
Abstract
Experimental autoimmune-orchitis (EAO), a rodent model of chronic testicular inflammation and fibrosis, replicates pathogenic changes seen in some cases of human spermatogenic disturbances. During EAO, increased levels of pro-inflammatory and pro-fibrotic mediators such as TNF, CCL2, and activin A are accompanied by infiltration of leukocytes into the testicular parenchyma. Activin A levels correlate with EAO severity, while elevated CCL2 acting through its receptor CCR2 mediates leukocyte trafficking and recruits macrophages. CCR2 + CXCR4 + macrophages producing extracellular matrix proteins contribute widely to fibrogenesis. Furthermore, testicular macrophages (TMs) play a critical role in organ homeostasis. Therefore, we aimed to investigate the role of the activin A/CCL2-CCR2/macrophage axis in the development of testicular fibrosis. Following EAO induction, we observed lower levels of organ damage, collagen deposition, and leukocyte infiltration (including fibronectin+, collagen I+ and CXCR4+ TMs) in Ccr2−/− mice than in WT mice. Furthermore, levels of Il-10, Ccl2, and the activin A subunit Inhba mRNAs were lower in Ccr2−/− EAO testes. Notably, fibronectin+ TMs were also present in biopsies from patients with impaired spermatogenesis and fibrotic alterations. Overexpression of the activin A antagonist follistatin reduced tissue damage and collagen I+ TM accumulation in WT EAO testes, while treating macrophages with activin A in vitro increased the expression of Ccr2, Fn1, Cxcr4, and Mmp2 and enhanced migration along a CCL2 gradient; these effects were abolished by follistatin. Taken together, our data indicate that CCR2 and activin A promote fibrosis during testicular inflammation by regulating macrophage function. Inhibition of CCR2 or activin A protects against damage progression, offering a promising avenue for therapeutic intervention.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Approximately 8–12% of couples worldwide suffer from infertility. Male factors account for ~ 50% of cases, while testicular inflammation induced by bacteria, viruses, or sterile inflammation (including autoimmune diseases) is a significant cause of male infertility [1,2,3,4]. Experimental autoimmune orchitis (EAO), a rodent model of chronic testicular inflammation, mimics the pathological changes observed in human testicular biopsies from infertile patients [5,6,7]. In the mouse, EAO leads to fibrosis and subsequent infertility [6, 8]. Pathological features of EAO include the destruction of the testicular structure, infiltration of the interstitium by leukocytes, elevated levels of pro-inflammatory cytokines (including C–C motif chemokine ligand 2 [CCL2], tumor necrosis factor [TNF], and activin A), as well as the loss of germ cells. Furthermore, levels of C–C motif chemokine receptor type 2 (CCR2), the receptor for CCL2, are significantly increased in interstitial mononuclear cells in EAO testis [9]. Testicular macrophages (TMs) constitute the major immune cell population in the testis and play a critical role in supporting steroidogenesis, promoting spermatogenesis, and possibly also maintaining immune privilege [10, 11]. During testicular inflammation, numbers of infiltrating monocytes and macrophages are increased, while a reduction in TMs correlates with decreased incidence and severity of testicular damage [12,13,14].
Fibrosis is a hallmark of progressive and severe EAO, characterized by the accumulation of extracellular matrix (ECM) proteins such as collagen and fibronectin and thickening of the α-smooth muscle actin (αSMA)-positive peritubular cell (PTC) layer of the seminiferous tubules [13]. Studies in other organs have indicated that ECM-producing cells may originate not only from resident mesenchymal fibroblasts but also from circulating hematopoietic cells or cells undergoing epithelial/endothelial-to-mesenchymal transition [15]. Monocytes and macrophages that are resident or recruited to injury sites produce a variety of factors, including transforming growth factor-β (TGF-β), platelet-derived growth factors (PDGFs), and matrix metalloproteinases (MMPs). These proteins stimulate the proliferation, differentiation, and activation of fibroblasts, thereby promoting fibrotic remodeling [16, 17]. Tissue inhibitors of metalloproteinases (TIMPs) also play a role in fibrosis by inhibiting MMPs, thereby suppressing ECM proteolysis, TGF-β release, and neutrophil chemotaxis [18].
Activin A, a homodimeric member of the TGF-β superfamily of cytokines, is produced mainly by Sertoli cells (SCs) in the normal postnatal testes and plays multiple biological roles in inflammation, immunity, and fibrosis [19, 20]. In EAO, serum and testicular activin A levels are increased [13, 21]. Activin A exerts both pro- and anti-inflammatory effects and stimulates monocytes and macrophages to produce many inflammatory mediators, including interleukin (IL)-1β, IL-6, and TNF [22, 23]. Furthermore, activin A promotes pro-fibrotic gene expression in many cell types, including PTCs and NIH 3T3 fibroblasts [24]. Activin A also drives the chemotactic migration and adhesion of L929 fibroblasts in vitro [25]. Activin activity is inhibited by follistatin (FST), an endogenous high-affinity activin A binding antagonist [26].
