
Abstract
The exposure to an unhealthy environment in utero can lead to the occurrence of cardiovascular diseases in the offspring. Glucocorticoids (GC) are essential for normal development and maturation of fetal organs and is a first-line treatment for pregnant women affected by autoimmune diseases. However, excess prenatal GC exposure might program the development of fetal organs and cause a number of chronic diseases in later life. Our previous studies indicated that cardiac functions were significantly compromised in rat offspring prenatally exposed to the synthetic glucocorticoid dexamethasone (DEX), only after ischemia–reperfusion. In the present study, we further observed that DNA hypermethylation of bone morphogenetic protein 4 (Bmp4) promoter in cardiomyocytes caused by prenatal DEX exposure substantially dampened the binding activity of transcription factor HIF-1α induced by cardiac ischemia. Therefore, prenatal DEX exposure inhibits the induction of BMP4 upon I/R and attenuates the protective effects of BMP4 in cardiomyocytes, which eventually manifests as malfunction of the adult heart. Moreover, we employed two cardiac-specific Bmp4 knock-in mouse models and found that in vivo BMP4 overexpression could rescue the cardiac dysfunction caused by prenatal GC exposure. In depth mechanistic research revealed that BMP4 protects the cardiomyocytes from mitophagy and apoptosis by attenuating mitochondrial PGC-1α expression in a p-Smad and Parkin-dependent manner. These findings suggest that prenatal GC exposure increases the susceptibility of the offspring’s heart to a “second strike” after birth, due to the failure of hypoxia-induced HIF-1α transactivation of the hypermethylated Bmp4 promoter in cardiomyocytes. Pretreatment with the DNA methylation inhibitor, 5-Aza-2′-deoxycytidine, could be a potential therapeutic method for this programming effect of GC exposure during pregnancy on neonatal cardiac dysfunction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Among non-communicable diseases, cardiovascular diseases (CVDs) are the major cause of morbidity and mortality worldwide [1]. In recent years, accumulating evidence indicates that exposure to an unhealthy environment in utero can lead to the occurrence of CVDs in the offspring, even into adulthood. Previously, Barker et al. discovered the association of an adverse intrauterine environment with an increased risk of hypertension and ischemic heart disease in adulthood [2]. This is now referred to as the Developmental Origins of Health and Disease (DOHAD). Thereafter, more and more influences of the intrauterine environment on CVDs have been discovered. Maternal glucocorticoid (GC) overexposure [3, 4], preeclampsia, diabetes [5,6,7], maternal obesity or excessive weight gain during pregnancy [8, 9], as well as maternal undernutrition [10], tobacco smoking [11, 12], maternal stress or alcohol consumption [13] during pregnancy, all greatly enhance risks of CVDs for offspring in their later life. Therefore, scientists worldwide have tried to answer the question of how to mitigate the cardiac damages caused by risk factors within the intrauterine environment.
Endogenous GC provides a critical developmental trigger, which is essential for normal development and maturation of many organs, including thyroid, kidney, brain, pituitary, and the fetal lung [14,15,16]. This makes GC a widely used therapy for women at risk of preterm birth to accelerate the maturation of fetal organs. Meanwhile, GC is also a first-line and compulsory treatment for pregnant women with asthma [17] or an autoimmune disease [18, 19], throughout the entire pregnancy until labor.
However, prenatal exposure to excess GC might cause a number of chronic diseases in later life, including diabetes mellitus, obesity, psychiatric diseases and cardiovascular diseases [15, 20]. In our recent study, we found that the cardiac functions were significantly compromised in male rat offspring prenatally exposed to the synthetic glucocorticoid dexamethasone (DEX) compared to those exposed to normal saline (NS), due to an increased infarction and apoptosis of myocardium, especially after myocardial ischemia and reperfusion injury (IRI) [21]. MeDIP-sequencing analysis revealed that the regions of bone morphogenetic protein 4 (Bmp4) promoter were hypermethylated due to prenatal exposure to DEX, which subsequently lead to the decreased Bmp4 expression [21]. The cardiac dysfunction was associated with a breakdown of mitochondrial membrane potential, a decrease in ATP production and the dramatically elevated release of reactive oxygen species (ROS), which could be rescued by the addition of exogenous Bmp4 [21]. However, why the cardiac functions were abnormal in the DEX-exposed offspring only after IRI has not been elucidated.
BMP4 is a secretory protein which belongs to transforming growth factor-beta (TGF-β) superfamily [22]. BMP4 is required for the development of the atrioventricular canal [23], branchial-arch artery remodeling [24], endocardial cushion development and for normal separation of the outflow tract [25]. Although BMP4 is very important for heart development during embryogenesis, it is almost undetectable in the adult heart [26]. However, BMP4 levels are elevated with myocardial injury in adult humans or mice. Plasma BMP4 levels in heart failure patients are found to be significantly higher in heart failure patients than that of subjects without heart failure [26]. Bmp4 mRNA was increased in left ventricular biopsies of patients with coronary artery disease with good cardiac function [27]. BMP4 expression was markedly increased in mouse hearts after 2- and 4-week transverse aortic constriction (TAC) [26] or 24 h post-infarction by permanent coronary artery ligation [27]. All of these findings suggest that BMP4 may exert protective effects against myocardial ischemic injury. Signaling of BMP4 through BMP receptors (BmpRI and BmpRII) is involved in two pathways: Smad and non-Smad pathways [28, 29], whereas the signaling pathway and molecular mechanisms underlying the protective effect of BMP4 in IRI have yet been fully illustrated.
In the present study, we aimed to investigate the protective effects and underlying signaling pathways of BMP4 on myocardium in vivo, and to dissect the molecular mechanisms mediating the upregulation of BMP4 under hypoxia which is attenuated by the prenatal GC exposure. We employed a cardiac specific Bmp4 knock-in mouse model in which BMP4 expression was specifically induced in the myocardium and found that BMP4 could protect the cardiomyocytes from apoptosis by attenuating mitochondrial PGC-1α expression in a p-Smad and Parkin-dependent manner. The ischemia-induced transcription factor HIF-1α bound to the Bmp4 promoter and activated Bmp4 transcription. DEX treatment caused the hypermethylation of HIF-1α binding sites on the Bmp4 promoter and impaired the transactivation of Bmp4, which led to the failure of increase in BMP4 upon IRI and attenuates the protective effects of BMP4 in the cardiomyocytes, and eventually manifests the malfunction of adult heart.
Results
GC exposure during pregnancy causes hypermethylation of the Bmp4 promoter which inhibits transactivation by HIF-1α in offspring’s cardiomyocytes
In a previous MeDIP-sequencing analysis, it was found that the methylation level of the Bmp4 promoter in cardiomyocytes of prenatally GC-exposed male offspring was significantly increased, along with the decrease in mRNA and protein expression of Bmp4 [21]. To verify effects of exposure of male rat offspring to DEX vs. NS during pregnancy on DNA methylation of the Bmp4 promoter, genomic DNA was extracted from myocardium and analyzed using MSP. The results showed that the DNA methylation level of Bmp4 promoter was significantly higher in the DEX-treated group compared to that treated with NS, indicated by the increased ratio of methylated PCR product (M) vs. unmethylated PCR product (U) between DEX-treated group and NS-treated group (Fig. 1A). In accord with our previous findings, cardiac function (indicated by ± dp/dt max) of adult offspring manifested no significant differences between DEX and NS group, but was significantly decreased in adult male rats after ischemia–reperfusion (I/R) of myocardium of the DEX-treated, compared with the NS group [21]. Therefore, we speculated that hypoxia might cause an increase in Bmp4 expression, which was attenuated by the promoter hypermethylation due to prenatal DEX exposure.

Excess GC exposure during pregnancy causes hypermethylation of the Bmp4 promoter; this inhibits transactivation by HIF-1α in cardiomyocytes of male rat offspring. The DNA methylation level of the Bmp4 promoter in cardiomyocytes of male rat offspring was analyzed by methylation-specific PCR, and the methylated PCR product and unmethylated PCR product was labeled as M and U, respectively (A, n = 3 per treatment group). Protein levels of BMP4 in NRCMs were significantly increased upon H/R treatment compared with normoxia treatment. Representative Western blot (B) and the corresponding histogram (C) are shown separately. n = 5 per treatment group, *p < 0.05, compared with normoxia treatment. mRNA levels of Bmp4 in NRCMs were also significantly increased upon H/R treatment (D). n = 5, **p < 0.01, compared with normoxia treatment. Bioinformatics prediction of the HIF-1α binding site on Bmp4 promoter was presented in panel E. The putative binding sequence was 5′-GCACGTCT-3′ (E). The binding ability of HIF-1α to the Bmp4 promoter upon I/R treatment was significantly decreased in the myocardium of rat offspring prenatally exposed to dexamethasone (DEX, F). n = 4, **p < 0.01, compared with rat offspring prenatally exposed to normal saline (NS). The putative binding site as well as the core sequences for DNA methylation on Bmp4 promoter were mutated or deleted (G). The transcriptional activity of luciferase was significantly increased after pGL4.23-Bmp4 transfection into H9C2 cells upon hypoxia treatment, which was diminished by the transfection of pGL4.23-Bmp4-mut or pGL4.23-Bmp4-del plasmids (H). n = 3, *p < 0.05, compared with pGL4.23-basic plasmid transfection; #p < 0.05, compared with pGL4.23-Bmp4 plasmid transfection
To verify this hypothesis, we analyzed the mRNA and protein expression of Bmp4 in neonatal rat cardiomyocytes (NRCMs) treated with hypoxia/reoxygenation (H/R) or under normoxic conditions. It was shown that both mRNA and protein levels of Bmp4 were significantly increased upon H/R exposure (Fig. 1B–D). Hypoxia can significantly increase expression of transcription factor HIF-1α, which binds the promoter regions and activate transcription of target genes. We employed the LASAGNA online tool to analyze the promoter region of Bmp4 and found a putative binding site for HIF-1α within the methylation area of Bmp4 promoter (Supplemental Table 1, Fig. 1E). ChIP-qPCR analysis showed that HIF-1α was significantly enriched in Bmp4 promoter region in the myocardium upon I/R from prenatally NS-exposed rat offspring, while the binding ability of HIF-1α to the Bmp4 promoter region upon I/R was significantly decreased in the myocardium of rat offspring prenatally exposed to DEX (Fig. 1F).
