psi. In QTRAP 6500+, each ion pair is scanned and detected based on the optimized declustering potential (DP) and collision energy (CE).
Construction and verification of the lactate oxidase (LOx) overexpression vector
The LOx overexpression lentiviral vector was constructed according to previous reports [34]. The gene sequence of LOx (GenBank: D50611.1) was optimized by Thermo Fisher Scientific Company (Shanghai, China), and then, the pCMV-LOx-IRES-EGFP plasmid was constructed based on the GV208 plasmid. The pCMV-IRES-EGFP plasmid was used as a control, and then, the lentiviral vectors (LVV) were packaged in H293T cells after the cotransfection of the plasmids to generate LVV-LOx-IRES-EGFP and the corresponding control virus LVV-IRES-EGFP. Then, the Escherichia coli strain DH5α was used to amplify LVV. The above services were provided by GeneChem Gene Company (Shanghai, China). Next, according to the manufacturer’s instructions, we selected an MOI of 20 to transfect lentiviral vectors into NPCs. The fluorescence intensity of green fluorescent protein (GFP) was observed and the relative level of LOx was observed by Western blotting to determine the virus transfection effect.
Lentivirus treatment of PIDD rats
Six healthy male SD rats aged 8 weeks were randomly selected for the animal experiments. Four consecutive intervertebral discs between coccyx 4 and coccyx 8 were selected for the experiments. After the rats were effectively anaesthetized with 2% pentobarbital (50 mg/kg), Co 5/6, Co 6/7, and Co 7/8 were punctured according to the method described above to establish PIDD. Co 4/5 was not operated and served as a reference control. A microsyringe (Hamilton, Switzerland) was used to inject lentivirus or normal saline into the intervertebral discs of rats in each group. Immediately after the puncture, Co 5/6 was injected with 2 µl of normal saline, Co 6/7 was injected with LVV-IRES-EGFP (LVV-control, 1 × 109 PFU, diluted in 2 µl of normal saline), and Co 7/8 was injected with LVV-LOx-IRES-EGFP (LVV-LOx, 1 × 109 PFU, diluted in 2 µl of normal saline). The surgical areas were disinfected after the injection was completed. Four weeks after treatment, the rats were sacrificed after effective anaesthesia and analgesia, and the intervertebral discs of the rats were harvested for subsequent experiments.
Magnetic resonance imaging (MRI) analysis
Rats were anaesthetised by inhalation of 4% sevoflurane. A magnetic resonance imager (PHILIPS Ingenia 3.0T) was used to scan the coccyx of the rats, and the parameters were set as follows: TR time: 2000 ms, TE time: 80 ms, incentive time: 2, scan time: 3 min 20 s, fat reduction technology: SPAIR, scan matrix: frequency encoding 368 and phase encoding 288, layer thickness: 2.5 mm, echo chains: 12, and spin echo sequence: TSE sequence. Pfirrmann grading of rat intervertebral discs was calculated according to the Pfirrmann grading criteria (Table S1).
Microcomputed tomography (µCT) analysis
After the rats were effectively anaesthetized, the coccyx of the rats was scanned with a µCT scanner (Bruker, SkyScan 1176) at a source voltage of 90 kV and a current of 160 µA, and the scanned image pixel was 73 μm. According to a previously reported method [31], the length of the intervertebral disc and the adjacent superior and inferior vertebral bodies were measured to calculate the disc height index (DHI), and the percentage of DHI (DHI%) was calculated as post-DHI/0 W-DHI.
Histological analysis of human nucleus pulposus tissues and rat caudal intervertebral discs
The human NP tissues removed during surgery were incubated in 4% paraformaldehyde solution (Beyotime) for 48 h, and then the samples were dehydrated and embedded in paraffin and sliced into 6 μm paraffin sections. The caudal vertebrae and intervertebral discs of the rats were incubated in 4% paraformaldehyde solution (Beyotime) for 48 h and then decalcified with 10% ethylenediaminetetraacetic acid (EDTA; pH 7.2) for 14 days. The samples were dehydrated and embedded in paraffin and then sliced into 6 μm paraffin sections. The prepared sections were stained using a Haematoxylin-Eosin (HE) Staining Kit (G1120, Solarbio) and a Modified Safranine O-Fast Green (SO & FG) Staining Kit (G1371, Solarbio) according to the manufacturer’s instructions, and then, the sections were fixed with neutral resin and observed by an optical microscope (BX53, OLYMPUS). The histology grading of the rat intervertebral discs was calculated according to the histological grading criteria of disc degeneration [35].
Immunofluorescence staining assay
Cell samples: NPCs were seeded in 6-well plates or confocal dishes, and after receiving different treatments, the cells were fixed with 4% paraformaldehyde, permeabilized with immunostaining permeabilizer (P0097, Beyotime), and blocked with QuickBlock immunostaining blocking solution (P0260, Beyotime). Then, the primary antibody was added and incubated overnight at 4 °C. A fluorescently labeled secondary antibody was added to the sample. Then, the results were visualized using fluorescence or confocal microscopy (LSM880, ZEISS, Germany). The mean fluorescence intensity of the samples was quantified using ImageJ (National Institute of Health, USA). Information about the antibodies used in the experiments is provided in Table S3.
Tissue samples: Tissue sections were prepared as previously described. The sections were deparaffinized with xylene and rehydrated with graded ethanol solutions, followed by antigen retrieval with sodium citrate-EDTA antigen retrieval solution (P0086, Beyotime). The sections were permeabilized, blocked, and incubated with primary antibodies overnight at 4 °C. Then, the sections were incubated with fluorescently labeled secondary antibodies. The fluorescence staining results were obtained using a confocal microscope. ImageJ was used to quantify the fluorescence intensities. Information about the antibodies used in the experiments is provided in Table S3.
Cell counting Kit-8 (CCK-8) assay
A cell counting kit-8 (C0038, Beyotime) was used to determine the effect of lactic acid on NPCs proliferation. NPCs were seeded into 96-well plates at a density of 2 × 103 cells. Then, lactic acid was added to the plates and incubated at concentrations of 2, 6, and 10 mM. After changing to fresh complete medium, 10 µl of CCK-8 reagent was added to each well and incubated at 37 °C for 2 h. A spectrophotometer (M2, MOLECULAR, USA) was used to measure the optical density (OD) values at a wavelength of 450 nm at 1, 2, 3, and 4 days.
EdU staining assay
Cell proliferation activity was measured using an EdU555 assay kit (C0075S, Beyotime). NPCs were seeded into 6-well plates at a density of 2 × 105 cells per well. After treatment with different concentrations of lactic acid, 2 ml of 10 µM EdU reagent was added to each well of NPCs and incubated for 2 h. The cells were fixed at room temperature for 15 min with 2 ml of 4% paraformaldehyde. Two milliliters of immunostaining strong permeability solution (P0097, Beyotime) were added to each well and incubated for 15 min. Two milliliters of click additive solution were added to each well and incubated for 30 min at room temperature (in the dark). The nuclei were stained with Hoechst 33,258 solution (C0021, Solarbio, Beijing, China). Immunofluorescence staining images were captured by a fluorescence microscope (IX73, Olympus, Tokyo, Japan).
Cell cycle assay
The effect of lactic acid on the cell cycle of NPCs was evaluated using PI/RNase staining buffer (550,825, BD) as well as a FITC-labeled Ki67 antibody (11,882 S, CST). Briefly, NPCs were seeded into T25 cell culture flasks at a density of 4 × 105 cells. The NPCs were treated with different concentrations of lactic acid when they reached 70% confluence. The cells were then fixed in precooled 70% ethanol for 24 h at -20 ℃ after collection in centrifuge tubes and centrifuged at 400×g for 5 min at 4 °C. The supernatants were discarded, and the cells were mixed with Ki67 (1:50 dilution in PBS). After 1 h of incubation in the dark, the cells were incubated with PI/RNase staining buffer for 15 min. Cell cycle progression was analysed by flow cytometry.
Senescence-associated β-galactosidase (SA-β-gal) staining assay
A SA-β-gal Staining kit (C0602, Beyotime) was used to assess the effect of lactic acid on NPCs senescence. The cells were seeded into 6-well plates at a density of 2 × 105 cells per well. The NPCs were treated with different concentrations of lactic acid, and 1 ml of SA-β-gal was added to the plates and incubated overnight in a CO2-free environment at 37 °C. Five fields were randomly captured to calculate the number of SA-β-gal-positive NPCs.
Reactive oxygen species (ROS) level assay
The ROS content in NPCs was measured using an ROS assay kit (CA1410, Solarbio). NPCs were seeded into 6-well plates at a density of 2 × 105 cells per well, and the NPCs were treated with different concentrations of lactic acid. Two milliliters of 10 µM DCFH-DA probe (diluted in serum-free medium) were added to each well and incubated at 37 °C for 20 min in the dark. The unbound probe was removed with PBS, the cells were harvested and resuspended in PBS in the dark, and the ROS content was measured by flow cytometry.
Measurement of mitochondrial membrane potential (MMP)
MitoTracker Red CMXRos (C1049B, Beyotime), a mitochondrial red fluorescent probe, is a cell permeable X-rosamine derivative that can specifically label bioactive mitochondria in cells and detect MMP [36]. We used it to measure the MMP to assess mitochondrial function in NPCs. Cells were seeded in confocal dishes and treated with different concentrations of lactic acid. Then 1 ml of MitoTracker Red CMXRos working solution was added to each dish and incubated at 37 °C for 30 min in the dark. The nuclei were stained with Hoechst 33,258 (1:1000, diluted in PBS). The fluorescence intensity in the different groups was observed using a laser confocal microscope (LSM880).
Malondialdehyde (MDA) measurement
MDA, a reactive aldehyde that is a byproduct of lipid peroxidation caused by ROS, is considered a marker of oxidative stress [37]. An MDA assay kit (S0121, Beyotime) was used to measure the MDA content in NPCs. Lysis buffer was added to the cell samples, and the samples were centrifuged at 12,000 × g for 10 min to extract cellular proteins. TBA detection solution was added to the supernatants, and then, the absorbance at 532 nm was measured using a spectrophotometer. The MDA content of each sample was calculated according to the standard curve, and then, the protein content of the sample was determined by the BCA method. All the experiments were normalized by the protein content of each sample.
Transmission electron microscopy (TEM)
NPCs treated with different concentrations of lactic acid were digested with trypsin and collected in a centrifuge tube. TEM fixative solution (G1102, Servicebio, Wuhan China) was added to the samples. The cell pellets were isolated from the bottom of the centrifuge tubes, resuspended in fixative solution and then fixed at 4 °C in the dark for 24 h. The samples were dehydrated with gradient concentrations of acetone, and osmotic embedding medium was added. The samples were cut into sections with a thickness of 60 nm and then stained with 2% uranium acetate saturated alcohol solution and 2.6% lead citrate. The stained sections were observed under a TEM (HT7800, Hitachi, Tokyo).