CCL2 and CCR2 play a critical role in the trafficking of lymphocytes, monocytes, macrophages, and bone marrow-derived fibroblasts to sites of injury [27, 28]. In addition to mediating chemotaxis, CCL2 stimulates the production of collagens by fibroblasts and TGF-β by macrophages [29]. CCR2+ cells also contribute to fibrogenesis by producing TIMP1, which inhibits collagen degradation [30]. In the EAO model, increased levels of CCL2 protein were observed in mononuclear and endothelial cells, Leydig cells, and PTCs as well as in testicular fluid and conditioned medium from cultured TMs; furthermore, elevated expression of CCR2 was identified in mononuclear cells [9]. Of note, stimulation of Sertoli cells with TNF led to upregulated levels of Ccl2 and activin A (Inhba) mRNA expression [24, 31].
At present, the role of the activin A/CCL2-CCR2/macrophage axis in this inflammatory and fibrotic response in the testis is unknown. We hypothesized that the interaction of activin A and CCR2 may influence the development of testicular fibrosis by regulating the properties of macrophages, a potentially important source of pro-fibrotic factors during orchitis. To verify our hypothesis, we compared the progress of EAO in wild type (WT) and Ccr2−/− mutant mice, which lack peripheral blood monocytes, as well as in mice overexpressing follistatin, which display reduced activin bioactivity. Furthermore, we investigated the influence of activin A on CCR2 and fibrotic mediator expression in macrophages, using bone marrow-derived macrophages (BMDMs) matured in the presence of macrophage-colony stimulating factor (M-CSF) as a surrogate for TMs.
Materials and methods
Animals
In this study, 10- to 12-week-old WT C57BL/6J (Charles River Laboratories, Sulzfeld, Germany) and B6.129P2-Ccr2tm1Mae/tm1Mae (Ccr2−/−) mice were housed in specific pathogen-free conditions (12 h light/dark cycle, 20–22 °C) at the animal facility of Justus Liebig University, Giessen, Germany [32]. The mice had access to water and food ad libitum. Femurs and tibias were collected from male adult WT and Ccr2−/− mice for the isolation of bone marrow progenitor cells, while testes were obtained from 21-day-old immature WT mice for the isolation of SCs.
Animal experiments were approved by the responsible local ethics committees on animal care (Regierungspraesidium Giessen GI 58/2014—Nr. 735-GP and the Monash Medical Centre Animal Experimentation Committee). All experiments involving animals were carried out in strict accordance with the recommendations in the guide for the Care and Use of Laboratory Animals of the German animal welfare law and the Australian Code for the Care and Use of Animals for Scientific Purposes.
Induction of EAO in WT and Ccr2−/− mice
Peri-procedural analgesia in mice was provided by supplying tramadol (STADApharm GmbH, Bad Vilbel, Germany) in drinking water (2.5 mg/ml), commencing 24 h before each immunization and continuing for the following 3 days.
For the induction of EAO, adult male C57BL/6 J and Ccr2−/− mice (n = 29) were first anesthetized by administration of 3–5% isoflurane and then actively immunized with testicular homogenate (TH) in complete Freund’s adjuvant (CFA; Sigma-Aldrich, St. Louis, USA), as previously described [13]. The homogenate was prepared from decapsulated testes collected from adult WT mice and homogenized in sterile 0.9% NaCl at a ratio of 1:1. Animals were immunized dorsally three times every 14 days with a mixture of TH in CFA; a total volume of 200 µl was delivered as four subcutaneous injections (50 µl per injection site). This was followed by intraperitoneal injection of 100 ng Bordetella pertussis toxin (Calbiochem, Darmstadt, Germany) in 100 µl Munõz Buffer (25 mM Tris, 0.5 M NaCl, 0.017% Triton X-100, pH 7.6) [33].
Adjuvant control animals (n = 22) received CFA mixed with 0.9% NaCl instead of TH, following the same injection regimen. Age-matched untreated mice (n = 21) were also included in the study. Animals were sacrificed by cervical dislocation 50 days after the first immunization (following deep anesthesia induced with 5% isoflurane). A schematic diagram illustrating the time course of immunizations and time points for organ collection is shown in Fig. 1A. The testes were removed, weighed, and either snap-frozen in liquid nitrogen or fixed in Bouin’s solution for embedding in paraffin. Freshly collected testes were used for flow cytometric analysis.