We next asked whether DNA hypermethylation of the Bmp4 promoter affects the binding of HIF-1α and subsequently attenuates Bmp4 transcriptional activity. To assess this, a dual luciferase reporter activity experiment was conducted. The promoter region of Bmp4 containing binding sites for HIF-1α was amplified from genomic DNA and subcloned into pGL4.23 plasmid to construct pGL4.23-Bmp4 vector. Meanwhile, pGL4.23-Bmp4-mut and pGL4.23-Bmp4-del vectors were also constructed by mutating and deleting the hypermethylation core sequences of Bmp4 promoter region, respectively (Fig. 1G). We observed that activation of HIF-1α by exposure to 1% O2 for 24 h significantly increased the transcriptional activity of luciferase in H9C2 cells transfected with pGL4.23-Bmp4 plasmid, compared with those transfected with a control vector lacking the Bmp4 promoter (Fig. 1H). However, the increased transcriptional activity was blunted by transfection of pGL4.23-Bmp4-mut or pGL4.23-Bmp4-del vectors (Fig. 1H).
Myocardial-specific BMP4 overexpression protects cardiac functions in male offspring of mice exposed to DEX during pregnancy
In order to explore the role of BMP4 in myocardial protection from ischemia–reperfusion in vivo, we constructed conditional Bmp4 overexpression (OE) mice in which BMP4 was specifically overexpressed in myocardium, with or without induction of tamoxifen (Bmp4flox/flox-αMyh6-Cre/Esr1* or Bmp4flox/flox-αMyh6-Cre) (Fig. 2A, Supplemental Fig. 1A, Supplemental Fig. 2A). Compared with oil-injection groups, the expression levels of BMP4 mRNA and protein in Bmp4flox/flox-αMyh6-Cre/Esr1* mice (Bmp4OE) were significantly increased after two weeks of tamoxifen treatment (Supplemental Fig. 1B, C). The FLAG protein linked to BMP4 also was present in the tamoxifen-injected group, which further confirmed the overexpression of transgenic Bmp4 (Supplemental Fig. 1C).

Cardiac function in male offspring of pregnant mice exposed to DEX during pregnancy is protected by BMP4 overexpression specifically in myocardium. Schematic figure showing strategy for the construction of BMP4 overexpression mice (A). All mice were subjected to ischemia/reperfusion (I/R, 30 min/24 h). Compared with oil-injection groups, the expression levels of Bmp4 mRNA (B) and protein (C, D) in Bmp4flox/flox-αMyh6-Cre/Esr1* (Bmp4OE) mice were significantly increased after two weeks of tamoxifen injection (40 mg/kg, once a week). n = 6 for each group. The expression of FLAG protein linked with BMP4 was presented as well (C, E). Representative echocardiographic images of the adult male offspring in Bmp4WT and Bmp4OE mice prenatally exposed to NS or DEX were shown (F). BMP4 overexpression restored left ventricular fractional shortening (FS) and ejection fraction (EF) in Bmp4OE mice prenatally exposed to DEX (G, H). Representative photographs of cardiac sections (I) and quantified data (J) demonstrate that BMP4 overexpression decreased infarct size in Bmp4OE mice prenatally exposed to DEX subjected to I/R (n = 7–12). *p < 0.05, compared with Bmp4WT mice prenatally exposed to NS; #p < 0.05, compared with Bmp4WT mice prenatally exposed to DEX
Consistent with the findings in rats, expression levels of both BMP4 mRNA (Fig. 2B) and protein (Fig. 2C,D) were decreased in mouse offspring prenatally treated with DEX and restored in Bmp4OE mouse offspring. The FLAG expression was significantly evident in the myocardium of Bmp4OE mouse offspring but not in the WT mouse offspring (Fig. 2C,E). In mice subjected to cardiac I/R injury, we found that BMP4 overexpression reversed cardiac dysfunction caused by prenatal DEX exposure, evidenced by recovered fractional shortening (FS, Fig. 2F,G) and ejection fraction (EF, Fig. 2H). Other parameters, including left ventricular internal dimensions (LVID), left ventricular anterior wall thickness (LVAW), left ventricular posterior wall thickness (LVPW), heart rate (HR), left ventricular volume (EDV, ESV), stroke volume (SV) and cardiac output (CO) in mice, are shown in Supplemental Table 2. LVID, LVAW, LVPW and ESV were notably restored in Bmp4OE mice prenatally exposed to DEX.
TTC staining was used to assess the infarction area in hearts of mice in each group. It was shown that after I/R, the percentage of cardiac infarction area in mouse offspring prenatally exposed to DEX was significantly increased compared with those prenatally exposed to NS (27.11 ± 8.106% vs. 63.17 ± 6.857%) (Fig. 2I,J). The induced expression of BMP4 in myocardium significantly decreased the percentage of cardiac infarction area in the mice offspring prenatally exposed to DEX by more than 40.81% (63.17 ± 6.857% vs. 22.36 ± 2.593%) (Fig. 2I,J).
Taken together, these data suggest that BMP4 exerted an in vivo protective effect on the cardiac function from I/R injury in prenatally GC-exposed mice offspring.
Cardiac protection by BMP4 is mediated by the intracellular Smad signaling pathway
Both Smad and non-Smad signaling pathways are involved in the regulatory effects of Bmp4 on the target genes. So we next investigated which pathway BMP4 uses to exert its protective effects on cardiac function. It was shown that the phosphorylation level of Smad1/5/8 (p-Smad1/5/8) was reduced remarkably in the myocardium from male mice offspring prenatally exposed to DEX compared with NS control group (Fig. 3A,B). Intriguingly, decreased p-Smad1/5/8 was restored in Bmp4OE mice prenatally exposed to DEX (Fig. 3A,B). The expression level of Smad1 was comparable among these groups (Fig. 3A,C). By contrast, the protein levels of non-Smad pathway signaling components, i.e., P85 (Fig. 3D–F), JNK (Fig. 3D,G,H), ERK1/2 (Fig. 3D,I,J) and p38 (Fig. 3D,K,L) manifested no significant differences either in the WT offspring or in the Bmp4OE offspring prenatally exposed to DEX. These results collectively suggested that endogenously overexpressed BMP4 activates the Smad1/5/8 signaling pathway, which subsequently leads to the protection from cardiac dysfunction caused by excess DEX exposure during pregnancy.

BMP4 exerts its cardiac protective effects via the intracellular Smad signaling pathway. The phosphorylation level of Smad1/5/8 (p-Smad1/5/8) was reduced remarkably in the myocardium from male mice offspring prenatally exposed to DEX compared with NS control group, which was restored in Bmp4OE mice (A, B). The expression level of Smad1 was comparable among these groups (A, C). The protein levels of non-Smad pathway, i.e., P-85, JNK, ERK1/2 and p38 manifested no significant differences either in the WT offspring or in the Bmp4OE offspring prenatally exposed to DEX (D–L). n = 6, **p < 0.01, compared with Bmp4WT mice prenatally exposed to NS; #p < 0.05, compared with Bmp4WT mice prenatally exposed to DEX
BMP4 overexpression in vivo protects cardiomyocytes from apoptosis induced by prenatal DEX exposure in male offspring mice
Our previous work showed that DEX exposure increased cardiomyocyte apoptosis caused by I/R injury, which could be reversed by pretreatment of BMP4 in cultured rat cardiomyocytes [21]. To further investigate the effect of BMP4 on cardiomyocyte apoptosis in vivo, we detected the apoptosis in myocardium after I/R in both WT and Bmp4OE mice prenatally exposed to DEX. We found that the cleaved-caspase 3 was decreased from myocardium in which BMP4 was specifically overexpressed, compared with that from WT offspring, both of which were prenatally exposed to DEX (Fig. 4A,B). Meanwhile, the expression of Bcl-2 and Bax showed no differences among the three groups (Fig. 4A,C,D). Moreover, the increased number of apoptotic cardiomyocytes in WT offspring prenatally exposed to DEX was significantly reduced in Bmp4OE mice offspring prenatally exposed to DEX (Fig. 4E,F). To further verify the anti-apoptotic effect of BMP4 in cardiomyocytes in vitro, we employed another mouse model in which BMP4 was constitutively overexpressed in myocardium, without need of tamoxifen induction (Bmp4αMyh6-Cre) (Supplemental Fig. 2A). The constitutive overexpression of BMP4 in Bmp4αMyh6-Cre transgenic mice was validated by qRT-PCR, Western Blot and fluorescence of EGFP in neonatal NMCMs (Supplemental Fig. 2B–D). We isolated and cultured neonatal mouse cardiomyocytes (NMCMs) from WT neonatal mice either exposed to NS or DEX, and Bmp4αMyh6-Cre neonatal mice exposed to DEX. NMCMs were double stained with Annexin V and PI after H/R treatment and were analyzed by Flow Cytometry. The results showed that the apoptotic rate of cardiomyocytes in WT offspring prenatally exposed to DEX was significantly higher than that of those prenatally exposed to NS (Fig. 4G,H). However, the apoptosis of cardiomyocytes was significantly reversed in the Bmp4αMyh6-Cre mice offspring prenatally exposed to DEX (Fig. 4G,H).