RNA sequencing and data analysis
NPCs were harvested after 4 days of treatment with lactic acid at a concentration of 10 mM. RNA sequencing analysis was performed by Bioguoke Biotechnology Co., Ltd. (Beijing, China). Total RNA was extracted from the samples on ice using TRIzol (15,596,018, Thermo Fisher, Wilmington, DE). The RNA concentration and purity were measured using a NanoDrop 2000 (Thermo Fisher). RNA integrity was assessed using the RNA nano 6000 assay kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). A cDNA library was constructed, and the library quality was assessed using the Agilent Bioanalyzer 2100 system. The Illumina NovaSeq platform was used for RNA sequencing. Clean data were obtained by removing reads containing adapters, poly-N and low-quality reads from the raw data. Transcripts were reconstructed using StringTie, and HISAT2 tools software was used to map the clean data to the reference Rattus norvegicus genome. Gene expression levels were estimated based on fragments per kilobase of transcript per million fragments mapped (FPKM). Differential expression analysis of the 2 groups was performed using the DESeq R package (1.10.1). The corrected p value (FDR < 0.05) and |log2foldchange| (|FPKM| ≥ 1) were used as the thresholds to identify significantly differential expression. KEGG pathway enrichment analysis was performed using KOBAS software (KOBAS, Surrey, UK).
Reverse transcription-quantitative PCR (RT-qPCR) analysis
After treatment with lactic acid (10 mM) for 4 days, NPCs were washed 3 times with PBS and then treated with TRIzol (9109, Takara, Japan) on ice to extract total RNA from cells. The quality and quantity of RNA were analysed by a NanoDrop spectrophotometer (ND-1000, Thermo Scientific, USA). According to the manufacturer’s protocols, a PrimeScript RT reagent kit (RR047A, Takara) was used for RNA reverse transcription, and a Premix Ex Taq II Kit (RR390A, Takara) was used for amplification and detection of the relative mRNA expression of target genes with the ViiA 7 RealTime PCR System (Thermo Scientific). The primer sequences are presented in Table S5. β-actin was used to normalize target gene mRNA and relative mRNA expression was calculated using the 2−ΔΔCt calculation method.
Akt kinase activity assay
Akt activity was measured using an Akt kinase activity assay kit (ab139436, Abcam, China). NPCs were lysed using cell lysis buffer supplemented with PMSF and phosphatase inhibitors, and the protein concentration of each sample was measured by the BCA method. The cell lysates were centrifuged at 12,000 × g for 10 min at 4 °C, and then, the supernatants were diluted to 30 µl according to the instructions. Each sample contained 2 mg of total protein. A spectrophotometer was used to measure the OD value at 450 nm, and then, the standard curve and Akt kinase activity for each sample were calculated. The data from all the samples were normalized using the Akt kinase activity of the control group (0 mM lactic acid).
Site-directed mutation of Akt and purification of mutant protein
The experimental design of site-directed mutation and subsequent lactic acid-Akt binding assay processes were referenced from previous studies [38]. The construction of mutant plasmids (AktWT, AktK39A, AktL52A and AktK39A, L52A) and shRNA and purification of mutant proteins were performed by Tsingke Biotech Co., Ltd. (Beijing, China). Akt-shRNA hairpins (Sequence: GGACGGGCACATCAAGATAAC) or Ctrl-shRNA was cloned and inserted into the lentiviral vector pLk0.1-U6-ShRNA-Puro. Lentiviral shRNA was produced by transfecting the shRNA, envelope, and packaging vectors into 293T cells. NPCs was transfected with lentiviral shRNA to obtain Akt-downregulated NPCs. Akt-downregulated NPCs were then transfected with AktWT, AktK39A, AktL52A, or AktK39A, L52A pGEX-4T2 mutant plasmids to achieve site-directed mutation of Akt. To achieve the purification of mutant Akt, the pGEX-4T2 plasmid expressing N-terminal GST-tagged AktWT, AktK39A, AktL52A, or AktK39A, L52A was transformed into BL21-CodonPlus (DE3)-RIL cells, and 300 µM isopropyl β-D-thiogalactopyranoside (IPTG) was added to induce protein production when the bacterial culture was grown to a 600 nm optical density (OD600) of approximately 0.6. Cells were lysed after centrifugation and the supernatant was collected. Purification of GST-tagged mutant proteins was performed after Ni-affinity chromatography, ion-exchange chromatography, and gel filtration. The GST tag was cut with PreScission Protease to obtain AktWT, AktK39A, AktL52A or AktK39A, L52A mutant proteins.
Microscale thermophoresis (MST) assay
The Kd value of the binding of lactic acid to different kinds of Akt was measured by Topscience Co. Ltd (Shanghai, China). Purified AktWT, AktK39A, AktL52A, and AktK39A,L52A were labeled with the NT-647-NHS (Thermo Fisher) fluorescent probe for 30 min at room temperature in the dark. Then, lactic acid was serially diluted in reaction buffer (50 mM HEPES buffer, pH = 7.4, containing 0.05% Tween 20). Next, 100 nM labeled Akt was added to the serial dilution. After incubation for 30 min at room temperature, the compounds were examined with a Monolith NT.115 instrument (NanoTemper). Experiments were performed in triplicate, and the Kd value was determined using Monolith software (NanoTemper).
Western blotting
Rat nucleus pulposus tissues or NPCs were lysed using RIPA lysis buffer (R0010, Solarbio) supplemented with 1% PMSF (P0100, Solarbio) and then centrifuged at 12,000 × g for 5 min at 4 °C. A bicinchoninic acid (BCA) assay kit (P0010s, Beyotime) was used to measure the protein concentration. Loading buffer (P1040, Solarbio) was added to the samples, and the samples were placed in a boiling water bath for 5 min. Sodium dodecyl sulfate‒polyacrylamide gel electrophoresis (SDS‒PAGE) technology was used to separate the proteins of different molecular weights in the samples, and then, the proteins were transferred to polyvinylidene fluoride membranes (PVDF; Merck, Germany). After blocking the membranes with QuickBlock Western blocking solution (P0252, Beyotime), antibodies were incubated with the membranes overnight at 4 °C. TBST was used to wash off the unbound antibodies, and the membranes were incubated with the secondary antibodies of the corresponding species. The bands were imaged using an ECL assay kit (1,705,060, Bio-Rad, CA, USA) and a ChemiDoc imaging system (Bio-Rad). ImageJ software was used to measure and analyse the gray value of the strip. The relative expression of the proteins was normalized to that of β-actin. The antibodies used in this experiment are shown in supplementary material Table S3.
Extraction of cytoplasmic and nuclear proteins
Proteins were extracted from NPCs using a nuclear and cytoplasmic protein extraction kit (P0028, Beyotime). A cell scraper was used to scrape the NPCs, which were then coincubated with PMSF-supplemented cytoplasmic protein extraction reagent A for 10 min on ice and coincubated with cytoplasmic protein extraction reagent B for 1 min on ice. The mixtures were centrifuged at 12,000 × g at 4 °C for 5 min. The supernatants were aspirated and considered the cytoplasmic protein. The precipitates were coincubated with PMSF-supplemented nuclear protein extraction reagent and collected in an ice bath for 30 min. The mixtures were centrifuged at 12,000 × g at 4 °C for 5 min. The supernatants were aspirated, and these supernatants were considered nuclear proteins.
Molecular docking
The two-dimensional structure of lactic acid was downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) [39] and saved in “SDF” format. Chem 3D was used to convert the “SDF” format to mol2 structures as small molecule ligands. The rat Akt (ID: P47196) protein structure was retrieved from the UniProt database (https://www.uniprot.org/) [40]. Water molecules were removed using PyMOL 2.3.6 software, and the original ligand was isolated from the core target protein. The processed protein targets were imported into AutoDock 4.2.0 software [41] for hydrogenation, calculation of total charge and setting of atom types. Molecular docking was performed with AutoDock-Vina1.1.2 software [42] to assess the affinity of the receptor‒ligand complexes, and a comprehensive score was provided. PyMOL was used to visualize the docking patterns as 3D structures. The docking effect was evaluated according to its affinity value.
Statistical analysis
The results are presented as the mean ± standard deviation (SD). Each experimental result was calculated from three biological replicates in technical triplicates. Statistical comparisons between groups were performed using unpaired Student’s t tests, and statistical comparisons among multiple groups were performed using one-way analysis of variance (ANOVA) or two-way repeated-measures ANOVA. p < 0.05 was considered statistically significant. GraphPad Prism 8.0 was used for statistical analysis and plotting histograms.
Results
Lactic acid accumulation in degenerated intervertebral discs
Nucleus pulposus tissues from patients with different Pfirrmann grades were collected and we found that the texture of nucleus pulposus tissue became coarse and lusterless in the severely degenerated intervertebral disc (Fig. 1A). HE staining showed that the amount of NPCs in the severely degenerated intervertebral disc was significantly reduced (Fig. 1B). The lactic acid content in nucleus pulposus tissues of patients with severe intervertebral disc degeneration (Pfirrmann IV/V) was significantly higher than that of patients with mild intervertebral disc degeneration (Pfirrmann II/III) (Fig. 1C). Furthermore, animal experiments were designed to investigate the relationship between IVDD and lactic acid content (Fig. 1D). The Pfirrmann grade of the caudal intervertebral discs of the PIDD group was significantly higher than that of the sham group (Fig. 1E-F), and the histological scores were significantly elevated compared with those of the normal group (Fig. 1G-H), indicating the establishment of experimental IVDD. During IVDD, there are degeneration and senescence-related anabolic and catabolic disorders of the extracellular matrix in nucleus pulposus tissue, manifested by the degradation of collagen II and accumulation of matrix metalloproteinases such as matrix metalloproteinases 9 (MMP9) and matrix metalloproteinases 13 (MMP13) [43,44,45]. Western blotting results showed that compared with the normal group, the expression levels of MMP9 and MMP13 were significantly increased, while the expression level of collagen II was significantly decreased in the PIDD group (Fig. 1I-J). The lactic acid levels in the PIDD discs were significantly higher than those in the normal group (Fig. 1K). Metabolomics analysis was performed in the NP tissues of normal rats and PIDD rats, and the differentially abundant metabolites identified under the condition of Log2FC > 1 (the logarithm of the difference fold was taken as base 2) are shown in Fig. 1L. The results showed that the lactic acid content was significantly increased in the PIDD group (Fig. 1M). The mass spectrum of the lactic acid content in each sample is shown in Fig. S1.