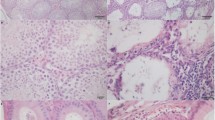
Tissue damage and collagen deposition caused by EAO is reduced in Ccr2−/− mouse testes. Schematic diagram representing the time points for EAO induction and organ collection in C57BL/6J (WT) and B6.129P2-Ccr2tm1Mae/tm1Mae (Ccr2−/−) mice. Mice were immunized three times every other week starting at day 0, and testes were collected at day 50 (A). Paired testis weight (B; n = 20–30) and total collagen content determined by hydroxyproline assay (C; n = 5–6) in untreated, adjuvant control, and EAO testes from WT and Ccr2−/− mice. Representative photomicrographs of picro-sirius red-stained paraffin sections (D) of untreated (I, IV), adjuvant control (II, V), and EAO (III, VI) testes from WT (I–III) and Ccr2−/− mice (IV–VI); asterisk in (VI) indicates seminiferous tubule damage. Values are mean ± SEM; statistical analyses used the Kruskal–Wallis test followed by Dunn’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Induction of EAO in mice with elevated follistatin levels
Thirty days before the first immunization with TH, C57BL/6J mice were injected intramuscularly with non-replicative recombinant adeno-associated viral (rAAV) vector serotype 6 carrying a gene cassette of the circulating form of follistatin (rAAV-FST315; n = 13) or an empty gene cassette (empty vector; EV; n = 18), as previously described [21]. Adjuvant-only and non-immunized controls were included. The immunization protocol was modified according to Monash University Animal Ethics committee requirements regarding the use of adjuvants: mice were immunized once with TH in CFA, followed by two further immunizations with TH in incomplete Freund’s adjuvant with B. pertussis toxin. Approximately 75% of mice immunized by this protocol developed EAO, ranging from mild to severe, within 50 days. Treatment with rAAV-FST315 resulted in a fivefold increase in serum follistatin levels by the time of the first immunization [21]. After collection, testes were either snap-frozen in liquid nitrogen for RNA analysis or fixed in Bouin’s solution for paraffin embedding and subsequent immunofluorescence [21]. A schematic diagram illustrating the time course of vector injection, immunizations, and time points for organ collection is shown in Fig. 4A.
Human testicular biopsies
Bouin’s-fixed paraffin-embedded testicular tissue samples obtained by routine open testicular biopsy (using the testicular sperm extraction [TESE] protocol) were provided by the University Hospital Zagreb, (Department of Urology, in cooperation with the School of Medicine) at the University of Zagreb. According to current guidelines, three pieces of tissue were obtained through corresponding incisions of the tunica albuginea (upper and lower testis pole and equatorial plane). Biopsied tissues were divided into two parts to provide an accurate diagnosis: one half for cryopreservation and the other half for histological analysis [34, 35]. This study was performed in line with the principles of the Declaration of Helsinki. Each patient provided written consent for the TESE procedure as well as the histological analysis/current study. Approval was granted by the Ethics Committee of the School of Medicine at the University of Zagreb (380-59-10106-20-111/171). The specimens (selected from infertile men with non-obstructive azoospermia) showed histological diagnoses of focal inflammatory lesions associated with disturbed spermatogenesis (mixed atrophy or Sertoli cell-only phenotype) and fibrotic remodeling (thickening of the lamina propria, interstitial accumulation of collagen; tubular fibrosis; n = 5). The control group comprised three patients suffering from obstructive azoospermia with fully preserved testicular parenchyma, revealing normal intact spermatogenesis without inflammatory signs.
Picro-sirius red staining
Testicular histology and collagen distribution were assessed in paraffin sections by picro-sirius red staining [36].
Hydroxyproline assay
Total testicular collagen content was quantified by chromogenic determination of hydroxyproline concentration using the QuickZyme Total Collagen Assay Kit (QuickZyme Biosciences, Leiden, Netherlands) according to the manufacturer’s instructions. Total testicular collagen content was normalized to total testicular protein using the QuickZyme Total Protein Assay Kit (QuickZyme Biosciences).
Isolation and treatment of Sertoli cells
SCs were isolated from 21-day-old C57BL/6 J mice (8 animals/isolation) according to an established protocol [24]. Two days after isolation, SCs were treated with 20 mM hypotonic Tris-hydrochloride solution (Sigma-Aldrich) for 2 min to remove germ cell contamination. One day after hypotonic shock, SCs were treated with 50 ng/ml recombinant mouse TNF (PromoCell, Heidelberg, Germany), 250 ng/ml human recombinant follistatin 288 (FST288; purified from HEK-293 cells transfected with a human FST 288 transgene), or a combination of both [37]. After 24 h in culture, Sertoli cell-conditioned medium (SCCM) was collected. The purity of SCs (> 85%) was determined by double staining for SOX9 (SC marker) and αSMA (PTC marker) [24].