BMP4 overexpression protects against myocardial apoptosis upon I/R in male offspring mice prenatally exposed to DEX. Representative immunoblot showing the expression of cleaved-caspase 3, Bax and Bcl-2 in myocardium of male offspring prenatally exposed to NS or DEX (A). Quantitative analysis of cleaved-caspase 3, Bax and Bcl-2 expression is shown in panels B, C and D, respectively. n = 6, *p < 0.05, compared with Bmp4WT mice prenatally exposed to NS; #p < 0.05, compared with Bmp4WT mice prenatally exposed to DEX. Tunel staining showed that the percentage of apoptotic cardiomyocytes was decreased in the Bmp4OE mice, compared with those in Bmp4WT prenatally exposed to DEX (E, F). Original magnification: × 200; scale bar: 50 μm. n = 12, *p < 0.05, compared with Bmp4WT mice prenatally exposed to NS; #p < 0.05, compared with Bmp4WT mice prenatally exposed to DEX. The similar apoptotic phenotype of cardiomyocytes was observed by Flow Cytometry analysis (G, H). n = 6–9, **p < 0.01, compared with Bmp4WT mice prenatally exposed to NS; ##p < 0.01, compared with Bmp4WT mice prenatally exposed to DEX
BMP4 overexpression rescues mitochondrial damage and reverses oxidative stress by limiting the aberrant increase in PGC-1α in myocardium of offspring prenatally exposed to DEX
It has been reported that peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator-1α (PGC-1α) can be upregulated by DEX in primary fetal cardiomyocytes and act as a key mediator of GC-induced maturation of fetal cardiomyocytes [30]. However, overexpression of PGC-1α has also been shown to induce mitochondrial proliferation and promote the production of reactive oxygen species (ROS) in cardiomyocytes [31,32,33]. This has been shown to underlie the pathogenesis of myocardial dysfunction in rats prenatally exposed to DEX [21]. Therefore, we next investigated if BMP4 could exert its cardiac protective effects by downregulating the expression of PGC-1α in prenatally DEX-exposed mice model. As expected, we found that the PGC-1α mRNA expression was increased in the cardiomyocytes from prenatally DEX-exposed mice, which was reversed by the in vivo overexpression of BMP4 in cardiomyocytes (Fig. 5A). Interestingly, both mRNA (Fig. 5B) and protein levels (Fig. 5C,D) of PGC-1α decreased in the Bmp4αMyh6-Cre mice, whereas the mRNA expression of PGC-1α could be restored by knocking down BMP4 expression in cultured NMCMs infected with shBmp4 adenovirus (Supplemental Fig. 3, Fig. 5E).

In vivo BMP4 overexpression rescues mitochondrial damage and reverses oxidative stress caused by prenatal exposure of DEX by limiting the aberrant increase in PGC-1α in offspring myocardium. PGC-1α mRNA expression was increased in cardiomyocytes from offspring prenatally exposed to DEX. This could be prevented by BMP4 overexpression in Bmp4OE mice (A), n = 4–6. **p < 0.01, compared with Bmp4WT mice prenatally exposed to NS; #p < 0.05, compared with Bmp4WT mice prenatally exposed to DEX. Both the mRNA and protein levels of PGC-1α were reduced by constitutive BMP4 overexpression in Bmp4flox/flox-αMyh6-Cre (Bmp4αMyh6-Cre) mice as well (B–D). n = 4, *p < 0.05, compared with Bmp4WT mice. PGC-1α mRNA levels could be upregulated by knocking down Bmp4 in NMCMs of Bmp4αMyh6-Cre (E). n = 7, *p < 0.05, compared with NMCMs infected with control adenovirus. The increased ROS levels and decreased ATP levels in offspring cardiomyocytes upon I/R caused by prenatal DEX exposure could both be reversed by in vivo overexpression of cardiac BMP4 in Bmp4αMyh6-Cre mice, to a level comparable with that in WT mice prenatally exposed to NS (F, G). n = 6, *p < 0.05, compared with Bmp4WT mice prenatally exposed to NS; ##p < 0.01, compared with Bmp4WT mice prenatally exposed to DEX
In primary cultured NMCMs isolated from WT and Bmp4αMyh6-Cre mice, we found that the increased ROS level and decreased ATP level in offspring cause by prenatal DEX exposure could be both reversed by in vivo overexpression of cardiac BMP4 in Bmp4αMyh6-Cre mice, to a level comparable with that in WT mice prenatally exposed to NS (Fig. 5F,G).
BMP4 overexpression inhibited DEX-enhanced mitophagy in cardiomyocytes caused by H/R
A growing number of studies suggest that accumulation of mitochondrial ROS promoted mitochondria-specific autophagy (mitophagy), and induced cell apoptosis [34]. Meanwhile, it was reported that DEX could promote Parkin-mediated mitophagy during maturation of embryonic stem cell-derived cardiomyocytes [35]. To further investigate the cellular mechanisms underlying the cardiac protective effect of BMP4 from H/R injury, we analyzed Parkin-mediated mitophagy in cardiomyocytes among different groups. It was found that the mRNA levels of Parkin were increased in cardiomyocytes from offspring prenatally exposed to DEX, compared to those of offspring prenatally exposed to NS (Fig. 6A). The upregulation of Parkin was reversed by cardiac specific overexpression of BMP4 in Bmp4OE mice (Fig. 6A). Intriguingly, in vivo overexpression of cardiac BMP4 significantly decreased the basal mRNA (Fig. 6B) and protein expression (Fig. 6C,E) of Parkin in Bmp4αMyh6-Cre mice. Moreover, the ratio between LC3II and LC3I was reduced in the Bmp4αMyh6-Cre mice as well (Fig. 6D,E), while the protein levels of P62 were not different in the myocardium between the WT and Bmp4αMyh6-Cre mice (Fig. 6E,F). Knocking down the BMP4 expression in cultured NMCMs by infection with shBmp4 adenovirus could restore the upregulation of Parkin (Supplemental Fig. 3, Fig. 6G). These results suggested that BMP4 could protect the cardiomyocytes from mitophagy through downregulation of Parkin.

BMP4 inhibited DEX-enhanced mitophagy in cardiomyocytes caused by H/R in a Parkin-dependent manner. Parkin mRNA expression was increased in cardiomyocytes from prenatally DEX-exposed mice; this which was reversed by BMP4 overexpression in Bmp4OE mice (A). n = 4–6. *p < 0.05, compared with Bmp4WT mice prenatally exposed to NS; #p < 0.05, compared with Bmp4WT mice prenatally exposed to DEX. Both the mRNA and protein levels of Parkin decreased in Bmp4αMyh6-Cre mice (B, C, E). n = 4. *p < 0.05, **p < 0.01, compared with Bmp4WT mice. The ratio between LC3II and LC3I was reduced in the Bmp4αMyh6-Cre mice (D, E). n = 4, **p < 0.01, compared with Bmp4WT mice. The protein levels of P62 manifested no differences in the myocardium between the WT and Bmp4αMyh6-Cre mice (E, F). n = 4. Parkin mRNA levels also could be upregulated by knocking down Bmp4 in NMCMs of Bmp4αMyh6-Cre mice (G). n = 7, *p < 0.05, compared with NMCMs infected with control adenovirus
BMP4 functions in physiological or pathological situations are generally mediated by two types of transmembrane receptors, which are type I and type II serine/threonine kinases [36]. We detected the mRNA and protein levels of BMP4 type I receptor, AVCRI, type II receptors, ACVRIIA and BMPRII, and found no significant differences in mRNA and protein expression in the myocardium among the three groups (Supplemental Fig. 4A–G). However, the inhibition of BMPRII by its specific antagonist, DMH1, significantly increased the production of ROS as well as the expression of PGC-1α and Parkin, and caused decreased production of ATP in NRCMs cells (Supplemental Fig. 4H–K). This suggests that the protective effects of BMP4 on the myocardium may be mediated by its specific receptor BMPRII.