Lactic acid accumulation in degenerative discs. (A) Magnetic resonance imaging of patients with different Pfirrmann grades and the morphology of the nucleus pulposus tissue removed during surgery. (B) HE staining of nucleus pulposus tissues from people with different degrees of degeneration. Red arrows indicate human NPCs (scale bar = 500 μm). (C) Measurement of lactic acid content in human NP tissues with different degrees of degeneration using a lactic acid detection kit (n = 12). (D) Flow chart and timeline of in vivo experiments. (E-F) MRI detection and Pfirrmann grades of rats after different treatments (n = 9). (G-H) HE staining, SO&FG staining, and histological scores of the intervertebral disc in different groups (scale bar = 500 μm; n = 6). (I-J) Western blotting analysis showing collagen II, MMP9 and MMP13 expression in the nucleus pulposus of the normal and PIDD groups and quantification of the results. (K) Measurement of lactic acid content in rat NP tissues of normal rats and rats with PIDD using a lactic acid detection kit (n = 6). (L) Heatmap of differentially abundant metabolites in the NP tissues of normal rats and rats with PIDD. (M) Metabolomics detection of lactic acid content in the NP tissue of normal rats and rats with PIDD (n = 6). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
LOx overexpression can alleviate PIDD in rats
The flow chart of the in vivo experiment is shown in Fig. 2A. LOx is a bacterial enzyme that irreversibly converts lactic acid to pyruvate [46]. Immunofluorescence showed high expression of GFP in the LVV-control group and LVV-LOx group (Fig. S2A), and western blotting showed that LOx expression in the LVV-LOx group was significantly higher than that in the other groups (Fig. S2B), indicating that LVV-LOx had been successfully transfected into the cells in the intervertebral disc. The lactic acid content of the NP tissue in the LVV-LOx group was higher than that in the saline group (Fig. S2C), indicating the successful elimination of lactic acid in the intervertebral disc by LOx. The MRI results showed that the Pfirrmann grades of the LVV-LOx group were significantly lower than those of the saline groups (Fig. 2B-C). µCT results showed that the LVV-LOx group had a higher DHI% after treatment than the saline group (Fig. 2D-E). Histological staining showed that the structure of the annulus fibrosus in the saline group and the LVV-control group was disordered, and the NP tissue and the height of the intervertebral space were reduced; however, the structure of the annulus fibrosus in the LVV-LOx group was more well aligned, the NP was more complete, the height of the intervertebral space was improved and the histological score was significantly lower than that in the saline groups (Fig. 2F-G). Western blotting results showed that compared with those in the saline group, the expression levels of MMP9 and MMP13 were significantly decreased, and the expression level of collagen II was significantly increased in the LVV-LOx group (Fig. 2H-I).

Reduced degree of IVDD after eliminating lactic acid. (A) Flow chart and timeline of in vivo experiments. (B-C) MRI detection and Pfirrmann grades of rats after different treatments (n = 6). (D-E) Changes in the DHI% were evaluated by µCT detection (n = 6). (F-G) HE staining, SO&FG staining, and histological scores of the intervertebral disc in different groups (scale bar = 500 μm. n = 3). (H-I) Western blotting analysis showing collagen II, MMP9 and MMP13 expression in different groups and quantification of the results. *p < 0.05; **p < 0.01; ****p < 0.0001; ns represents no statistical significance
Lactic acid inhibits NPCs proliferation and promotes NPCs senescence
The intracellular lactic acid content in NPCs was measured after treatment with different concentrations of exogenous lactic acid (2, 6, or 10 mM). We found that the lactic acid content in NPCs gradually increased with increasing lactic acid concentration (Fig. 3A). The CCK8 experiment showed that the OD value decreased significantly after treatment with 6 mM and 10 mM lactic acid for 2 d and above (Fig. 3B). Lactic acid treatment for 4 days was selected as the experimental condition in the subsequent experiment. The EdU experiment showed that 6 mM and 10 mM lactic acid treatment could significantly reduce the percentage of EdU-positive NPCs compared to the control group (Fig. 3C-D). Flow cytometry results showed that the number of cells in the G0 phase was significantly increased in the 6 mM and 10 mM groups compared with the control group (Fig. 3E-F). γ-H2AX is located in cell nuclei and is considered to be a typical marker of DNA double-strand breaks [7, 47]. The γ-H2AX staining results showed that NPCs in the 6 mM and 10 mM groups exhibited more severe DNA damage (Fig. 3G-H). The SA-β-galactosidase assay results showed that the number of β-galactosidase-positive cells was significantly increased in the 6 mM and 10 mM groups compared to the control group (Fig. 3I-J). Western blotting results showed that compared with those in the control group, the expression levels of MMP9 and MMP13 were significantly decreased, and the expression level of collagen II was significantly increased in the 6 mM and 10 mM lactic acid-treated groups (Fig. 3K-L).

High concentrations of lactic acid inhibited NPCs proliferation. (A) Measurement of lactic acid content in NPCs using a lactic acid detection kit (n = 3). (B) The effect of different concentrations or treatment times of lactic acid on the proliferation of NPCs as determined by CCK-8 assay (n = 3). (C-D) The effect of different concentrations of lactic acid on the proliferation of NPCs as determined by EdU staining. Proliferating NPCs appeare red, and nuclei were counterstained with Hoechst 33,258 (blue) (scale bar = 100 μm. n = 3). (E-F) The effect of different concentrations of lactic acid on the cell cycle of NPCs; the box positions represent negative controls (n = 3). (G-H) The effect of different concentrations of lactic acid on DNA double stranded as determined by γ-H2AX; DNA double-stranded-damaged cells appear red, and nuclei were counterstained with DAPI (blue) (scale bar = 50 μm. n = 3). (I-J) The effect of different concentrations of lactic acid on the NPCs senescence as determined by SA-β-gal assay; senescent cells appear blue (scale bar = 200 μm. n = 3). (K-L) Western blotting analysis showing the effect of different lactic acid concentrations on collagen II, MMP9 and MMP13 expression and quantification of the results. La represents lactic acid; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns represents no statistical significance
Furthermore, LOx was successfully expressed in NPCs (Fig. S3A-B). When exogenous lactic acid was added, LOx overexpression effectively reduced the lactic acid content in NPCs (Fig. S3C). Compared to the LVV-control group, LOx overexpression could significantly increase the percentage of EdU-positive NPCs (Fig. S3D-E). In addition, the number of cells in the G0 phase was significantly decreased in the LOx overexpression groups (Fig. S3F-G). Furthermore, the number of β-galactosidase-positive cells was significantly decreased in the LOx-overexpression groups compared to the LVV-control group (Fig. S3H-I).
Lactic acid promotes the accumulation of ROS and mitochondrial damage in NPCs
The DCFH-DA probe results showed that compared with the control-treated NPCs, 2 mM, 6 mM, and 10 mM lactic acid-treated NPCs exhibited significantly increased intracellular ROS levels (Fig. 4A-B). The CMXRos probe test showed that 6 mM and 10 mM lactic acid caused a significant decrease in the MMP of NPCs compared with the control group (Fig. 4C-D). Intracellular MDA measurement results showed that compared with the control-treated cells, 2 mM, 6 mM, and 10 mM lactic acid-treated NPCs exhibited significantly elevated intracellular MDA (Fig. 4E). TEM observation results showed that lactic acid can lead to increased mitochondrial volume and decreased mitochondria number in NPCs, as well as the enhanced disorder of mitochondrial structure and the formation of vacuoles (Fig. 4F). LOx overexpression also reduced intracellular ROS levels in NPCs compared to the LVV-control group (Fig. S3J-K); LOx overexpression caused a significant increase in the MMP of NPCs compared to the LVV-control group (Fig. S3L-M).

High concentrations of lactic acid damage mitochondria and induce oxidative stress in NPCs. (A-B) The effect of different concentrations of lactic acid on ROS production in NPCs. The FL1 subset was set according to the ROS intensity of the negative control (n = 3). (C-D) The effect of different concentrations of lactic acid on the MMP of NPCs as determined by the CMXRos probe (red); nuclei were counterstained with Hoechst 33,258 (blue) (scale bar = 50 μm. n = 3). (E) The effect of different concentrations of lactic acid on the MDA content of NPCs (n = 3). (F) The effect of different concentrations of lactic acid on the mitochondrial morphology of NPCs detected by TEM. **p < 0.01; ***p < 0.001; ****p < 0.0001
Transcriptomics revealed that lactic acid affects the function of NPCs by inhibiting the PI3K/Akt signaling pathway
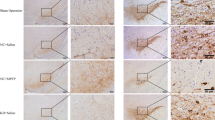
High-throughput sequencing was performed in 3 samples of 10 mM lactic acid-treated NPCs and 3 samples of control NPCs to further explore the underlying mechanisms by which lactic acid affects NPCs senescence and oxidative stress. A total of 2674 genes were upregulated in NPCs treated with lactic acid, and 3685 genes were downregulated (Fig. 5A). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that the differentially expressed genes were significantly enriched in the PI3K-Akt pathway (Fig. 5B). The RT-qPCR results showed that the mRNA expression levels of 8 major genes in the PI3K-Akt pathway in NPCs were downregulated after treatment with lactic acid (Fig. S4). Western blotting results verified that the levels of phosphorylated Akt were significantly reduced after 6 mM or 10 mM lactic acid treatment of NPCs (Fig. 5C-E). In vivo tests demonstrated that p-Akt (Thr308) and p-Akt (Ser473) expression was decreased in the NP tissue of the saline group and the LVV-control group compared with the LVV-LOx group (Fig. 5F-H).