Generation and treatment of BMDMs
Bone marrow progenitor cells were isolated from adult C57BL/6J and Ccr2−/− mice (n = 28) according to a published protocol [38] and cultured in a complete RPMI-1640 medium (Gibco). BMDMs were generated by treating the progenitor cells with 50 ng/ml mouse recombinant (M-CSF (Miltenyi Biotec) and then stimulating with 25 or 50 ng/ml human recombinant activin A (Miltenyi Biotec), 250 ng/ml FST288, or a combination of both for 6 days.
For gelatin zymography, after 6 days in culture, BMDMs were treated with 50 ng/ml M-CSF and 50 ng/ml activin A or 250 ng/ml FST288 or a combination of both activin A and FST288 in serum-free RPMI-1640 medium. BMDM-conditioned medium was collected 24 h later.
For SC-conditioned medium (SSCM) treatment, BMDMs were generated as above (50 ng/ml M-CSF for 3 days) and subsequently incubated with SCCM for an additional 3 days.
Flow cytometry
Testes were decapsulated and digested in phosphate-buffered saline (PBS; Gibco, Bleiswijk, Netherlands) containing 1.2 mg/ml collagenase A (Roche Diagnostic, Mannheim, Germany) and 15 U/ml DNase I (Roche Diagnostic) in a 34 °C water bath for 15 min, with agitation. After the sedimentation of the seminiferous tubules, supernatants containing interstitial cells were collected. BMDMs growing in Nunc UpCell 6-well Multidishes (Thermo Fisher Scientific, Roskilde, Denmark) were detached at room temperature (RT) for 30 min. All incubation steps were performed at 4 °C. A total of 1 × 106 cells were blocked with mouse FcR blocking reagent (Miltenyi Biotec, Bergisch-Gladbach, Germany) for 10 min, followed by incubation with the appropriate antibodies (see Table 1 for details) for 15 min. Afterwards, cells were permeabilized using the Fix/Permeabilization Staining Buffer Set (Miltenyi Biotec) for 30 min and blocked again. Subsequently, the cells were incubated with anti-fibronectin or anti-collagen I antibody for 45 min and the respective secondary antibodies for 30 min at 4 °C. Data were collected using the MACSQuant Analyzer 10 flow cytometer (Miltenyi Biotec) and analyzed using FlowJo V10 software (FlowJo LLC, Oregon, USA). The flow cytometry gating strategy is shown in Supplementary Fig. S1.
Immunofluorescence
Frozen testicular sections (10 μm) or fresh BMDMs were fixed in ice-cold methanol for 10 min. BMDMs were permeabilized using 0.5 M glycine for 30 min at RT. Subsequently, specimens were blocked in 5% bovine serum albumin (BSA; Sigma-Aldrich, Steinheim, Germany) and 10% goat serum (BioLegend, San Diego, USA) in Tris-buffered saline containing 0.1% Tween-20 (TBST) at RT for 2 h. After incubating with the appropriate primary antibodies overnight (see Table 2 for details), the sections were washed with TBST and incubated with the corresponding secondary antibodies at RT for 1 h. Nuclei were counterstained with Topro 3 (Life Technologies, Darmstadt, Germany). Finally, the slides were mounted in ProLong™ Gold Antifade Mountant containing DAPI (Invitrogen, Oregon, USA). Fluorescence images were captured on a confocal laser scanning microscope 710 (Carl Zeiss, Göttingen, Germany).
Paraffin-embedded testicular sections (8 μm) were deparaffinized in xylene twice for 10 min and rehydrated by immersing in a series of ethanol solutions of gradually decreasing concentrations. Afterwards, sections were treated with pre-warmed proteinase K solution (20 μg/ml; Sigma-Aldrich) for 3 min at RT. Sections were then blocked in 5% BSA and 10% goat serum in TBS at RT for 1.5 h and incubated with primary antibodies in blocking solution at RT for 4 h (see Table 2 for details). Subsequently, sections were washed with TBS and incubated with the corresponding secondary antibodies at RT for 1 h, followed by treatment with 0.3% Sudan Black B (Sigma-Aldrich) for 5 min to reduce autofluorescence. Finally, after several washes with TBS, sections were mounted using ProLong™ Gold Antifade Mountant containing DAPI and photographed using a confocal laser scanning microscope, as above.