Pretreatment of cultured NMCMs with 5-AZA may counteract the adverse effects on cardiac function caused by DEX exposure during pregnancy
Finally, to assess whether blocking DNA hypermethylation induced by prenatal DEX exposure could reverse the mitochondrial damage, cell apoptosis and cardiac dysfunction upon H/R, we pretreated NMCMs isolated from WT neonatal mice prenatally exposed to DEX with 5-AZA, a DNA methylation inhibitor, before H/R treatment in vitro. As shown in Fig. 7, treatment with 5-AZA significantly increased cardiac BMP4 protein expression (Fig. 7A) and reduced the apoptosis of cardiomyocytes in mice offspring prenatally exposed to DEX compared with control group (Fig. 7B,C). However, in the NMCMs isolated from WT neonatal mice prenatally exposed to NS, pretreatment of 5-AZA did not significantly affect the apoptosis of cardiomyocytes (Fig. 7B,D). In addition, the accumulation of ROS in NMCMs isolated from WT neonatal mice prenatally exposed to DEX upon H/R treatment was decreased (Fig. 7E), while ATP levels were increased after pretreatment with 5-AZA (Fig. 7F). These results indicated that pretreatment with 5-AZA may reverse the cardiac dysfunction induced by DEX exposure during pregnancy by restoring BMP4 production and protecting mitochondrial function from overwhelmed oxidative stress in cardiomyocytes.

Pretreatment of cultured NMCMs with 5-AZA can counteract the adverse effects on cardiac function caused by DEX exposure during pregnancy. Pretreatment with the DNA methylation inhibitor 5-AZA increased cardiac BMP4 protein expression in WT mouse offspring that were prenatally exposed to DEX (A, n = 4). The apoptosis of cardiomyocytes was not affected by 5-AZA pretreatment in mice prenatally exposed to NS (B, D), but was significantly reduced in mice prenatally exposed to DEX after H/R, compared with the cardiomyocytes pretreated with PBS (B, C). n = 3–4. The accumulation of ROS was decreased (E, n = 6), while ATP levels were increased (F, n = 6) after pretreatment of 5-AZA in NMCMs isolated from myocardium of offspring of mice prenatally exposed to DEX. *p < 0.05, compared with NMCMs treated with PBS control
Discussion
Embryonic development and early life are two major susceptibility windows during which epigenetic programming is sensitive to environmental influences, such as diet, temperature, environmental toxins, maternal behavior or childhood abuse [37]. Recently, a growing amount of evidence indicated that epigenetic mechanisms, including DNA methylation, histone modification and the actions of small non-coding RNAs, are important in developmental reprogramming. However, the precise mechanisms by which exposure to environmental factors can influence long-term outcomes are highly complex. In our previous studies, it was of great interest to determine the abnormal cardiac functions that were manifested in DEX-exposed offspring only after I/R but not in those maintained under normal oxygen conditions. Hypoxia inducible factor-1α (HIF-1α) is a key transcription factor that regulates the expression of hundreds of genes in the adaptive response to hypoxia [38]. Under normoxic conditions, HIF-1α expression is tightly regulated by ubiquitination and subsequent proteasomal degradation [39]. This regulation can be suppressed by hypoxia, which leads to elevated expression levels of active HIF-1α that translocates to the nucleus and transcriptionally regulates genes associated with physiological processes, including angiogenesis and metabolism [40]. Many studies have demonstrated that up-regulation of HIF-1α enhanced the cardioprotective effects against I/R [41,42,43].
In the present study, we observed that the mRNA and protein levels of Bmp4 in neonatal rat cardiomyocytes were enhanced upon H/R treatment in comparison with those cultured under normoxic condition. Intriguingly, HIF-1α is predicted by bioinformatics analysis to be a potential transcriptional factor binding to the Bmp4 promoter, suggesting that hypoxia or ischemia-induced HIF-1α may bind to the promoter region of Bmp4 and activate its transcription and expression to protect the myocardium from hypoxia/reoxygenation or ischemia/reperfusion injury. Our current findings using ChIP-qPCR confirmed that HIF-1α binding was indeed increased in the promoter region of Bmp4 upon hypoxia treatment, resulting in increased Bmp4 expression in rat H9C2 cardiomyocytes. These results were consistent with previous reports, showing that HIF-1α also upregulated transcriptional activation of Bmp4 expression in rat pulmonary arterial smooth muscle cells and mouse embryonic stem cells [44, 45]. This further indicates that hypoxia induction of HIF-1α and HIF-1α upregulation of BMP4 is a ubiquitous mechanism among various tissues.
However, chromatin states can substantially influence the binding abilities of transcription factors to the promoters of target genes and their subsequent transactivation [46]. Our present results, from MeDIP-sequencing, Q-PCR, Western Blot and MSP, demonstrated that the promoter region of Bmp4 in myocardium from rat male offspring exposed to GC during pregnancy is hypermethylated. Therefore, it makes perfect sense that we speculated that hypermethylation of Bmp4 promoter impairs its binding of HIF-1α and attenuates the transactivation of Bmp4. We designed the ChIP-qPCR primers flanking the hypermethylated sequences on Bmp4 promoter containing the HIF-1α binding site and found that HIF-1α enrichment on Bmp4 promoter was significantly decreased after GC exposure during pregnancy, mostly due to the hypermethylation of Bmp4 promoter. Moreover, mutation or deletion of core sequences within the hypoxia response element (HRE) of the Bmp4 promoter that were hypermethylated caused by prenatal GC exposure prevented the induction of luciferase activity. This further indicates that hypermethylation of Bmp4 promoter hampered its ability to bind HIF-1α, resulting in prevention of Bmp4 transactivation and a reduced protection by BMP4 of cardiac function against H/R or I/R. Mechanistically, these results revealed that hypoxia acts as a “second hit” exaggerating the pathological changes on myocardium and explains why cardiac function of rats prenatally exposed to DEX was decreased only after ischemia–reperfusion injury, but not under normoxic conditions.
BMP4 regulates downstream signaling pathways through its receptors. Although the DNA methylation level of Bmp4 type II receptor, ACVRIIb, is also increased according to our MeDIP-sequencing results [21], neither mRNA nor protein changes of AVCRI, ACVRIIa or BMPRII were observed in the myocardium from WT or Bmp4OE mice prenatally exposed to DEX or to NS. The abundance of ACVRIIb was too low to be detected by Western Blot. These results suggested that prenatal GC overexposure does not affect the expression of BMP4 receptors. There are generally two pathways downstream BMP4 receptor activation, namely Smad and non-Smad pathways. It was reported that BMP4 induced myocardial apoptosis by activating the JNK/MAPK pathway in Bmp4 heterozygous KO mice during I/R [29]. However, in the present study, it was shown that BMP4 performed its cardiac protective functions via activating p-Smad1/5/8 pathway, rather than non-Smad pathways in the hearts of male offspring prenatally exposed to DEX, suggesting that the different downstream pathways activated by BMP4 may mediate different subsequent biological functions. Through activation of p-Smad1/5/8 pathway, overexpression of BMP4 exerted the anti-apoptotic effect in cardiomyocytes rather than pro-apoptotic effect mediated by non-Smad pathway.
In the present study, we found that the ROS level was increased and the ATP level was decreased in NMCMs isolated from male mouse offspring of pregnant mothers exposed to DEX. This is consistent with our previous findings in rats [21], suggesting again that mitochondrial damage and oxidative stress in cardiomyocytes caused by H/R were further negatively affected by prenatal DEX exposure. Intriguingly, these phenotypic changes could be reversed by either in vivo or in vitro overexpression of cardiac BMP4. It has been reported that PGC-1α is an essential molecule in normal mitochondrial development, function, dynamics, and in maturation of cardiomyocytes [47]. Heart-specific PGC-1α KO induces metabolic, functional, and structural abnormalities leading to dilating cardiomyopathy and heart failure [48]. However, overexpression of PGC-1α has been shown to induce mitochondrial proliferation and promote the production of ROS in cardiomyocytes [49], suggesting the fine-tuning of PGC-1α is critical for normal mitochondrial functions.
Members of the TGF-β superfamily exert different effects on the regulation of PGC-1α expression. BMP-7 significantly increased the expression of PGC-1α in a brown preadipocyte cell line [50], while the treatment of differentiated C2C12 myotubes with TGF-β suppressed PGC-1α mRNA and protein expression [51]. As similar with TGF-β, BMP4 was shown to reverse the upregulation of PGC-1α in cardiomyocytes of male offspring of DEX-treated pregnant mothers, which is consistent with the previous report that DEX can induce PGC-1α mRNA in a dose-dependent manner in primary fetal cardiomyocytes [30] and collectively suggest that DEX may induce PGC-1α by reducing BMP4 expression. In the canonical Smad-dependent pathway, phosphorylated Smad1/5/8 hetero-oligomerize with Smad4 and translocate to the nucleus, where they interact with transcription factors to regulate gene expression [52], such as suppressing Pentraxin 3 [53] and StAR [54] expression in human granulosa-lutein cells. Therefore, we speculate that the p-Smad1/5/8-Smad4 complex may suppress PGC-1α mRNA transcription and mediate the suppressive effect of BMP4 on PGC-1α. However, the specific regulatory mechanisms for these effects need to be further investigated in future studies.