High concentrations of lactic acid affected the phosphorylation of Akt in NPCs. (A) Volcano map of differentially expressed genes in NPCs after treatment with lactic acid. (B) KEGG pathway analysis of differentially expressed genes in NPCs after treatment with lactic acid. (C-E) Western blotting analysis showing the effect of different lactic acid concentrations on p-Akt (Thr308), and p-Akt (Ser473) expression and quantification of the results (n = 3). (F-G) Immunofluorescence staining of p-Akt (Thr308) and p-Akt (Ser473) in NP tissues in different groups (n = 3). **p < 0.01; ****p < 0.0001
Lactic acid inhibits akt kinase activity by binding to the PH domain
The addition of lactic acid (6 mM and 10 mM) during NPCs culture significantly inhibited the activity of intracellular Akt kinase (Fig. 6A). Furthermore, as predicted by molecular docking, lactic acid may bind to the Lys39 and Leu52 residues, which are located in the PH domain of Akt, thus curtailing Akt phosphorylation at Thr308 and Ser473 and corresponding Akt kinase activity (Fig. 6B-D). After knocking down Akt with shRNA, plasmids AktWT, AktK39A, AktL52A, and AktK39A,L52A were transfected into NPCs respectively (Fig. 6E). The Akt kinase activity assay showed that mutations at these sites had no significant effect on Akt enzyme activity compared with the control group (Fig. 6F). Both the AktK39A, AktL52A, and AktK39A,L52A mutations significantly reduced the proliferation rate of lactic acid in NPCs (Fig. 6G-H). Meanwhile, β-gal positive cells were decreased in the AktK39A, AktL52A, and AktK39A,L52A groups (Fig. 6I-J). In addition, the AktK39A, AktL52A, and AktK39A,L52A groups exhibited decreased intracellular ROS levels (Fig. 6K-L). Furthermore, the expression levels of MMP9 and MMP13 were decreased, and the expression level of collagen II was enhanced in the AktK39A, AktL52A, AktK39A,L52A group (Fig. 6M-Q). Simultaneous AktK39A,L52A mutations resulted in better beneficial effects than single AktK39A or AktL52A mutations, although lactic acid-induced oxidative stress and senescence in AktK39A,L52A mutated NPCs were not reversed to the normal degree. Solutions containing lactic acid and different kinds of Akt proteins were analysed using MST. We found that simultaneous AktK39A,L52A mutations nearly completely disrupted the interaction of lactic acid with Akt, and individual AktK39A or AktL52A mutations also partly hindered the interaction of lactic acid with Akt. (Fig. 6Q-T).

Lactic acid regulates the function of NPCs by binding to specific sites of Akt. (A) Detection of Akt kinase activity in NPCs after treatment with different concentrations of lactic acid (n = 3). (B) Domain structure of Akt. (C-D) Molecular docking diagram of lactic acid and Akt. (E) Western blotting analysis showed Akt expression after knocking down Akt with shRNA and transfecting AktWT, AktK39A, AktL52A and AktK39A,L52A into NPCs respectively. (F) Detection of Akt kinase activity in NPCs of different groups (n = 3). (G-H) The effect of site mutation of Akt on the proliferation of NPCs (red) as determined by EdU staining. Nuclei were counterstained with Hoechst 33,258 (blue) (scale bar = 100 μm. n = 3). (I-J) The effect of lactic acid (10 mM) on the senescence of Akt site-mutated NPCs (blue) as determined by SA-β-gal assay (scale bar = 200 μm. n = 3). (K-L) The effect of lactic acid (10 mM) on ROS production of Akt site-mutated NPCs as determined by a DCFH-DA probe. The FL1 subset was set according to the ROS intensity of the negative control (n = 3). (M-P) Western blotting analysis showing the effect of lactic acid (10 mM) on collagen II, MMP9 and MMP13 expression in Akt site-mutated NPCs and quantification of the results (n = 3). (Q-T) MST analysis of lactic acid in the interaction of NT-647-NHS-labeled Akt. Kd represents the dissociation constant. La represents lactic acid; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns represents no statistical significance
Lactic acid affects the proliferation and senescence of NPCs by inhibiting the Akt/p21/p27/cyclin D1 pathway
Western blotting results showed that after lactic acid treatment, the expression levels of p21 and p27 were significantly increased, which in turn inhibited the downstream expression of cyclin D1 (Fig. 7A-D), while the Akt activator SC79 reversed this process (Fig. 7E-H). EdU staining showed that cotreatment of lactic acid-treated NPCs with SC79 reversed the detrimental effects of lactic acid on cell proliferation (Fig. 7I-J). Cotreatment with SC79 also significantly reduced the number of cells in the G0 phase (Fig. 7K-L) and the number of β-galactosidase-positive cells (Fig. 7M-N).

High concentrations of lactic acid inhibited the proliferation of NPCs via the Akt/p21/p27/cyclin D1 pathway. (A-D) Western blotting analysis showing the effect of different lactic acid concentrations on p21, p27, and cyclin D1 expression and quantification of the results (n = 3). (E-H) Western blotting analysis showing the effect of lactic acid and/or SC79 on p21, p27, and cyclin D1 expression and quantification of the results (n = 3). (I-J) The effect of lactic acid and/or SC79 on the proliferation of NPCs as measured by EdU staining. Proliferating NPCs appear red, and nuclei were counterstained with Hoechst 33,258 (blue) (scale bar = 100 μm. n = 3). (K-L) The effect of lactic acid and/or SC79 on the cell cycle of NPCs; the box positions represent negative controls (n = 3). (M-N) The effect of lactic acid and/or SC79 on the senescence of NPCs as determined by SA-β-gal assay. The senescent cells appeared blue (scale bar = 200 μm. n = 3). **p < 0.01; ***p < 0.001; ****p < 0.0001
Lactic acid promotes ROS accumulation and mitochondrial damage by inhibiting the Akt/Nrf2/HO-1 pathway
Western blotting results showed that lactic acid reduced the content of Nrf2 in the nucleus (n-Nrf2), the content of Nrf2 in the cytoplasm (c-Nrf2), and the expression of HO-1 (Fig. 8A-D), while SC79 reversed these effects (Fig. 8E-H). Immunofluorescence staining showed that cotreatment with SC79 enhanced the fluorescence intensity of Nrf2 in the nuclei of lactic acid-treated NPCs (Fig. 8I-J). Cotreatment with SC79 also reduced the content of intracellular ROS (Fig. 8K-L) and intracellular MDA content (Fig. 8M). The CMXRos probe staining results showed that SC79 cotreatment significantly increased the intracellular mean fluorescence intensity (Fig. 8N-O). The TEM results showed that SC79 rescued the mitochondrial damage caused by lactic acid in nucleus pulposus cells (Fig. 8P). A schematic of the comprehensive mechanisms considered in the current study is shown in Fig. 9.

High concentrations of lactic acid promote oxidative stress in NPCs via the Akt/Nrf2/HO-1 pathway. (A-D) Western blotting analysis showing the effect of different lactic acid concentrations on Nrf2 in the nucleus (n-Nrf2), Nrf2 in the cytoplasm (c-Nrf2), and HO-1 expression and quantification of the results (n = 3). (E-H) Western blotting analysis showing the effect of lactic acid and/or SC79 on n-Nrf2, c-Nrf2, and HO-1 expression and quantification of the results (n = 3). (I-J) The effect of lactic acid and/or SC79 on the Nrf2 nuclear translocation of NPCs as determined by immunofluorescence. Nrf2 in NPCs appeared red, and nuclei were counterstained with DAPI (blue) (n = 3). (K-L) The effect of lactic acid and/or SC79 on the ROS content of NPCs as determined by flow cytometry. The FL1 subset was set according to the ROS intensity of the negative control (n = 3). (M) The effect of lactic acid and/or SC79 on the MDA content of NPCs as determined by flow cytometry (n = 3). (N-O) The effect of lactic acid and/or SC79 on the MMP of NPCs as determined by the CMXRos probe (red); nuclei were counterstained with Hoechst 33,258 (blue) (scale bar = 50 μm. n = 3). (P) The effect of lactic acid and/or SC79 on the mitochondrial morphology of NPCs detected by TEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001

Schematic diagram of the signaling pathways involved in the promotion of senescence and oxidative stress in NPCs by and corresponding IVDD by lactic acid
Discussion
A high concentration of lactic acid is a hallmark and cause of IVDD. However, the underlying mechanisms by which lactic acid promotes IVDD are not clear. In this study, we explored the role of high concentrations of lactic acid in NPCs senescence and oxidative stress and investigated the potential mechanisms. There are several reasons that may contribute to lactic acid accumulation during IVDD. First, glycolysis in NPCs may be enhanced in degenerated intervertebral discs, elevating the production of lactic acid [48]. In addition, reduced lactic acid efflux by impaired monocarboxylate transporters 4 expression can result in inadequate lactic acid clearance during IVDD [11]. In addition, cartilage endplate calcification and degeneration result in decreased permeability, which may diminish oxygen and nutrient concentrations and elevate the accumulation of lactic acid in degenerated intervertebral discs [49]. We found that the concentration of lactic acid significantly increased in the degenerative intervertebral discs of humans and rats with PIDD. A high concentration of lactic acid promoted primary rat NPCs senescence and oxidative stress. Eliminating the accumulation of lactic acid in the rat intervertebral disc alleviated the progression of IVDD. Reducing the lactic acid content in NPCs by LOx overexpression could reduce cellular senescence and oxidative stress in NPCs. High concentrations of lactic acid induced NPCs senescence and oxidative stress by inhibiting the PI3K/Akt pathway via binding to Lys39 and Leu52 residues in the PH domain of Akt and then suppressed downstream Akt/p21/p27/cyclin D1 signaling and Akt/Nrf2/HO-1 signaling. This study reveals the role of lactic acid in NPCs senescence and oxidative stress during IVDD and may facilitate the effects of current IVDD therapies.
IVDD, which is a common degenerative disease, is characterized by early onset and severe conditions compared with the degenerative diseases that occur in other organs and tissues. This is associated with the earlier senescence and oxidative stress of intervertebral disc cells, especially NPCs. Anatomical and metabolic peculiarities of these cells contribute significantly to this susceptibility [53, 54]. The intervertebral disc is the largest avascular tissue in the whole body, and it has poor oxygen and nutrition supply. Previous research verified that many metabolites, including amino acids, nucleosides, and bioenergetic metabolites, accumulated in the intervertebral disc [5, 55]. We then performed metabolomics analysis to compare the discrepancy between normal and degenerative discs. We found that lactic acid, pyruvate, isocitric acid, α-glutanic acid, UMP, guanine, and UDP-GlcNAc were the distinctly upregulated metabolites in degenerated intervertebral discs. Among these metabolites, pyruvic acid [56], α-glutanic acid [57, 58], UMP [59], and UDP-GlcNAc [60] have all been reported to alleviate cellular senescence or oxidative stress, whereas lactic acid has been proven to accelerate cellular senescence and oxidative stress [25, 26, 29, 30]. Moreover, oxidative stress is considered an important cause of IVDD [61]. Therefore, we hypothesized that lactic acid is a factor that promotes NPCs senescence and oxidative stress and thus participates in IVDD progression.
We then treated NPCs with a series of lactic acid concentrations and evaluated the changes in the biological function of NPCs using a series of cellular senescence and oxidative stress assessment assays. We proved that a high concentration of lactic acid inhibits cell proliferation and promotes NPCs senescence. Moreover, a high concentration of lactic acid promotes the accumulation of ROS and mitochondrial damage in NPCs. These results are similar to previous reports that a high concentration of lactic acid inhibits proliferation and exacerbates ROS-related oxidative stress damage in chondrocytes [62, 63], which are cells that have characteristics that are analogous to those of NPCs. In our experiments, the time point for evaluating the senescence-related effects of lactic acid on NPCs was 4 days, so some of the altered phenotypes, such as p21, EdU, and Ki67, may be partly attributed to the acute response of NPCs to lactic acid apart from the direct induction of senescence, which may require more time to establish detrimental effects after sublethal oxidative stress [64]. Further long-term evaluation of the senescence-related effects of lactic acid on NPCs will be explored in future research.