RNA extraction and qRT-PCR
Total RNA was isolated from frozen testes using the RNeasy Fibrous Tissue Mini Kit (Qiagen, Hilden, Germany) and from cells using the RNeasy Mini Kit (Qiagen), according to the manufacturer’s instructions. Contaminating DNA was removed using the RNase-Free DNase Set (Qiagen). Total RNA was reverse transcribed as described previously [21, 24]. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed using iTaq Universal SYBR Green Supermix (Bio-Rad, Munich, Germany) for self-designed primers or QuantiTect SYBR Green PCR Master Mix (Qiagen) for QuantiTect primer assays (Qiagen) in a CFX96 Touch thermal cycler (Bio-Rad). Primer sequences and annealing temperatures are listed in Table 3. Hypoxanthine–guanine-phosphoribosyltransferase (Hprt) and 18S rRNA were selected as stably expressed housekeeping genes. All samples were run as duplicates. Relative mRNA expression was calculated using the 2−ΔΔCt method [39].
Bulk RNA sequencing
Total RNA was isolated from BMDMs using the RNeasy Mini Kit (Qiagen) combined with on-column DNase digestion (RNase-Free DNase Set, Qiagen) to avoid contamination by genomic DNA. RNA and library preparation integrity were verified with the LabChip Gx Touch 24 (Perkin Elmer, Waltham, USA). Library preparation was carried out with 4 µg of total RNA as input using the VAHTS Stranded mRNA-seq Library Prep kit according to the manufacturer’s protocol (Vazyme, Nanjing, China). Sequencing was performed on the NextSeq500 System (Illumina, San Diego, USA) using v2 chemistry with a 1 × 75 bp single-end setup. The resulting raw reads were assessed for quality, adapter content, and duplication rates using FastQC [40]. Trimmomatic version 0.39 was employed to trim reads after a quality drop below a mean of Q20 in a window of 5 nucleotides [41]. Only reads between 30 and 150 nucleotides were cleared for further analysis. Trimmed and filtered reads were aligned against Ensembl mouse genome version mm10 (ensemble release 101) using STAR 2.7.9a with the parameter “–outFilterMismatchNoverLmax 0.1” to increase the maximum ratio of mismatches to mapped length to 10% [42]. The number of reads aligning to genes was counted using the featureCounts 2.0.2 tool of the Subread package [43]. Only reads mapping at least partially inside exons were admitted and aggregated per gene; reads overlapping multiple genes or aligning to multiple regions were excluded. Differentially expressed genes (DEGs) were identified using DESeq2 version 1.30.0 [44]. Only genes with a minimum fold change of ± 2 (log2 = ± 1), a maximum Benjamini-Hochberg-corrected p-value of 0.05, and a minimum combined mean of five reads were deemed to be significantly differentially expressed. Ensemble annotation was enriched using UniProt data (release 06.06.2014) based on Ensembl gene identifiers [45].
Protein extraction and Western blot
BMDMs were lysed on ice in RIPA buffer (150 mM NaCl, 0.1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 50 mM Tris–HCl pH 8.0, 2 mM EDTA) supplemented with protease inhibitor cocktail (1:100; Sigma-Aldrich) and centrifuged at 13,000×g for 30 min at 4 °C. The RC DC Protein Assay (Bio-Rad, Hercules, USA) was used to measure total protein concentration. Protein samples diluted in Laemmli buffer (30 µg/lane) were resolved by 10% (SMADs only) or 12.5% SDS–polyacrylamide gel electrophoresis, transferred onto nitrocellulose membrane (GE Healthcare, Darmstadt, Germany), blocked with 5% non-fat milk in TBST for 1 h at RT, and subsequently washed in TBST. Incubation with primary antibodies was performed overnight at 4 °C (Table 2). Afterwards, membranes were washed three times for 10 min at RT in TBST before incubation with the appropriate secondary antibodies for 1 h at RT (Table 2). Finally, the membranes were probed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA) and subsequently developed using the Fusion FX Western Blot and chemiluminescence imaging system (Vilber Lourmat, Eberhardzell, Germany). The results were quantified using FusionCapt Advance Solo 4 16.07 software (Vilber Lourmat).
BMDM migration assay
BMDM generation and treatment with activin A and/or FST288 were performed as described above, and cells were cultivated in a Nunc UpCell 6-well dish before harvesting. For the migration assay, cells were gently washed with PBS and incubated on ice for 15 min before collection. After seeding the BMDMs (3 × 105) in Transwell inserts (8 µm pore size; Corning Life Sciences, Tewksbury, USA) in serum-free RPMI-1640 medium, 750 µl serum-free RPMI-1640 medium containing 500 ng/ml CCL2 was added to the lower chamber. Cells were allowed to migrate for 3 h at 37 °C. Migrated cells were collected from the lower chamber and counted using the MACSQuant Analyzer 10 flow cytometer (Miltenyi Biotec).