Mitophagy plays an important role in cardiac I/R through Parkin-dependent and Parkin-independent processes, and dysregulation of mitophagy confers cardiac mitochondrial damage [55]. Parkin, an ubiquitin E3 ligase, adds ubiquitin to many substrates leading to interactions with LC3, a key component of autophagosomes [56]. The expression of Parkin in cardiomyocytes was found to be elevated by treatment with exogenous H2O2 or inhibited by ROS inhibitors [57]. Our present findings suggest that Parkin mRNA levels are increased in cardiomyocytes treated with H/R. This was further enhanced by prenatal DEX exposure, probably caused by increased expression of PGC-1α and ROS. However, the mRNA and protein levels of Parkin and the ratio of LC3II/LC3I were decreased in cardiomyocytes by BMP4 overexpression. Taken together, these findings strongly suggest that BMP4 may inhibit mitophagy in cardiomyocytes by blocking the PGC-1α-ROS-Parkin pathway.
Conclusion
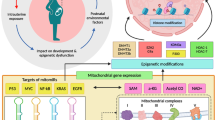
In summary, this study demonstrates that DNA hypermethylation of the Bmp4 promoter in cardiomyocytes caused by prenatal DEX exposure substantially dampens promoter binding of transcription factor HIF-1α induced by cardiac ischemia and hypoxia. The resulting decrease in BMP4 expression relieves its inhibitory effects on PGC-1α, and the elevated PGC-1α increases ROS production which leads to Parkin-dependent mitophagy and enhances cardiomyocyte apoptosis upon I/R or H/R. Pretreatment with the DNA methylation inhibitor, 5-AZA, could serve as a potential therapeutic for the programming effect of GCs during pregnancy on neonatal cardiac dysfunction (Fig. 8).

Working model. In normal cardiomyocytes, hypoxia due to ischemia–reperfusion induces the expression of transcription factor HIF-1α, which can bind to the promoter region of Bmp4 and substantially increase BMP4 expression. BMP4 exerts cardiac protection effects by inhibiting PGC-1α expression and attenuating mitophagy of cardiomyocytes caused by hypoxia. Prenatal DEX exposure causes DNA hypermethylation of the HIF-1α binding site within the Bmp4 promoter in cardiomyocytes, which dampens the binding activity of HIF-1α and decreases the transcription and expression of Bmp4. The decrease in BMP4 expression relieves its inhibitory effects on PGC-1α, which is further enhanced by DEX exposure. The elevated PGC-1α increases ROS production, leading to Parkin-dependent mitophagy. This enhances apoptosis of cardiomyocytes upon I/R or H/R, eventually leading to cardiac dysfunction in offspring of mice prenatally exposed to DEX
Methods and materials
Animal model
Bmp4flox/flox mice on C57BL/6 background were generated using the CRISPR-Cas9 gene editing method by the Shanghai Model Organisms (Shanghai, China). Bmp4flox/flox mice were crossed with αMyh6-Cre/Esr1* (Shanghai Model Organisms) transgenic mice to generate Bmp4flox/flox-αMyh6-Cre/Esr1* mice. Cre recombinase (MerCreMer) induced by tamoxifen cleavage of the stop codon upstream of the Bmp4 coding sequence, resulted in mRNA and protein overexpression of Bmp4 in juvenile and adult cardiac myocytes. In our study, tamoxifen (40 mg/kg, #T5648, Sigma-Aldrich) or corn oil (c8267, Sigma-Aldrich) was injected intraperitoneally into mice every 7 days, from the age of 8 weeks for two consecutive weeks before performing the subsequent experiments. In another set of experiments, the Bmp4flox/flox mice were crossed with αMyh6-Cre transgenic mice (gifted by Dr. Junjie Xiao, Shanghai University) to generate Bmp4flox/flox-αMyh6-Cre mice, in which Bmp4 was constitutively overexpressed in the myocardium without the need of induction by tamoxifen.
All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Committee on Ethics of Medicine, Second Military Medical University, Shanghai. Mice were housed in the SPF animal center of Second Military Medical University, under a 12-h-dark/12-h-light cycle, and allowed free access to a standard pellet chow and water. Males and females were mated at 18:00 h and separated the following morning at 06:00 h. Pregnancy was indicated by the presence of a vaginal plug, which was designated as 0.5 days post-coitus (dpc). The time to labor was documented upon delivery of the first pup or by the presence of a litter. The pregnant mice were randomly divided into DEX-treated group and NS-injected groups, which were subcutaneously injected with DEX (0.1 mg/kg/day) or the same volume of NS daily from 12.5 dpc until labor. The offspring were maintained in the same environment to adulthood (12 weeks).
The in vivo ischemia/reperfusion (I/R) model
After Bmp4 expression in heart was induced for 2 weeks in male mouse offspring, myocardial ischemia–reperfusion (I/R) was performed in vivo according to a previously described protocol [21]. Briefly, mice were initially anesthetized with 3% isoflurane inhalation and narcosis was maintained at 1.5–2% isoflurane, using a gas anesthesia machine (CWE Incorporated, USA). Mice were placed in a supine position, and the hair was removed from the sternal epidermis. The skin around the heart was removed, and the third and fourth ribs and intercostal muscles were punctured on the left side. The pericardium was then dissected, and the heart was squeezed out. The left coronary artery (LCA) was ligated with a 6–0 silk suture at a point 1–2 mm inferior to the left auricle. Successful ligation was confirmed when the anterior wall of the LV turned pale. The thoracotomy site was then stitched in layers with a 4–0 silk suture. After 30 min of ischemia, the silk suture was loosened and reperfusion was allowed for another 24 h.
Primary cardiomyocyte isolation, culture, and treatment
Neonatal mouse cardiomyocytes (NMCMs) and neonatal rat cardiomyocytes (NRCMs) were isolated and cultured, as previously reported [21, 58]. The cells were cultured for 4-days before the hypoxia/reoxygenation model (H/R) was established according to the protocol described previously [21]. Briefly, cardiomyocytes cultured in serum-free and glucose-free DMEM were placed in a hypoxic chamber (HF100, Heal Force, China) containing 94% N2/5% CO2/1% O2 for 5 h, after which cells were subjected to reoxygenation in a standard incubator (5% CO2/95% air) in normal medium for 2 h.
5-Aza-2′-deoxycytidine (5-AZA, 189826, Sigma) was used as a DNA methylation inhibitor. The solution of 5-AZA was prepared with saline, and the final working concentration in the culture medium was 1 µM, as described in a previous study [59]. The control cells were cultured in the same volume of saline. After 48 h of treatment with 5-AZA or saline, NMCMs were subjected to H/R, as well.
DMH1 (T1942, Topscience, China) was used as a BMPRI antagonist and was dissolved in DMSO. The NMCMs were treated with DMH1 at a final concentration of 10 nmol/l, and the same volume of DMSO was given as the vehicle control [26].
Echocardiography
Echocardiography of the I/R mice was performed using an ultrasound imaging system (Esaote MyLab Touch, SL3116, Italy). In short, mice were anesthetized with 2% isoflurane inhalation and scanned by two-dimensional M-mode echocardiography with a 22 MHz cardiac probe to record physiological data. Measurement techniques were consistent with the American Society of Echocardiography conventions [60]. Briefly, the probe was placed on the left side of sternum with 30-degree angle to the sternal midline, showing the left ventricular long axis section; then, the probe was rotated 90 degrees clockwise to show the left ventricular short axis section. Specifically, the left ventricular end–diastolic anterior wall thickness (AWT), posterior wall thickness (PWT), the left-ventricular end diastolic dimension (LVEDD), the left-ventricular end systolic dimension (LVESD) and heart rate were measured and recorded. Left-ventricular fractional shortening (LVFS), an index of LV systolic function, was calculated using the following equation: LVFS = (LVEDD − LVESD)/LVEDD × 100%. Left ventricular end-diastolic volume (LVEDV) = 1.04 × LVEDD3; left ventricular end-systolic volume (LVESV) = 1.04 × LVESD3. Left ventricular ejection fraction (LVEF) = (LVEDV − LVESV)/LVEDV × 100%. All ultrasound measurements were taken on an average of 5 cardiac cycles, and all investigators performing echocardiographic acquisition or analysis were blinded to treatment group.