We then evaluated whether eliminating lactic acid accumulation in the intervertebral disc can alleviate IVDD. There are many enzymes involved in lactic acid metabolism in mammalian cells, the most important enzymes are lactate dehydrogenase (LDH). LDH catalyses a redox reaction, the reversible conversion between pyruvate and lactic acid [50]. However, LDH is part of an orchestrated regulation network of glycolysis and oxidative phosphorylation, and alterations of LDH may bring in uncertain implications. Lox is a bacterial enzyme that irreversibly converts lactic acid to pyruvate and subsequently reduces the lactic acid content, which may have mild implications on glycolysis and oxidative phosphorylation. LOx has been proven to effectively reduce the content of lactic acid in tumor, brain, and intervertebral disc to treat relevant diseases [46, 51, 52, 65]. Therefore, we utilized a lentivirus-mediated LOx overexpression system to treat cultured NPCs and rats PIDD. We found that eliminating the accumulation of lactic acid significantly mitigated NPCs senescence, oxidative stress, and degenerative alterations. These results verified that lactic acid promotes NPCs senescence and oxidative stress to aggravate IVDD. In addition, LOx can catalyze lactic acid to generate pyruvate, which has previously been reported to have certain antioxidant effects [66,67,68]. Therefore, although the alleviation of cellular senescence and oxidative stress are attributed to the LOx-mediated elimination of lactic acid, there exists the possibility that LOx-mediated elevation of pyruvate may bring in some beneficial effects, which need further exploration in the future.
To determine the potential mechanism by which high concentrations of lactic acid stimulate NPCs senescence and oxidative stress, RNA-seq and bioinformatics analysis were performed. We found that the PI3K/Akt pathway was the most highly enriched signaling pathway, which may underlie lactic acid-induced NPCs senescence and oxidative stress. The PI3K/Akt pathway is a crucial signaling pathway in regulating cellular biological processes, including senescence, apoptosis, cell proliferation, and oxidative stress resistance [69,70,71,72]. Activating the PI3K/Akt signaling pathway exerts protective effects in IVDD [73, 74], and PI3K/Akt activation can ameliorate NPCs senescence and oxidative stress [75,76,77]. Subsequent Western blotting experiments demonstrated that lactic acid inhibits the PI3K/Akt pathway by suppressing the phosphorylation of Akt at Thr308 and Ser473, which are two common phosphorylation sites of Akt [78, 79]. This evidence proved that a high concentration of lactic acid contributes to NPCs senescence and oxidative stress by suppressing PI3K/Akt signaling.
Finally, we explored the molecular mechanism by which lactic acid regulates the PI3K/Akt pathway. Previous studies have shown that lactic acid can regulate NPCs cellular processes by directly entering NPCs through monocarboxylate transporters [80, 81] or indirectly creating an acidic microenvironment and affecting NPCs through acid-sensing ion channels [20]. Two recent studies have proven that lactic acid can directly interact with intracellular proteins, including SENP1 and PHD2, to regulate biological processes [82, 83]. We then surmised that the inhibition of the PI3K/Akt pathway by lactic acid may be associated with lactic acid-protein interactions. Using molecular docking, we found that two residues (Lys39 and Leu52) in the PH domain of Akt can form hydrogen bonds with lactic acid. The modular structure of the Akt protein is composed of the PH domain at the N-terminus, the kinase catalytic domain in the middle and the AGC-kinase C-terminal domain at the C-terminus [84]. The activation of Akt commonly requires membrane translocation and subsequent phosphorylation by the kinase recruited to the membrane [85, 86]. The PH domain has affinity for PI(3,4,5)P3, which is a second messenger of the PI3K/Akt pathway. PI(3,4,5)P3 binds to the PH domain, which is indispensable for the plasma membrane targeting of Akt, resulting in the phosphoactivation of Akt by PDK1 [87, 88]. Several small molecule compounds have been proven to inactivate Akt by binding to the PH domain [89,90,91]. For Lys39 and Leu52, it has been reported that genipin binds to Leu52 in the PH domain and inhibits Akt activation [92]. Moreover, Abrus Agglutinin can bind to Lys39 in the PH domain to inactivate Akt [93].
To verify the function of Lys39 and Leu52 in the interaction of lactic acid and Akt, site-directed mutation was implemented. Further MST results revealed that both Lys39 and Leu52 are crucial in the interaction of lactic acid with Akt. The simultaneous site-directed mutation of Lys39 and Leu52 nearly destroys the binding of lactic acid to Akt, while the single mutation of Lys39 or Leu52 partly hampers the binding of lactic acid to Akt. This evidence indicates that lactic acid may suppress Akt phosphoactivation by binding to Lys39 and Leu52 in the PH domain of Akt. The effects of lactic acid on oxidative stress and senescence were decreased after simultaneous Lys39 and Leu52 mutation or single mutation. We also noticed that even after simultaneous Lys39 and Leu52 mutation, lactic acid-induced oxidative stress and senescence were not reversed to the normal degree, when the interaction of lactic acid with Akt was nearly totally destroyed. We surmised that Akt may have other unrevealed binding sites with lactic acid, and there exists a certain possibility that other signaling pathways may contribute to lactic acid-induced cellular senescence and oxidative stress apart from the Akt pathway. Other potential underlying mechanisms and signaling pathways warrant further exploration.
Akt can regulate various downstream pathways to mediate biological effects. Western blotting experiments proved that lactic acid may affect the proliferation and senescence of NPCs by inhibiting the Akt/p21/p27/cyclin D1 pathway. Treating NPCs with the Akt-specific activator SC79 distinctly elevated the expression levels of p21, p27 and cyclin D1 in lactic acid-treated NPCs and alleviated lactic acid-induced NPCs senescence. p21, p27, and cyclin D1 are crucial cell cycle regulators [94], and the Akt/p21/p27/cyclin D1 pathway has been reported to be a positive regulator of cell proliferation and a negative regulator of senescence [95, 96], which is similar to our results. Akt can phosphorylate and inhibit GSK3β to reduce P21 and alleviate cellular senescence [97]. In addition, synergistic Akt and Erk action can suppress P21 expression and reduce cellular senescence [98]. For P27, Akt can suppress P27 expression and alleviate senescence by FOXO3/P27 or P53/P27 signaling [99, 100]. Akt can also downregulate P27 by increasing the expression of the p27 ubiquitin mediator Skp2 [101]. Western blotting and immunofluorescence results proved that lactic acid may promote ROS accumulation and mitochondrial damage by inhibiting the Akt/Nrf2/HO-1 pathway. Treating NPCs with SC79 distinctly elevated the expression levels of Nrf2 and HO-1 and Nrf2 nuclear translocation in lactic acid-treated NPCs and alleviated ROS accumulation and mitochondrial damage. Nrf2 and HO-1 are well-known crucial antioxidant proteins in the body [102, 103]. The Akt/Nrf2/HO-1 pathway has been reported to be associated with oxidative stress defense and mitochondrial protection [33, 104, 105], which partly verified our results. Nrf2 can be regulated by the Akt/GSK3β/Nrf2 pathway to alleviate cellular senescence [106, 107]. In addition, Akt/Erk signaling activation can enhance the expression of Nrf2 and reduce cellular senescence [108]. Therefore, a high concentration of lactic acid may promote NPCs senescence by suppressing the Akt/p21/p27/cyclin D1 pathway and promote NPCs oxidative stress by suppressing the Akt/Nrf2/HO-1 pathway. The concrete regulatory networks between Akt and P21, P27, and Nrf2 in lactic acid-treated NPCs deserve further exploration in the future.
Taken together, our findings prove that lactic acid accumulates in degenerative intervertebral discs. High concentrations of lactic acid inhibit NPCs proliferation, promote ROS accumulation and mitochondrial damage and accelerate NPCs senescence and oxidative stress to stimulate IVDD. These alterations are mediated by inhibiting PI3K/Akt signaling and the downstream Akt/p21/p27/cyclin D1 and Akt/Nrf2/HO-1 pathways. Mechanistically, lactic acid may suppress Akt phosphoactivation by binding to Lys39 and Leu52 in the PH domain of Akt. Research that explores potential therapeutics that target lactic acid in the treatment of IVDD is worth further investigation in the future.
Conclusions
Lactic acid accumulation is associated with the progression of IVDD. Lactic acid promotes NPCs senescence by binding to the PH domain of Akt and inhibiting PI3K/Akt signaling and the downstream Akt/p21/p27/cyclin D1 and Akt/Nrf2/HO-1 pathways. This study is the first to explore the effects of lactic acid on NPCs senescence as well as the underlying mechanism, which may provide a new strategy that targets lactic acid in IVDD treatment.