Gelatin zymography
The enzymatic activity levels of MMP2 and MMP9 were measured in BMDM-conditioned medium (generated as described above); gelatin zymography was performed as reported [46]. BMDM-conditioned medium was loaded and gels were stained with Coomassie Brilliant blue solution. Band intensity was analyzed using ImageJ software (National Institutes of Health, Bethesda, USA). Relative enzymatic activity was calculated by normalizing the band intensity of the untreated group to 1.0.
Statistical analysis
Statistical data analysis was performed using GraphPad Prism 7 software (GraphPad Software, San Diego, USA). Data are presented as mean ± standard error of the mean (SEM) and were tested for normal distribution using the Kolmogorov–Smirnov test. The statistical significances of differences in multiple groups were calculated using one-way or two-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparisons test (Gaussian distribution) or the Kruskal–Wallis test followed by Dunn’s multiple comparisons test (non-normal distribution). Correlation significance between two groups was analyzed using the Pearson coefficient (normal distribution) or Spearman coefficient (non-normal distribution). Statistical significance was set at P < 0.05.
Results
Ccr2 deficiency protects the testis from damage caused by EAO
To determine the role of CCR2 in EAO-induced fibrosis, we compared EAO in WT and Ccr2−/− mice. Measurement of testicular weight 50 days after the first immunization showed that EAO caused a reduction in mean testis weight in both WT and Ccr2−/− mouse strains, compared with untreated and adjuvant control groups (Fig. 1B). However, mean testis weight was significantly higher in the Ccr2−/− EAO mice (0.089 ± 0.004 g) than in the WT EAO mice (0.045 ± 0.004 g) (Fig. 1B), indicating lower levels of testicular damage in the Ccr2 null mice. To quantify the fibrotic response, testicular collagen content was analyzed by hydroxyproline assay and picro-sirius red staining and found to be significantly elevated in WT EAO testes, compared with WT untreated testes (Fig. 1C). In contrast, EAO did not result in significantly increased levels of collagen in Ccr2−/− mice, and total collagen content was 2.7-fold lower in EAO testes than in WT testes (Fig. 1C). Histological examination of testicular sections revealed that normal testicular architecture was destroyed in 94% of WT EAO testes, with germ cell sloughing, atrophy of seminiferous tubules, thickening of the lamina propria of seminiferous tubules, and an increase in the density of collagen fibers (Fig. 1D). In contrast, although a few disrupted seminiferous tubules were visible in 3 of 15 mice (20%), testicular structure was largely intact in Ccr2−/− EAO testes, indicating a crucial role for CCR2 in inflammation and damage following EAO (Fig. 1D).
Ccr2 deficiency reduces inflammatory responses in EAO testes
Increased numbers of CD45+ leukocytes and elevation of inflammatory cytokines such as CCL2, TNF, IL-1 and IL-10, and activin A are important hallmarks of the inflamed testis in EAO [6, 13]. To investigate whether depletion of Ccr2 affects the testicular inflammatory response, the presence and number of immune cells and expression of cytokines and activins in testes were analyzed. Flow cytometry demonstrated significantly lower numbers of total CD45+ leukocytes (Fig. 2A and B), CD45+Ly6G+ neutrophils (Fig. 2C), and CD45+Ly6C−CD11b+CD64+ macrophages (Fig. 2E), as well as an approximately 14-fold reduced population of CD45+Ly6C+ monocytes (Fig. 2D) in Ccr2-deficient EAO testes, compared with EAO WT testes. These results imply that the depletion of CCR2 inhibits the recruitment of infiltrating immune cells in the EAO testis.