Quantification of I/R injury size using TTC staining
After I/R, the suture on the LCA was tightened again, and the heart was filled with 1% Evans blue (A602025, BBI, Sangon Biotech, St. Louis, China) by intravenous injection. When the skin of chest wall became blue, the heart was immediately excised and quickly frozen on dry ice. Then, the heart was cut into 2-mm-thick slices parallel to the atrioventricular groove; these were subsequently incubated with 1% 2,3,5-triphenyltetrazolium chloride (TTC, BBI, Sangon Biotech, China) in phosphate buffer (pH 7.4) at 37 °C for 3 min. Finally, the slices were immersed into 10% paraformaldehyde for 20 min. The heart sections not stained with Evans blue were identified as the risk area. The infarct area was identified as pale in color after TTC staining, since the infarct area does not contain dehydrogenase enzymes which can convert TTC into formazan and stain viable myocardium with dark red. The area at risk and the area of infarct were quantified using ImageJ (https://imagej.nih.gov/ij/). The quantification of I/R injury was expressed as a ratio of the infarct area and the area at risk.
Methylation-specific PCR
In a previous study, we found that the methylation level of the Bmp4 promoter region (rn5_dna range = chr15:24738861–24739100) was significantly increased in prenatally DEX-exposed rat male offspring using MeDIP sequencing. To further confirm this result, we performed methylation-specific PCR (MSP) using bisulfite-modified DNA isolated from the heart of male mouse offspring prenatally exposed to DEX. The primer pairs for the methylated (M) DNA and unmethylated (U) DNA sequences were designed using an online tool from The Li Lab (http://www.urogene.org), and the sequences of primer pairs are listed in Supplemental Table 3. Genomic DNA from the mouse hearts of the DEX or NS groups was extracted using a Mag-MK Animal Genomic DNA Extraction Kit (B518721-0100, Sangon Biotech, China). DNA bisulphite conversion was performed with a commercially available kit (EpiMark® Bisulfite Conversion Kit, #E3318S, New England Biolabs). DNA Amplification was achieved in a 25 μl reaction volume containing 0.5 μl of each primer, 2 μl of modified DNA, 0.125 μl EpiMark Hot Start Taq DNA Polymerase (#M0490S, New England Biolabs). The PCR reaction conditions were optimized according to the manufacturer's instructions. The PCR products (134 bp) were visualized on a 2% agarose gel stained with 4S GelRed (Sangon Biotech, China) which can be illuminated by UV.
RNA extraction and quantitative real-time PCR analysis
Total RNA was isolated from heart tissues or cells using TRIzol reagent (15596018, Invitrogen, America). The concentration of total RNA was determined by measuring absorbance at OD260 on a Microplate Reader. cDNA synthesis was performed using PrimeScript™ RT reagent Kit (RR047A, Takara, China), and 0.5 μg of mRNA from each sample was reverse transcribed into cDNA, according to the manufacturer’s instructions. Quantitative analysis of mRNA was performed on a CFX Connect Real-Time PCR Detection System (Bio-Rad) using SYBR Green Real-time PCR Master Mix (QPK-201, TOYOBO, Japan). The cycling conditions were 95 °C for 3 min, followed by 40 cycles at 95 °C for 15 s, 50 °C for 30 s, and 72 °C for 15 s. The primers are listed in Supplemental Table 4. As a negative control for all of reactions, distilled water was used in place of cDNA. The specificity of primers was verified by examining the melting curve, and amplification of the GAPDH was determined for sample loading and normalization. Each sample was amplified in triplicate, and the mean Ct value of each sample was calculated. The comparative Ct method (2−ΔΔCt) was employed to compare the relative fold changes among different samples [61].
Western Blot
Total proteins were extracted from tissues and cells using RIPA buffer (Cell Signaling). Equivalent amounts of protein determined by BCA assay were resolved by 10% Bis–Tris gel electrophoresis and blotted to Immobilon PVDF membranes (Millipore). The following primary antibodies and dilutions were used: anti-BMP4 (1:1000; ab39973, Abcam); anti-GAPDH (1:2000; sc-32233, Santa Cruz);anti-Flag (1:1000; F1804, Sigma); anti-ACVRIIA (1:1000; ab134082, Abcam); anti-ACVRI (1:1000; 4398 s, Cell Signaling); anti-BMPRII (1:1000; 6979 s, Cell Signaling); anti-p-Smad1/5/8 (1:1000; 13820 s, Cell Signaling); anti-Smad1 (1:1000; 9743 s, Cell Signaling); anti-P85 (1:1000; 4292S, Cell Signaling); anti-Phospho-SAPK/JNK (Thr183/Tyr185) (1:1000; 4668S, Cell Signaling); anti-SAPK/JNK (1:1000; 9252 s, Cell Signaling); anti-P-ERK1/2 (1:1000; 8544S, Cell Signaling); anti-ERK1/2 (1:1000; 4695S, Cell Signaling); anti-P-P38 (1:1000; 4511S, Cell Signaling); anti-P38 (1:1000; 8690S, Cell Signaling); anti-Capspase3 (1:1000; 9662S, Cell Signaling); anti-LC3B (1:1000; ab192890, Abcam); anti-Parkin (1:1000; ab77924, Abcam); anti-P62 (1:1000; ab109012, Abcam); anti-PGC1 alpha + beta (1:1000; ab188102, Abcam). The secondary antibody was horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Jackson). The ratio of band intensity of target protein to that of GAPDH was calculated as the relative expression level of protein.
Transcription factor-binding site analysis
In order to identify the putative transcription factor (TF) binding sites within the methylated regions of the Bmp4 promoter, bioinformatics analysis was performed using LASAGNA-Search 2.0 online tool (https://biogrid-lasagna.engr.uconn.edu/lasagna_search/) with TRANSFAC matrices and aligned models.
Chromatin immunoprecipitation (ChIP)-qPCR
ChIP assays were performed using a SimpleChIP® Plus Enzymatic Chromatin IP Kit (#9005, CST, USA) following the manufacturer’s instructions. Briefly, tissues (25 mg each sample) were crosslinked using formaldehyde (#12606, CST, USA) and digested into a single-cell suspension using a Tissuelyser (#12606, CST, USA). The nuclear extracts from pelleted cells were then sonicated, and the genomic DNA was fragmented to an average length of 200–1000 bp using a Bioruptor®Pico (Diagenode, America). Immunoprecipitation was conducted with an anti-HIF-1α antibody (Abcam) or IgG (Beyotime, China). Crosslinking was reversed by incubation in Elution buffer at 62 °C for 2 h. DNA was then extracted from samples using column purification and amplified by Real-time PCR.
Cell line culture and transfection
Rat myocardium cell line H9C2 cells (Chinese Academy of Science, Shanghai, China) were cultured in DMEM (11995065, Gibco, USA) containing 10% (v/v) fetal bovine serum (FBS, 10099-141C, Gibco, USA) and 1% (v/v) penicillin–streptomycin solution (15140-122, Thermo, USA) at 37 °C in an incubator (HFsafe1200LC) with 5% CO2 and 95% air. Lipofectamine™ 3000 transfection reagent (L3000-008, Invitrogen, USA) was utilized for transfection according to the manufacturer’s instructions.
Plasmids and luciferase assay
The methylated region of the rat Bmp4 promoter (rn5_dna range = chr15:24738861–24739100) was amplified and cloned into the pGL4.23-basic vector (Promega). The Hieff Mut™ Site-Directed Mutagenesis Kit (11003ES10, Yeasen) was used to mutate four nucleotides (gcaCGTCt to gcaAAGTt), or delete the four nucleotides (CGTC) in the putative HIF-1α binding site within the methylated region of the Bmp4 promoter, according to the manufacturer’s protocols. All constructs were confirmed by DNA sequencing at Sangon Biotech Inc. (Shanghai, China) using the primers listed in Supplemental Table 5.
H9C2 cells were seeded in 24-well plates and cultured for 24 h. pGL4.23-basic, pGL4.23-Bmp4 or pGL4.23-mutants were transfected into the H9C2 cells. Each plasmid was transfected in quadruplicate. pRL-TK plasmid (Renilla) was cotransfected as an endogenous control for normalization. After 24 h, the plate was removed into the tri-gas incubator (94%N2/5%CO2) for incubation in a hypoxic (1% oxygen) environment for another 24 h. The cells were then washed three times with cold phosphate-buffered saline (PBS) and lysed with luciferase assay buffer. Luciferase activity was analyzed using the dual-luciferase reporter assay system (E1960, Promega) according to the manufacturer’s instructions. Transcriptional activities were determined as the ratio of firefly luciferase activity to Renilla luciferase activity. This experiment was repeated three times.
TUNEL staining
After I/R, the hearts were washed with PBS and fixed with paraformaldehyde. Paraffin-embedded heart sections were deparaffinized, rehydrated and incubated with proteinase K at 37 °C for 20 min to retrieve the antigens. Apoptotic death of cardiomyocytes was determined using TUNEL kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Briefly, the cells were permeabilized with 0.1% Triton X-100 and incubated with 50 μl TUNEL reaction mixture in a humidified atmosphere for 60 min at 37 °C in the dark. To detect the nuclei, the cells were counter-stained with hematoxylin for 10 min at room temperature. The fraction of apoptotic cells was determined as the average ratio between the TUNEL-positive cells (brown staining) and the hematoxylin-positive cardiac myocyte nuclei (blue staining), from five randomly selected fields under the microscope (magnification 400 ×).