Data availability
The whole original Western blotting bands demonstrating the molecular weight of the targeted protein in the adjacent “protein ladder” are shown in Fig. S5. The metabolomics data used to support the findings of this study have been deposited in the Metabolights (https://www.ebi.ac.uk/metabolights/) repository (MTBLS5880). The RNA sequencing data used to support the findings of this study has been deposited in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/) repository (GSE219145). The other datasets described and analysed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- IVDD:
-
Intervertebral disc degeneration
- NPCs:
-
Nucleus pulposus cell
- PIDD:
-
Puncture-induced disc degeneration
- LOx:
-
Lactate oxidase
- RNA-seq:
-
RNA sequencing
- PH domain:
-
Pleckstrin homology domain
- NP:
-
Nucleus pulposus
- SD rat:
-
Sprague Dawley rat
- BMI:
-
Body mass index
- LVV:
-
Lentiviral vectors
- MRI:
-
Magnetic resonance imaging
- µCT:
-
Microcomputed tomography
- DHI:
-
Disc height index
- EDTA:
-
Ethylenediaminetetraacetic acid
- HE:
-
Haematoxylin-Eosin
- SO & FG:
-
Safranine O-Fast Green
- CCK-8:
-
Cell counting kit-8
- MMP9:
-
Matrix metalloproteinases 9
- MMP13:
-
Matrix metalloproteinases 13
- SA-β-gal:
-
Senescence-associated β-galactosidase
- MMP:
-
Mitochondrial membrane potential
- MDA:
-
Malondialdehyde
- TEM:
-
Transmission electron microscopy
- FPKM:
-
Fragments mapped
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MST:
-
Microscale thermophoresis
- BCA:
-
Bicinchoninic acid
- SDS‒PAGE:
-
Sodium dodecyl sulfate‒polyacrylamide gel electrophoresis
- PVDF:
-
Polyvinylidene fluoride membranes
- GFP:
-
Green fluorescent protein
- LDH:
-
lactate dehydrogenase
References
Andersson G (1999) Epidemiological features of chronic low-back pain. Lancet (London England) 354(9178):581–585. https://doi.org/10.1016/s0140-6736(99)01312-4
Wu A et al (2020) Global low back pain prevalence and years lived with disability from 1990 to 2017: estimates from the global burden of Disease Study 2017. Annals of Translational Medicine 8(6):299. https://doi.org/10.21037/atm.2020.02.175
Urban J, Smith S, Fairbank J (2004) Nutrition of the intervertebral disc, Spine, 29(23):2700-9, https://doi.org/10.1097/01.brs.0000146499.97948.52
Holm S, Maroudas A, Urban J, Selstam G, Nachemson A (1981) Nutrition of the intervertebral disc: solute transport and metabolism, Connective tissue research, 8(2):101–19, https://doi.org/10.3109/03008208109152130
Radek M et al (2016) Assessing the correlation between the degree of disc degeneration on the Pfirrmann scale and the metabolites identified in HR-MAS NMR spectroscopy, Magnetic resonance imaging, 34(4):376–80, https://doi.org/10.1016/j.mri.2015.12.005
Zhao K et al (2021) Acid-sensing ion channels regulate nucleus pulposus cell inflammation and pyroptosis via the NLRP3 inflammasome in intervertebral disc degeneration, (in eng), Cell proliferation, 54(1):e12941, https://doi.org/10.1111/cpr.12941
Xu X et al (2019) Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane, (in eng), Theranostics, 9(8):2252–2267, https://doi.org/10.7150/thno.30658
Song Y et al (Oct 2018) Sirtuin 3-dependent mitochondrial redox homeostasis protects against AGEs-induced intervertebral disc degeneration, (in eng). Redox Biol 19:339–353. https://doi.org/10.1016/j.redox.2018.09.006
Bibby S, Jones D, Ripley R, Urban J (2005) Metabolism of the intervertebral disc: effects of low levels of oxygen, glucose, and pH on rates of energy metabolism of bovine nucleus pulposus cells, Spine, 30(5):487–96, https://doi.org/10.1097/01.brs.0000154619.38122.47
Soukane D, Shirazi-Adl A, Urban J (2007) Computation of coupled diffusion of oxygen, glucose and lactic acid in an intervertebral disc. J Biomech 40(12):2645–2654. https://doi.org/10.1016/j.jbiomech.2007.01.003
Silagi E et al (2020) Lactate Efflux from intervertebral disc cells is Required for Maintenance of Spine Health. J bone Mineral Research: Official J Am Soc Bone Mineral Res 35(3):550–570. https://doi.org/10.1002/jbmr.3908
Silagi E, Schipani E, Shapiro I, Risbud M (2021) The role of HIF proteins in maintaining the metabolic health of the intervertebral disc. Nat Rev Rheumatol 17(7):426–439. https://doi.org/10.1038/s41584-021-00621-2
Shi J, Zhou X, Wang Z, Kurra S, Niu J, Yang H (2019) Increased lactic acid content associated with extracellular matrix depletion in a porcine disc degeneration induced by superficial annular lesion. BMC Musculoskelet Disord 20(1):551. https://doi.org/10.1186/s12891-019-2937-x
Huang C, Gu W (2008) Effects of mechanical compression on metabolism and distribution of oxygen and lactate in intervertebral disc. J Biomech 41(6):1184–1196. https://doi.org/10.1016/j.jbiomech.2008.02.002
Bartels E, Fairbank J, Winlove C, Urban J (1998) Oxygen and lactate concentrations measured in vivo in the intervertebral discs of patients with scoliosis and back pain, Spine, 23(1):1–7; discussion 8, https://doi.org/10.1097/00007632-199801010-00001
Jünger S, Gantenbein-Ritter B, Lezuo P, Alini M, Ferguson S, Ito K (2009) Effect of limited nutrition on in situ intervertebral disc cells under simulated-physiological loading, Spine, 34(12):1264-71, https://doi.org/10.1097/BRS.0b013e3181a0193d
Wang Z et al (2022) Inhibition of aberrant Hif1α activation delays intervertebral disc degeneration in adult mice, (in eng), Bone Res, 10(1):2, Jan 5 https://doi.org/10.1038/s41413-021-00165-x
Malandrino A, Noailly J, Lacroix D (2011) The effect of sustained compression on oxygen metabolic transport in the intervertebral disc decreases with degenerative changes, (in eng), PLoS computational biology, 7(8):e1002112, https://doi.org/10.1371/journal.pcbi.1002112
Shen J et al (2022) Exhausted local lactate accumulation via injectable nanozyme-functionalized hydrogel microsphere for inflammation relief and tissue regeneration. Bioactive Mater 12:153–168. https://doi.org/10.1016/j.bioactmat.2021.10.013
Zhao K et al (2021) Acid-sensing ion channels regulate nucleus pulposus cell inflammation and pyroptosis via the NLRP3 inflammasome in intervertebral disc degeneration. Cell Prolif 54(1):e12941. https://doi.org/10.1111/cpr.12941
Wang C, Hsieh M, Hu Y, Bit A, Lai P (2022) Monocarboxylate transporter 1-mediated lactate accumulation promotes nucleus pulposus degeneration under hypoxia in a 3D multilayered nucleus pulposus degeneration model. Eur Cells Mater 43:53–65. https://doi.org/10.22203/eCM.v043a06
Ohshima H, Urban J (1992) The effect of lactate and pH on proteoglycan and protein synthesis rates in the intervertebral disc, Spine, 17(9):1079-82, https://doi.org/10.1097/00007632-199209000-00012
Wu W et al (2014) Lactate down-regulates matrix systhesis and promotes apoptosis and autophagy in rat nucleus pulposus cells, J Orthop Res, 32(2):253–61, https://doi.org/10.1002/jor.22503
Zhao C, Wang L, Jiang L, Dai L (2007) The cell biology of intervertebral disc aging and degeneration, Ageing research reviews, 6(3):247–61, https://doi.org/10.1016/j.arr.2007.08.001
Iatsenko I, Boquete JP, Lemaitre B Microbiota-Derived Lactate Activates Production of Reactive Oxygen Species by the Intestinal NADPH Oxidase Nox and Shortens Drosophila Lifespan, Immunity, 49(5):929–942 e5, Nov 20 2018, https://doi.org/10.1016/j.immuni.2018.09.017
Ross J et al (2010) High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci USA 107(46):20087–20092. https://doi.org/10.1073/pnas.1008189107
Mullins R, Reiter D, Kapogiannis D (2018) Magnetic resonance spectroscopy reveals abnormalities of glucose metabolism in the Alzheimer’s brain. Ann Clin Transl Neurol 5(3):262–272. https://doi.org/10.1002/acn3.530
Wang Q et al (2022) Glucose Metabolism, Neural Cell Senescence and Alzheimer’s Disease. Int J Mol Sci 23(8). https://doi.org/10.3390/ijms23084351
Jia L et al Rheb-regulated mitochondrial pyruvate metabolism of Schwann cells linked to axon stability, (in eng), Developmental cell, 56(21):2980–2994.e6, Nov 8 2021, https://doi.org/10.1016/j.devcel.2021.09.013
Zhu Y et al (2019) Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy, (in eng), Cellular signalling, 58:53–64, https://doi.org/10.1016/j.cellsig.2019.03.006
Zhang YY et al (Jul 26 2022) Pretreatment of nucleus pulposus mesenchymal stem cells with appropriate concentration of H2O2 enhances their ability to treat intervertebral disc degeneration. Stem Cell Res Ther 13(1):340. https://doi.org/10.1186/s13287-022-03031-7
Lu TC, Wu YH, Chen WY, Hung YC (2022) Targeting Oxidative Stress and Endothelial Dysfunction Using Tanshinone IIA for the Treatment of Tissue Inflammation and Fibrosis, Oxid Med Cell Longev, 2022:2811789, https://doi.org/10.1155/2022/2811789
Hua W et al (Jan 1 2020) Icariin protects human nucleus pulposus cells from hydrogen peroxide-induced mitochondria-mediated apoptosis by activating nuclear factor erythroid 2-related factor 2. Biochim Biophys Acta Mol Basis Dis 1866(1):165575. https://doi.org/10.1016/j.bbadis.2019.165575
Vaccari Cardoso B et al (2021) May., Reducing l-lactate release from hippocampal astrocytes by intracellular oxidation increases novelty induced activity in mice, Glia, 69(5):1241–1250, https://doi.org/10.1002/glia.23960
Lai A et al (2021) Jun., Development of a standardized histopathology scoring system for intervertebral disc degeneration in rat models: An initiative of the ORS spine section, (in eng), JOR Spine, 4(2):e1150, https://doi.org/10.1002/jsp2.1150
Pan L et al (Dec 2020) Bufalin exerts antitumor effects in neuroblastoma via the induction of reactive oxygen species–mediated apoptosis by targeting the electron transport chain, (in eng). Int J Mol Med 46(6):2137–2149. https://doi.org/10.3892/ijmm.2020.4745
Dmitriev LF, Titov VN (2010) Lipid peroxidation in relation to ageing and the role of endogenous aldehydes in diabetes and other age-related diseases, (in eng), Ageing Res Rev, 9(2):200–10, https://doi.org/10.1016/j.arr.2009.09.004
Lv D et al (2023) Targeting phenylpyruvate restrains excessive NLRP3 inflammasome activation and pathological inflammation in diabetic wound healing, (in eng), Cell Rep Med, 4(8):101129, Aug 15 https://doi.org/10.1016/j.xcrm.2023.101129
Wang L et al (Mar 2021) Novel PF74-like small molecules targeting the HIV-1 capsid protein: balance of potency and metabolic stability, (in eng). Acta Pharm Sin B 11(3):810–822. https://doi.org/10.1016/j.apsb.2020.07.016