Immune cell infiltration and expression of inflammatory mediators caused by EAO are reduced in Ccr2−/− mouse testes. Representative density plots for CD45+ leukocytes in untreated, adjuvant control, and EAO testicular single-cell suspensions from WT and Ccr2−/− mice (A). After gating out doublets and nonviable cells, numbers of testicular CD45+ leukocytes (B), CD45+Ly6G+ neutrophils (C), CD45+Ly6C+ monocytes (D), and CD45+Ly6C−CD11b+CD64+ macrophages (E), were determined in untreated, adjuvant control, and EAO testicular single-cell suspensions from WT and Ccr2−/− mice by flow cytometry (n = 5–6). Relative expression (RE) of Ccl2 (F), Il-10 (G), Inhba (H), and Acvr2b (I) was determined in untreated, adjuvant control, and EAO testes from WT and Ccr2−/− mice by qRT-PCR analysis and normalization to 18S rRNA and Hprt (n = 4–6). Values are mean ± SEM; statistical analyses used the Kruskal–Wallis test followed by Dunn’s multiple comparison test or two-way ANOVA followed by Bonferroni’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Quantitative RT-PCR showed that Ccr2 deficiency led to significant suppression of testicular Ccl2 (Fig. 2F), Il-10 (Fig. 2G), and Il-1a (Supplementary Fig. S2A) mRNAs during EAO, compared with the levels observed in WT mice undergoing EAO. Moreover, levels of Ccl2 and Il-10 mRNA were not significantly changed in any Ccr2−/− experimental group (Fig. 2F and G). Absence of Ccr2 also inhibited the EAO-mediated increase in expression of the activin A subunit Inhba (Fig. 2H). Expression of follistatin (Fst) was significantly increased in EAO testes (Supplementary Fig. S2E), while expression of the activin A receptor subunit Acvr1b was reduced (Supplementary Fig. S2F). In addition, following EAO, expression of the Acvr2b subunit was significantly reduced in WT testes but remained in the normal range in Ccr2−/− testes (Fig. 2I). However, other inflammation-related genes, including Il-1b and other activin-related genes (Inhbb and Inha), were not significantly altered in untreated, adjuvant control, or EAO testes from WT or Ccr2−/− mice (Supplementary Fig. S2B and G-H). Furthermore, expression of Tnf and Il-6 mRNA was significantly increased in EAO WT testes, but unchanged in all Ccr2−/− groups (Supplementary Fig. S2C and D).
Ccr2 deficiency suppresses changes in the numbers of ECM-expressing immune cells and CXCL12/CXCR4 expression induced by EAO
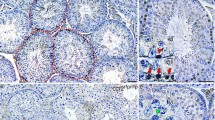
Since the above results indicated that loss of Ccr2 expression reduced immune cell accumulation and inhibited the fibrotic response in EAO testes, we wanted to investigate the involvement of immune cells (particularly macrophages) in the development of testicular fibrotic remodeling. Therefore, the numbers and localization of ECM-expressing immune cells in the testes during EAO were analyzed by flow cytometry and immunofluorescence, respectively; CXCR4+ cells were also quantified as CXCR4 plays a significant role in ECM-producing immune cells [47]. The proportions of CD45+fibronectin+ cells (Fig. 3A) and CD45+fibronectin+CXCR4+ cells (Fig. 3B) within the total CD45+ leukocyte population were significantly increased in WT EAO testes, while Ccr2 deficiency inhibited this effect. Similar results were observed for CD45+collagen I+ cells and CD45+collagen I+CXCR4+ cells (Supplementary Fig. S3A and B). Among immune cells, macrophages are an important source of ECM proteins [48]. The numbers of fibronectin+ and collagen I+ macrophages (identified by F4/80 and CD11b) were increased 117-fold and 70-fold, respectively, in WT EAO testes, compared with testes from untreated and adjuvant controls (Fig. 3C and Supplementary Fig. S3C). These results were confirmed by immunofluorescence (Fig. 3D and Supplementary Fig. S3D). In contrast, the numbers of fibronectin- and collagen I-expressing TMs detected in Ccr2−/− EAO testes were approximately sevenfold lower, compared with the numbers in WT EAO testes (Fig. 3C, D and Supplementary Fig. S3C, D). Furthermore, analysis of testicular biopsies from patients with impaired spermatogenesis, testicular fibrosis, and inflammation showed that fibronectin expression was increased in CD68+ macrophages. This indicated that similar to mouse EAO, macrophages express fibronectin under pathological conditions (Fig. 3E). These results suggest that infiltrating bone marrow CCR2+ cells play a role in modulating the levels of ECM proteins during testicular inflammation and thus also in fibrotic remodeling.