Flow cytometry analysis
Apoptosis of primary cardiomyocytes was determined using an APC Annexin V apoptosis detection kit with PI (640932, BioLegend) according to the manufacturer’s protocol. The cultured cells were dispersed using EDTA-trypsin and resuspended in 100 μl binding buffer. After incubation with 5 μl Annexin V-APC and 7 μl PI for 15 min in the dark, another 300 µl of binding buffer were added and the stained cells were examined on a BD FACSVerse (BD Biosciences, USA) within 1 h. Fragments of dead cells and debris were gated by forward- and side-scatter analysis with the FACSSuite software (BD Biosciences).
Determination of cellular reactive oxygen species (ROS)
The levels of ROS in cells were detected using 2′–7′-dichlorodihydrofluorescein diacetate (DCFH-DA; S0033S, Beyotime) following the manufacturer's instructions. Briefly, the cultured cells subjected to H/R treatment were washed with FBS-free culture medium and incubated with 10 μM oxidation-sensitive fluorescent probe DCFH-DA in the carbon dioxide incubator for 20 min at 37 °C. The stained cells were subjected to trypsin–EDTA digestion, and 2 × 104 cells in 100 μl DMEM were placed into a 96-well plate. The relative ROS levels generated by the cells were measured by a multifunctional microplate reader (Cytation5, BioTek, USA). The absorbance values were detected at the emission wavelength of 488 nm and the excitation wavelength of 525 nm.
Detection of cellular ATP levels
The ATP levels in cells were assessed using an Enhanced ATP Assay Kit (S0027, Beyotime, China). Briefly, cardiomyocytes in primarily-culture subjected to H/R treatment in 6-well plates were lysed in 200 μl lysis buffer on ice and collected by centrifugation. Twenty microliters of supernatant were added to a 96-well plate containing 100 μL ATP detection buffer in triplicate, and the luminescence was detected by multifunctional microplate reader (Cytation5, BioTek, USA). The concentration of ATP was calculated according to a standard curve. The protein concentration of each sample was also determined and used for normalization of the ATP levels.
Adenovirus construction and adenoviral infection
Knockdown of Bmp4 was accomplished using specific recombinant adenovirus small hairpin RNA (shRNA). The shRNA-Bmp4 recombinant adenovirus Adeno-U6-shRNA-CMV-mCherry (Ad-shBmp4-1 and Ad-shBmp4-2) and the empty control recombinant adenovirus (Ad-shNC) were purchased from Obio Technology Company (Shanghai, China). The target sequences of shRNA were as follows: shBmp4-1 (5′-GGATTACATGAGGGATCTTTA-3′), shBmp4-2 (5′-CCCTAGTCAACTCTGTTAATT-3′), and shNC (5′-TTCTCCGAACGTGTCACGT-3′). Adenoviral infection was performed according to the manufacturer’s instructions. Briefly, the NMCMs were cultured in serum-free and glucose-free DMEM for 48 h, before the cells were infected with recombinant adenovirus at a multiplicity of infection of 10 for 6 h. Then, the medium was removed, and the infected cells were allowed to recover in normal medium for another 24 h. Finally, the cells were harvested for the subsequent analysis.
Statistics
Statistical analysis was performed using GraphPad Prism 7 software, and all data are presented as mean ± SEM, or as the mean percentage of control ± SEM, in some cases. Differences between two groups were assessed by Student’s t test. Differences between multiple groups were analyzed by one-way ANOVA followed by a multiple comparisons test. p < 0.05 indicated a statistically significant difference.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Orlandi M, Graziani F, D’Aiuto F (2020) Periodontal therapy and cardiovascular risk. Periodontol 83(1):107–124
Barker DJ, Osmond C (1986) Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1(8489):1077–1081
Drake AJ, Tang JI, Nyirenda MJ (2007) Mechanisms underlying the role of glucocorticoids in the early life programming of adult disease. Clin Sci (Lond) 113(5):219–232
Langdown ML, Holness MJ, Sugden MC (2003) Effects of prenatal glucocorticoid exposure on cardiac calreticulin and calsequestrin protein expression during early development and in adulthood. Biochem J 371(Pt 1):61–69
Geelhoed JJM, Fraser A, Tilling K, Benfield L, Davey Smith G, Sattar N, Nelson SM, Lawlor DA (2010) Preeclampsia and gestational hypertension are associated with childhood blood pressure independently of family adiposity measures. Circulation 122(12):1192–1199
Lawlor DA, Macdonald-Wallis C, Fraser A, Nelson SM, Hingorani A, Davey Smith G, Sattar N, Deanfield J (2012) Cardiovascular biomarkers and vascular function during childhood in the offspring of mothers with hypertensive disorders of pregnancy: findings from the Avon Longitudinal Study of Parents and Children. Eur Heart J 33(3):335–345
Leybovitz-Haleluya N, Wainstock T, Landau D, Sheiner E (2018) Maternal gestational diabetes mellitus and the risk of subsequent pediatric cardiovascular diseases of the offspring: a population-based cohort study with up to 18 years of follow up. Acta Diabetol 55(10):1037–1042
Eriksson JG, Sandboge S, Salonen MK, Kajantie E, Osmond C (2014) Long-term consequences of maternal overweight in pregnancy on offspring later health: findings from the Helsinki Birth Cohort Study. Ann Med 46(6):434–438
Gaillard R, Steegers EA, Duijts L, Felix JF, Hofman A, Franco OH, Jaddoe VW (2014) Childhood cardiometabolic outcomes of maternal obesity during pregnancy: the Generation R Study. Hypertension 63(4):683–691
Victora CG, Adair L, Fall C, Hallal PC, Martorell R, Richter L, Sachdev HS (2008) Maternal and child undernutrition: consequences for adult health and human capital. Lancet 371(9609):340–357
Juonala M, Pitkanen N, Tolonen S, Laaksonen M, Sievanen H, Jokinen E, Laitinen T, Sabin MA, Hutri-Kahonen N, Lehtimaki T et al (2019) Childhood exposure to passive smoking and bone health in adulthood: the cardiovascular risk in Young Finns Study. J Clin Endocrinol Metab 104(6):2403–2411
Taal HR, de Jonge LL, van Osch-Gevers L, Steegers EA, Hofman A, Helbing WA, van der Heijden AJ, Jaddoe VW (2013) Parental smoking during pregnancy and cardiovascular structures and function in childhood: the Generation R Study. Int J Epidemiol 42(5):1371–1380
Zegkos T, Ntiloudi D, Giannakoulas G (2020) Parental alcohol exposure and congenital heart diseases in offspring: a causal link with controversial evidence. Eur J Prev Cardiol 27(4):407–409
Fowden AL, Li J, Forhead AJ (1998) Glucocorticoids and the preparation for life after birth: are there long-term consequences of the life insurance? Proc Nutr Soc 57(1):113–122
Moisiadis VG, Matthews SG (2014) Glucocorticoids and fetal programming part 1: outcomes. Nat Rev Endocrinol 10(7):391–402
Gao L, Rabbitt EH, Condon JC, Renthal NE, Johnston JM, Mitsche MA, Chambon P, Xu J, O’Malley BW, Mendelson CR (2015) Steroid receptor coactivators 1 and 2 mediate fetal-to-maternal signaling that initiates parturition. J Clin Invest 125(7):2808–2824
Namazy JA, Schatz M (2005) Treatment of asthma during pregnancy and perinatal outcomes. Curr Opin Allergy Clin Immunol 5(3):229–233
Grgic O, Ivanisevic M, Delmis J (2010) Treatment of idiopathic thrombocytopenic purpura in pregnancy with pulsed dose of dexamethasone. J Obstetr Gynaecol 30(8):864
Luo L, Li X, Yan R, Zhang H, Li C (2022) Risk factors for adverse pregnancy outcomes in women with rheumatoid arthritis and follow-up of their offspring. Clin Rheumatol 41:3135
Ninan K, Liyanage SK, Murphy KE, Asztalos EV, McDonald SD (2022) Evaluation of long-term outcomes associated with preterm exposure to antenatal corticosteroids: a systematic review and meta-analysis. JAMA Pediatr 176(6):e220483
Peng J, Zhou Y, Zhang Z, Wang Z, Gao L, Zhang X, Fang Z, Li G, Chen H, Yang H et al (2018) The detrimental effects of glucocorticoids exposure during pregnancy on offspring’s cardiac functions mediated by hypermethylation of bone morphogenetic protein-4. Cell Death Dis 9(8):834
Guo WT, Dong DL (2014) Bone morphogenetic protein-4: a novel therapeutic target for pathological cardiac hypertrophy/heart failure. Heart Fail Rev 19(6):781–788
Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BL (2003) An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev 17(19):2362–2367
Liu W, Selever J, Wang D, Lu MF, Moses KA, Schwartz RJ, Martin JF (2004) Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc Natl Acad Sci USA 101(13):4489–4494
McCulley DJ, Kang JO, Martin JF, Black BL (2008) BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev Dyn 237(11):3200–3209