Bahuguna A, Bharadwaj S, Chauhan AK, Kang SC Inhibitory insights of strawberry (Fragaria × ananassa var. Seolhyang) root extract on tyrosinase activity using computational and in vitro analysis, (in eng), International journal of biological macromolecules, 165(Pt B):2773–2788, Dec 15 2020, https://doi.org/10.1016/j.ijbiomac.2020.10.135
Morris GM et al (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, (in eng), Journal of computational chemistry, 30(16):2785-91, https://doi.org/10.1002/jcc.21256
Das S, Singh VK, Dwivedy AK, Chaudhari AK, Deepika, Dubey NK (Mar 1 2021) Eugenol loaded Chitosan nanoemulsion for food protection and inhibition of aflatoxin B(1) synthesizing genes based on molecular docking, (in eng). Carbohydr Polym 255:117339. https://doi.org/10.1016/j.carbpol.2020.117339
Yang H et al (2022) Eupatilin attenuates the senescence of nucleus pulposus cells and mitigates intervertebral disc degeneration via inhibition of the MAPK/NF-κB signaling pathway, (in eng). Front Pharmacol 13:940475. https://doi.org/10.3389/fphar.2022.940475
Du J et al (Apr 2022) CB2R attenuates intervertebral disc degeneration by delaying Nucleus Pulposus Cell Senescence through AMPK/GSK3β Pathway, (in eng). Aging Dis 13(2):552–567. https://doi.org/10.14336/ad.2021.1025
Li X, Yang S, Qin L, Yang S (2021) Type II collagen-positive embryonic progenitors are the major contributors to spine and intervertebral disc development and repair, (in eng), Stem Cells Transl Med, 10(10):1419–1432, https://doi.org/10.1002/sctm.20-0424
Jiang S, Chen X, Lin J, Huang P (May 2023) Lactate-Oxidase-Instructed Cancer diagnosis and therapy, (in eng). Adv Mater 35(19):e2207951. https://doi.org/10.1002/adma.202207951
Dong X et al (May 26 2020) A heterojunction structured WO(2.9)-WSe(2) Nanoradiosensitizer Increases Local Tumor Ablation and checkpoint blockade immunotherapy upon low Radiation Dose, (in eng). ACS Nano 14(5):5400–5416. https://doi.org/10.1021/acsnano.9b08962
Wang C, Gonzales S, Levene H, Gu W, Huang C (2013) Energy metabolism of intervertebral disc under mechanical loading. J Orthop Research: Official Publication Orthop Res Soc 31(11):1733–1738. https://doi.org/10.1002/jor.22436
Ruiz Wills C, Foata B, González Ballester M, Karppinen J, Noailly J (2018) Theoretical explorations Generate New hypotheses about the role of the cartilage endplate in early intervertebral disk degeneration, (in eng). Front Physiol 9:1210. https://doi.org/10.3389/fphys.2018.01210
Li X et al (2022) Lactate metabolism in human health and Disease. Signal Transduct Target Therapy 7(1):305. https://doi.org/10.1038/s41392-022-01151-3
Shen J et al (2022) Exhausted local lactate accumulation via injectable nanozyme-functionalized hydrogel microsphere for inflammation relief and tissue regeneration, (in eng), Bioact Mater, 12:153–168, https://doi.org/10.1016/j.bioactmat.2021.10.013
Vaccari Cardoso B et al (2021) Reducing l-lactate release from hippocampal astrocytes by intracellular oxidation increases novelty induced activity in mice, Glia, 69(5):1241–1250, https://doi.org/10.1002/glia.23960
Malandrino A, Noailly J, Lacroix D (2014) Numerical exploration of the combined effect of nutrient supply, tissue condition and deformation in the intervertebral disc. J Biomech 47(6):1520–1525. https://doi.org/10.1016/j.jbiomech.2014.02.004
Jackson A, Huang C, Brown M, Gu W (2011) 3D finite element analysis of nutrient distributions and cell viability in the intervertebral disc: effects of deformation and degeneration. J Biomech Eng 133(9):091006. https://doi.org/10.1115/1.4004944
Wu X et al (2021) Glycine-Serine-Threonine Metabolic Axis Delays Intervertebral Disc Degeneration through Antioxidant Effects: An Imaging and Metabonomics Study, Oxidative medicine and cellular longevity, 2021:5579736, https://doi.org/10.1155/2021/5579736
Kim J et al (2018) Pyruvate protects against Cellular Senescence through the Control of Mitochondrial and lysosomal function in dermal fibroblasts. J Invest Dermatol 138(12):2522–2530. https://doi.org/10.1016/j.jid.2018.05.033
Asadi Shahmirzadi A et al (2020) Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice, Cell metabolism, 32(3):447–456.e6, https://doi.org/10.1016/j.cmet.2020.08.004
Wang Y et al (2020) Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat Commun 11(1):5596. https://doi.org/10.1038/s41467-020-19360-1
Krylova IB et al (2006) The cardioprotective effect of uridine and uridine-5’-monophosphate: the role of the mitochondrial ATP-dependent potassium channel, (in eng), Experimental gerontology, 41(7):697–703, https://doi.org/10.1016/j.exger.2006.03.005
Yuzwa S, Vocadlo D (2014) O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer’s Disease and beyond. Chem Soc Rev 43:6839–6858. https://doi.org/10.1039/c4cs00038b
Seol D et al (2021) Targeting oxidative stress with amobarbital to prevent intervertebral disc degeneration: Part I. in vitro and ex vivo studies, (in eng), The spine journal: official journal of the North American Spine Society, 21(6):1021–1030, https://doi.org/10.1016/j.spinee.2021.02.008
Li Q, Chen H, Li Z, Zhang F, Chen L (2022) Glucocorticoid caused lactic acid accumulation and damage in human chondrocytes via ROS-mediated inhibition of Monocarboxylate Transporter 4, Bone, 155:116299, https://doi.org/10.1016/j.bone.2021.116299
Zhang X et al (2016) The effects of lactate and acid on articular chondrocytes function: implications for polymeric cartilage scaffold design. Acta Biomater 42:329–340. https://doi.org/10.1016/j.actbio.2016.06.029
Venkatachalam G, Surana U, Clément M (2017) Replication stress-induced endogenous DNA damage drives cellular senescence induced by a sub-lethal oxidative stress. Nucleic Acids Res 45(18):10564–10582. https://doi.org/10.1093/nar/gkx684
Gao F et al (2019) Intra/Extracellular lactic acid exhaustion for synergistic metabolic therapy and immunotherapy of tumors. Adv Mater (Deerfield Beach Fla) 31(51):e1904639. https://doi.org/10.1002/adma.201904639
Jia L et al (2021) Rheb-regulated mitochondrial pyruvate metabolism of Schwann cells linked to axon stability. Dev Cell 56(21):2980–2994. https://doi.org/10.1016/j.devcel.2021.09.013
Heather LC et al (2012) Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol 107(3). https://doi.org/10.1007/s00395-012-0268-2
Motori E et al (2020) Neuronal metabolic rewiring promotes resilience to neurodegeneration caused by mitochondrial dysfunction, (in eng), Sci Adv, 6(35):eaba8271, https://doi.org/10.1126/sciadv.aba8271
Wu S et al (2022) Telomerase RNA TERC and the PI3K-AKT pathway form a positive feedback loop to regulate cell proliferation Independent of telomerase activity. Nucleic Acids Res 50(7):3764–3776. https://doi.org/10.1093/nar/gkac179
Chan K et al (2020) A functional genetic screen defines the AKT-induced senescence signaling network. Cell Death Differ 27(2):725–741. https://doi.org/10.1038/s41418-019-0384-8
Matheny R, Adamo M (2010) PI3K p110 alpha and p110 beta have differential effects on akt activation and protection against oxidative stress-induced apoptosis in myoblasts. Cell Death Differ 17(4):677–688. https://doi.org/10.1038/cdd.2009.150
He J, Zhang N, Zhu Y, Jin R, Wu F (2021) MSC spheroids-loaded collagen hydrogels simultaneously promote neuronal differentiation and suppress inflammatory reaction through PI3K-Akt signaling pathway, Biomaterials, 265:120448, https://doi.org/10.1016/j.biomaterials.2020.120448
Luo L et al (2022) Injectable cartilage matrix hydrogel loaded with cartilage endplate stem cells engineered to release exosomes for non-invasive treatment of intervertebral disc degeneration. Bioactive Mater 15:29–43. https://doi.org/10.1016/j.bioactmat.2021.12.007
Shao Z et al (2020) Stachydrine ameliorates the progression of intervertebral disc degeneration via the PI3K/Akt/NF-κB signaling pathway: in vitro and in vivo studies. Food Funct 11(12):10864–10875. https://doi.org/10.1039/d0fo02323j
Chen H et al (2022) Proanthocyanidins inhibit the apoptosis and aging of nucleus pulposus cells through the PI3K/Akt pathway delaying intervertebral disc degeneration. Connect Tissue Res 1–13. https://doi.org/10.1080/03008207.2022.2063121
Kakiuchi Y et al (2019) Pharmacological inhibition of mTORC1 but not mTORC2 protects against human disc cellular apoptosis, senescence, and extracellular matrix catabolism through akt and autophagy induction. Osteoarthr Cartil 27(6):965–976. https://doi.org/10.1016/j.joca.2019.01.009
Qi S, Li C, Kong X, Zheng Q (2020) Dexmedetomidine suppresses oxidative stress and inflammation of nucleus pulposus cells by activating the PI3K/Akt signaling pathway, (in eng), Die Pharmazie, 75(10):505–509, https://doi.org/10.1691/ph.2020.9927
Ma K, Cheung S, Marshall A, Duronio V (2008) PI(3,4,5)P3 and PI(3,4)P2 levels correlate with PKB/akt phosphorylation at Thr308 and Ser473, respectively; PI(3,4)P2 levels determine PKB activity, Cellular signalling, 20(4):684 – 94, https://doi.org/10.1016/j.cellsig.2007.12.004
Khor T, Gul Y, Ithnin H, Seow H (2004) Positive correlation between overexpression of phospho-BAD with phosphorylated Akt at serine 473 but not threonine 308 in colorectal carcinoma, Cancer letters, 210(2):139–50, https://doi.org/10.1016/j.canlet.2004.01.017
Wang CY, Hsieh MK, Hu YJ, Bit A, Lai PL Monocarboxylate transporter 1-mediated lactate accumulation promotes nucleus pulposus degeneration under hypoxia in a 3D multilayered nucleus pulposus degeneration model, (in eng). Eur Cell Mater, 43:53–65, Feb 21 2022, https://doi.org/10.22203/eCM.v043a06
Silagi E et al (2020) Lactate Efflux from intervertebral disc cells is Required for Maintenance of Spine Health. J Bone Miner Res 35(3):550–570. https://doi.org/10.1002/jbmr.3908
Liu W et al (2023) Lactate regulates cell cycle by remodeling the anaphase promoting complex, (in eng). Nat Mar 15. https://doi.org/10.1038/s41586-023-05939-3
Feng T et al (Sep 5 2022) Adipocyte-derived lactate is a signalling metabolite that potentiates adipose macrophage inflammation via targeting PHD2, (in eng). Nat Commun 13(1):5208. https://doi.org/10.1038/s41467-022-32871-3
Kumar C, Madison V (2005) AKT crystal structure and AKT-specific inhibitors, Oncogene, 24(50):7493–501, https://doi.org/10.1038/sj.onc.1209087
Chu N et al (2020) The structural determinants of PH domain-mediated regulation of Akt revealed by segmental labeling, eLife, 9, https://doi.org/10.7554/eLife.59151
Chu N et al (2018) Akt Kinase Activation Mechanisms Revealed Using Protein Semisynthesis, Cell, 174(4):897–907.e14, https://doi.org/10.1016/j.cell.2018.07.003
Mahadevan D et al (2008) Discovery of a novel class of AKT pleckstrin homology domain inhibitors. Mol Cancer Ther 7(9):2621–2632. https://doi.org/10.1158/1535-7163.Mct-07-2276
Harris T (2003) PDK1 and PKB/Akt: ideal targets for development of new strategies to structure-based drug design, IUBMB life, 55(3):117–26, https://doi.org/10.1080/1521654031000115951
Berndt N et al (2010) The akt activation inhibitor TCN-P inhibits akt phosphorylation by binding to the PH domain of akt and blocking its recruitment to the plasma membrane. Cell Death Differ 17(11):1795–1804. https://doi.org/10.1038/cdd.2010.63
Agamasu C, Ghanam R, Xu F, Sun Y, Chen Y, Saad J (2017) The interplay between Calmodulin and membrane interactions with the pleckstrin homology domain of Akt. J Biol Chem 292(1):251–263. https://doi.org/10.1074/jbc.M116.752816
Miao B et al (2010) Small molecule inhibition of phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains. Proc Natl Acad Sci USA 107(46):20126–20131. https://doi.org/10.1073/pnas.1004522107
Jiao Y et al (2019) Genipin, a natural AKT inhibitor, targets the PH domain to affect downstream signaling and alleviates inflammation. Biochem Pharmacol 170:113660. https://doi.org/10.1016/j.bcp.2019.113660
Panda P et al (2017) Abrus Agglutinin, a type II ribosome inactivating protein inhibits Akt/PH domain to induce endoplasmic reticulum stress mediated autophagy-dependent cell death. Mol Carcinog 56(2):389–401. https://doi.org/10.1002/mc.22502
Sugimoto M et al (2002) Activation of cyclin D1-kinase in murine fibroblasts lacking both p21(Cip1) and p27(Kip1), Oncogene. 21(53):8067–8074. https://doi.org/10.1038/sj.onc.1206019
Axanova L, Chen Y, McCoy T, Sui G, Cramer S (2010) 1,25-dihydroxyvitamin D(3) and PI3K/AKT inhibitors synergistically inhibit growth and induce senescence in prostate cancer cells, The Prostate, 70(15):1658-71, https://doi.org/10.1002/pros.21201
Collado M et al (2000) Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27Kip1. J Biol Chem 275(29):21960–21968. https://doi.org/10.1074/jbc.M000759200
Kennedy A et al (2011) Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell 42(1):36–49. https://doi.org/10.1016/j.molcel.2011.02.020
He Q et al (2019) Dual inhibition of Akt and ERK signaling induces cell senescence in triple-negative Breast cancer. Cancer Lett 448:94–104. https://doi.org/10.1016/j.canlet.2019.02.004
Halkoum R et al (2022) Glyoxal Induces Senescence in Human Keratinocytes through Oxidative Stress and Activation of the Protein Kinase B/FOXO3a/p27 Pathway, The Journal of investigative dermatology, 142(8):2068–2078.e7, https://doi.org/10.1016/j.jid.2021.12.022
Bernhart E et al (2014) Silencing of protein kinase D2 induces glioma cell senescence via p53-dependent and -independent pathways, Neuro-oncology, 16(7):933–45, https://doi.org/10.1093/neuonc/not303
Chan KC et al (Oct 24 2013) A novel Hsp90 inhibitor AT13387 induces senescence in EBV-positive nasopharyngeal carcinoma cells and suppresses Tumor formation, (in eng). Mol Cancer 12(1):128. https://doi.org/10.1186/1476-4598-12-128
Szepanowski F et al (2017) Dimethyl fumarate accelerates peripheral nerve regeneration via activation of the anti-inflammatory and cytoprotective Nrf2/HO-1 signaling pathway. Acta Neuropathol 133(3):489–491. https://doi.org/10.1007/s00401-017-1676-z
Yang Y et al (2022) Targeting ferroptosis suppresses osteocyte glucolipotoxicity and alleviates diabetic osteoporosis. Bone Res 10(1):26. https://doi.org/10.1038/s41413-022-00198-w
Ali T et al (2018) Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease, Molecular neurobiology, 55(7):6076–6093, https://doi.org/10.1007/s12035-017-0798-6
Li H et al (2018) Neuroprotective effect of phosphocreatine on oxidative stress and mitochondrial dysfunction induced apoptosis in vitro and in vivo: Involvement of dual PI3K/Akt and Nrf2/HO-1 pathways, Free radical biology & medicine, 120:228–238, https://doi.org/10.1016/j.freeradbiomed.2018.03.014
Zhang J et al (2023) Mesencephalic astrocyte-derived neurotrophic factor (MANF) prevents the neuroinflammation induced dopaminergic neurodegeneration. Exp Gerontol 171:112037. https://doi.org/10.1016/j.exger.2022.112037
Bian Y et al (2021) vitroOxyphylla A ameliorates cognitive deficits and alleviates neuropathology via the Akt-GSK3β and Nrf2-Keap1-HO-1 pathways in and in murine models of Alzheimer’s Disease. J Adv Res 34:1–12. https://doi.org/10.1016/j.jare.2021.09.002
Wang Y et al (2019) Kinsenoside ameliorates intervertebral disc degeneration through the activation of AKT-ERK1/2-Nrf2 signaling pathway, Aging, 11(18):7961–7977, https://doi.org/10.18632/aging.102302
Acknowledgements
We thank MetWare Biotechnology Co., Ltd. (Wuhan, China) and Topscience Co. Ltd. (Shanghai, China) for their contributions and assistance in this study. Additionally, we thank all the peer reviewers for their opinions and suggestions.
Funding
This research was supported by the National Natural Science Foundation of China (Grant No. 81972113 and 82172488) and the Natural Science Foundation of Chongqing in China (Grant No. cstc2020jcyj-bshX0091).
Author information
Authors and Affiliations
Contributions
LCQ and ZY conceived and designed the experiments; ZYY, LLBX, and QYH performed the experiments; ZY, CYX and LJH participated in the creation of the experimental animal model. LC, LHY and CX revised the article and conducted the experimental complementation; HZL and LYY analysed the data; ZYY, LLBX and QYH edited the figures and wrote this article; ZY and FCC reviewed the article and polished the language. All the authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All human experimental protocols were scrutinized and approved by the Ethics Committee of Second Affiliated Hospital, Army Medical University (approval no. 2022-YD212-01). All of our human experiments were conducted in strict accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) and written informed consent was obtained from the patients before the experimental procedures were performed. Eight-week-old male Sprague Dawley (SD) rats were provided by the Animal Center of Xinqiao Hospital and used in this research. All experiments were approved by the Laboratory Animal Welfare and Ethics Committee of the Third Military Medical University (approval no. AMUWEC20212131).
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
: Table S1. Grading Criteria for Pfirrmann.
Supplementary Material 2
: Table S2. Details of the Patients Enrolled in This Study.
Supplementary Material 3
: Table S3. Antibody Information Sheet.
Supplementary Material 4
: Fig. S1. The mass spectrum of lactic acid content in the NP tissues in the normal group and the PIDD group (AREA: mass spectrum peak intensity, S/N: signal-to-noise ratio).
Supplementary Material 5
: Fig. S5. Molecular weight of targeted protein in the adjacent “protein ladder”.
Supplementary Material 6
: Table S4. Primer Sequence Used in the Quantitative RT-PCR. PTEN, phosphatase and tensin homolog; CDC37, cell division cycle 37; PDK1, pyruvate dehydrogenase kinase 1; CTMP, thioesterase superfamily member 4; PP2A, serine/threonine protein phosphatase 2A; PHLPP, PH domain leucine-rich repeat protein phosphatase; HSP90, heat shock protein 90; Akt1, serine/threonine kinase 1; Akt2, serine/threonine kinase 2; Akt3, serine/threonine kinase 3; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PIK3R1, phosphoinositide-3-kinase regulatory subunit 1; PIK3R2, phosphoinositide-3-kinase regulatory subunit 2; PIK3CG, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma.
Supplementary Material 7
: Fig. S2. LOx was successfully expressed in intervertebral disc cells and reduced the amount of lactic acid in the intervertebral disc. (A) Differences in the expression of GFP in the intervertebral discs of rats in each group after injection of lentiviral vector. GFP in cells appeared green, and nuclei were counterstained with DAPI (blue) (n = 3). (B) Western blotting analysis showed LOx expression after injection of the lentiviral vector. (C) Measurement of lactic acid content in the NP tissues of rats in each group after different treatments (n = 3). ****p < 0.0001
Supplementary Material 8
: Fig. S3. Effects of overexpression of LOx in NPCs. (A) Western blotting analysis showing the transfection efficiency of LOx. (B) The expression of GFP (green) in the NPCs in each group after transfection with lentiviral vector (scale bar = 200 μm). (C) Measurement of lactic acid content in NPCs using a lactic acid detection kit. (D-E) The effect of lactic acid on the proliferation of NPCs (red) with LOx-overexpression as determined by EdU staining. Nuclei were counterstained with Hoechst 33258 (blue) (scale bar = 100 μm). (F-G) The effect of lactic acid on the cell cycle of NPCs overexpressing LOx; the box positions represent negative controls. (H-I) The effect of lactic acid on the senescence of LOx-overexpressing NPCs as determined by SA-β-gal assay; senescent cells appear blue (scale bar = 200 μm). (J-K) The effect of lactic acid on ROS production in NPCs overexpressing LOx. The FL1 subset was set according to the ROS intensity of the negative control. (L-M) The effect of lactic acid on the MMP of NPCs with LOx-overexpression as determined by the CMXRos probe (red); nuclei were counterstained with Hoechst 33258 (blue) (scale bar = 50 μm). La represents lactic acid; n = 3; *p < 0.05; **p < 0.01; ****p < 0.0001; ns represents no statistical significance
Supplementary Material 9
: Fig. S4. Relative mRNA expression levels of PI3K/AKT signaling pathway-related genes in NPCs under lactic acid treatment. β-actin was used to normalize target gene mRNA, and the formula 2−∆∆Ct was utilized to measure the relative mRNA expression compared to the control group. Data are expressed as the mean ± SD from three independent experiments (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001; ns represents no statistical significance
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Zhang, Y., Liu, L., Qi, Y. et al. Lactic acid promotes nucleus pulposus cell senescence and corresponding intervertebral disc degeneration via interacting with Akt. Cell. Mol. Life Sci. 81, 24 (2024). https://doi.org/10.1007/s00018-023-05094-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-05094-y