Sun B, Huo R, Sheng Y, Li Y, Xie X, Chen C, Liu H-B, Li N, Li C-B, Guo W-T et al (2013) Bone morphogenetic protein-4 mediates cardiac hypertrophy, apoptosis, and fibrosis in experimentally pathological cardiac hypertrophy. Hypertension (Dallas, Tex: 1979) 61(2):352–360
Wu X, Sagave J, Rutkovskiy A, Haugen F, Baysa A, Nygard S, Czibik G, Dahl CP, Gullestad L, Vaage J et al (2014) Expression of bone morphogenetic protein 4 and its receptors in the remodeling heart. Life Sci 97(2):145–154
Liu Y, Du SY, Ding M, Dou X, Zhang FF, Wu ZY, Qian SW, Zhang W, Tang QQ, Xu CJ (2017) The BMP4-Smad signaling pathway regulates hyperandrogenism development in a female mouse model. J Biol Chem 292(28):11740–11750
Pachori AS, Custer L, Hansen D, Clapp S, Kemppa E, Klingensmith J (2010) Bone morphogenetic protein 4 mediates myocardial ischemic injury through JNK-dependent signaling pathway. J Mol Cell Cardiol 48(6):1255–1265
Rog-Zielinska EA, Craig MA, Manning JR, Richardson RV, Gowans GJ, Dunbar DR, Gharbi K, Kenyon CJ, Holmes MC, Hardie DG et al (2015) Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: a role for PGC-1alpha. Cell Death Differ 22(7):1106–1116
Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP (2000) Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Investig 106(7):847–856
Birket MJ, Casini S, Kosmidis G, Elliott DA, Gerencser AA, Baartscheer A, Schumacher C, Mastroberardino PG, Elefanty AG, Stanley EG et al (2013) PGC-1alpha and reactive oxygen species regulate human embryonic stem cell-derived cardiomyocyte function. Stem Cell Rep 1(6):560–574
Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, Attie AD, Muoio DM, Kelly DP (2014) A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res 114(4):626–636
Roca-Agujetas V, de Dios C, Leston L, Mari M, Morales A, Colell A (2019) Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid Med Cell Longev 2019:3809308
Zhou R, Li J, Zhang L, Cheng Y, Yan J, Sun Y, Wang J, Jiang H (2020) Role of Parkin-mediated mitophagy in glucocorticoid-induced cardiomyocyte maturation. Life Sci 255:117817
Hanna A, Frangogiannis NG (2019) The role of the TGF-beta superfamily in myocardial infarction. Front Cardiovasc Med 6:140
Cavalli G, Heard E (2019) Advances in epigenetics link genetics to the environment and disease. Nature 571(7766):489–499
Semenza GL (2000) HIF-1 and human disease: one highly involved factor. Genes Dev 14(16):1983–1991
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ et al (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (New York, NY) 292(5516):468–472
Gunton JE (2020) Hypoxia-inducible factors and diabetes. J Clin Invest 130(10):5063–5073
Nanayakkara G, Alasmari A, Mouli S, Eldoumani H, Quindry J, McGinnis G, Fu X, Berlin A, Peters B, Zhong J et al (2015) Cardioprotective HIF-1alpha-frataxin signaling against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 309(5):H867-879
Li X, Zhao H, Wu Y, Zhang S, Zhao X, Zhang Y, Wang J, Wang J, Liu H (2014) Up-regulation of hypoxia-inducible factor-1alpha enhanced the cardioprotective effects of ischemic postconditioning in hyperlipidemic rats. Acta Biochim Biophys Sin 46(2):112–118
Du M, Huang K, Huang D, Yang L, Gao L, Wang X, Huang D, Li X, Wang C, Zhang F et al (2015) Renalase is a novel target gene of hypoxia-inducible factor-1 in protection against cardiac ischaemia-reperfusion injury. Cardiovasc Res 105(2):182–191
Wang J, Fu X, Yang K, Jiang Q, Chen Y, Jia J, Duan X, Wang EW, He J, Ran P et al (2015) Hypoxia inducible factor-1-dependent up-regulation of BMP4 mediates hypoxia-induced increase of TRPC expression in PASMCs. Cardiovasc Res 107(1):108–118
Pramono A, Zahabi A, Morishima T, Lan D, Welte K, Skokowa J (2016) Thrombopoietin induces hematopoiesis from mouse ES cells via HIF-1alpha-dependent activation of a BMP4 autoregulatory loop. Ann N Y Acad Sci 1375(1):38–51
Deplancke B, Alpern D, Gardeux V (2016) The genetics of transcription factor DNA binding variation. Cell 166(3):538–554
Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, Attie AD, Muoio DM, Kelly DP (2014) A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res 114(4):626–636
Karkkainen O, Tuomainen T, Mutikainen M, Lehtonen M, Ruas JL, Hanhineva K, Tavi P (2019) Heart specific PGC-1alpha deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc Res 115(1):107–118
Zhu X, Shen W, Yao K, Wang H, Liu B, Li T, Song L, Diao D, Mao G, Huang P et al (2019) Fine-tuning of PGC1alpha expression regulates cardiac function and longevity. Circ Res 125(7):707–719
Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, Tran TT, Suzuki R, Espinoza DO, Yamamoto Y et al (2008) New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454(7207):1000–1004
Tiano JP, Springer DA, Rane SG (2015) SMAD3 negatively regulates serum irisin and skeletal muscle FNDC5 and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1alpha) during exercise. J Biol Chem 290(12):7671–7684
Martinez-Hackert E, Sundan A, Holien T (2021) Receptor binding competition: a paradigm for regulating TGF-β family action. Cytokine Growth Factor Rev 57:39–54
Bai L, Chang H-M, Cheng J-C, Chu G, Leung PCK, Yang G (2017) ALK2/ALK3-BMPR2/ACVR2A mediate BMP2-induced downregulation of pentraxin 3 expression in human granulosa-lutein cells. Endocrinology 158(10):3501–3511
Fang L, Jia Q, Liu B, Yan Y, Han X, Guo Y, Cheng J-C, Sun Y-P (2021) BMP-9 downregulates StAR expression and progesterone production by activating both SMAD1/5/8 and SMAD2/3 signaling pathways in human granulosa-lutein cells obtained from gonadotropins induced ovarian cycles. Cell Signal 86:110089
Yang M, Linn BS, Zhang Y, Ren J (2019) Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis 1865(9):2293–2302
Lee SH, Du J, Stitham J, Atteya G, Lee S, Xiang Y, Wang D, Jin Y, Leslie KL, Spollett G et al (2016) Inducing mitophagy in diabetic platelets protects against severe oxidative stress. EMBO Mol Med 8(7):779–795
Won KJ, Lee KP, Yu S, Lee D, Lee D-Y, Lee HM, Kim J, Jung SH, Baek S, Kim B (2015) Ketoconazole induces apoptosis in rat cardiomyocytes through reactive oxygen species-mediated parkin overexpression. Arch Toxicol 89(10):1871–1880
Hou Z, Qin X, Hu Y, Zhang X, Li G, Wu J, Li J, Sha J, Chen J, Xia J et al (2019) Longterm exercise-derived exosomal miR-342-5p. Circ Res 124(9):1386–1400
Chen Z, Gong L, Zhang P, Li Y, Liu B, Zhang L, Zhuang J, Xiao D (2019) Epigenetic down-regulation of sirt 1 via DNA methylation and oxidative stress signaling contributes to the gestational diabetes mellitus-induced fetal programming of heart ischemia-sensitive phenotype in late life. Int J Biol Sci 15(6):1240–1251
Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I et al (1989) Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr 2(5):358–367
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408
Acknowledgements
We thank Dr. Junjie Xiao (Shanghai University) for his kind gift of αMyh6-Cre transgenic mice. We thank Dr. Yin Wang in the Department of Ultrasonography at Shanghai Pulmonary Hospital for the echocardiography and cardiac function analysis. We thank Dr. Carole Mendelson in the Department of Biochemistry and Department of Obstetrics and Gynecology, University of Texas Southwestern Medical Center, for her enlightening suggestions and careful revisions on the language of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China no. 82120108011 and 81771608 (LG); Major Project of Shanghai Municipal Education Commission's Scientific Research and Innovation Plan No. 2021-01-07-00-07-E00144 (LG); Strategic Collaborative Research Program of the Ferring Institute of Reproductive Medicine FIRMA200502 (LG); General Project of Shanghai Municipal Health Commission no. 201740046 (ZYZ).
Author information
Authors and Affiliations
Contributions
LG contributed to study concept and design; LTG, JQY, ZYZ and HMZ contributed to development of methodology and performed all the experiments; LTG, ZYZ and LG analyzed and interpreted the data; LTG and LG wrote, reviewed and revised the manuscript. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, LT., Yuan, JQ., Zhang, ZY. et al. Hypermethylation of the Bmp4 promoter dampens binding of HIF-1α and impairs its cardiac protective effects from oxidative stress in prenatally GC-exposed offspring. Cell. Mol. Life Sci. 80, 58 (2023). https://doi.org/10.1007/s00018-023-04703-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-04703-0