The number of fibronectin+ immune cells is increased in mouse testes with EAO and human testes with impaired spermatogenesis. After gating out doublets and nonviable cells, the numbers of testicular CD45+fibronectin+ cells (A), CD45+fibronectin+CXCR4+ cells (B), and CD45+CXCR4+ cells (F) within the CD45+ leukocyte population and the number of testicular CD45+fibronectin+F4/80+CD11b+ cells (C) within the live cell population were analyzed in untreated, adjuvant control, and EAO testicular single-cell suspensions from WT and Ccr2−/− mice by flow cytometry (n = 5–8). Representative photomicrographs of fibronectin (green), F4/80 (red), and DAPI (blue) triple immunofluorescence staining in frozen sections (D) from untreated (I, IV), adjuvant control (II, V) and EAO (III, VI) testes in WT (I–III) and Ccr2−/− mice (IV–VI); arrows indicate fibronectin+ macrophages. Representative photomicrographs of CD68 (green), fibronectin (orange), and DAPI (blue) triple immunofluorescence staining in paraffin sections from human testicular biopsies with normal and impaired spermatogenesis (E); arrows indicate triple-positive cells. Relative expression (RE) of Cxcr4 (G) and Cxcl12 (H) in untreated, adjuvant control, and EAO testes from WT and Ccr2−/− mice was determined by qRT-PCR analysis and normalization to 18S rRNA and Hprt (n = 7–13). Values are mean ± SEM; statistical analyses used the Kruskal–Wallis test followed by Dunn’s multiple comparison test or two-way ANOVA followed by Bonferroni’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Because CXCR4 and its ligand CXCL12 contribute to the recruitment of circulating progenitors of active fibroblasts [49], we also analyzed the testicular expression of CXCR4 by flow cytometry, qRT-PCR, and immunofluorescence in WT and Ccr2−/− EAO mouse models. While increased numbers of CD45+CXCR4+ cells (within the total CD45+ leukocyte population) were detected in WT testes in response to EAO, this increase was abrogated in the absence of Ccr2 expression (Fig. 3F). Furthermore, reduced levels of Cxcr4 (Fig. 3G) and Cxcl12 (Fig. 3H) mRNA were detected in Ccr2−/− EAO testes, compared with WT EAO testes. Immunofluorescence revealed elevated numbers of CXCR4+ macrophages (as well as total numbers of CXCR4+ cells) in WT EAO testes, while infiltration by CXCR4+ macrophages was not increased in Ccr2−/− EAO testes (Supplementary Fig. S3E). These data suggest that CCR2 signaling is also involved in the recruitment of CXCR4+ cells in EAO testis.
Activin A contributes to the production of collagen I by macrophages and increases MMP expression during EAO
We previously reported a positive correlation between the severity of disease and testicular activin A concentration [13, 21]. In addition, inhibition of activin A through overexpression of follistatin (using a viral vector expressing FST315) significantly reduced the fibrotic response in EAO [21, 24]. In the present study, the activation of Inhba during EAO was suppressed in Ccr2-deficient mice (see Fig. 2H), accompanied by decreased macrophage infiltration. Transfection with FST315 also clearly reduced testicular infiltration by collagen I+ TMs in mice undergoing EAO (compared with empty vector [EV]-treated control mice; Fig. 4A, B). Collagen I was almost undetectable in TMs from untreated and adjuvant controls (Fig. 4B). These data suggest that activin A is involved in regulating the expression of collagen I by TMs in EAO testis. In addition, as MMPs and their inhibitors TIMPs are key regulators of ECM component degradation and fibrotic remodeling [50], we quantified the levels of Mmp and Timp1 mRNAs by qRT-PCR. Subsequently, correlation analysis demonstrated significant associations between Inhba and Mmp2, Mmp9, Mmp14, and Timp1 mRNA levels in testes from mice treated with control EV or FST315 after induction of EAO (Fig. 4C–F). Altogether, these data suggest that activin A influences the expression of MMPs and TIMP1 by macrophages.

Inhibition of activin A by FST315 reduces collagen I production by macrophages and expression of Mmps in EAO testes. Schematic diagram representing the time points for rAAV (adeno-associated viral vector) injection, EAO induction, and organ collection in C57BL/6 J (WT) mice. Mice were injected with rAAV carrying the FST315 gene cassette (rAAV-FST315) or an empty vector (rAAV-EV) 30 days before the first EAO immunization. Subsequently, mice were immunized three times every other week starting at day 0, and testes were collected at day 50 (A). Representative photomicrographs of collagen I (green), F4/80 (red), and DAPI (blue) triple immunofluorescence staining (B) in paraffin sections from untreated (I, IV), adjuvant control (II, V), and EAO (III, VI) testes in rAAV-EV (I–III) and rAAV-FST315 (IV–VI) mice; arrows indicate triple-positive cells. The correlations between Inhba and Mmp2 (C), Mmp9 (D), Mmp14 (E), and Timp1 (F) relative expression (RE) were determined in rAAV-EV and rAAV-FST315 EAO testes (n = 14); red dots represent immunized mice that did not develop EAO. Correlation significance was determined using the Pearson coefficient. *P < 0.05, **P < 0.01, ****P < 0.0001
Activin A affects BMDMs and their transcriptome
To investigate how regulation of macrophage function by activin A may contribute to inflammatory and fibrotic responses, mouse BMDMs were stimulated with activin A in vitro (Fig. 5A). FST288 was used as an antagonist of activin A. Phosphorylation of SMAD2 was analyzed to confirm the effect of activin A in BMDMs as activin A downstream signaling is induced by phosphorylation of SMAD2/3 and translocation of SMAD2/3-SMAD4 to the nucleus [22, 24]. Western blotting demonstrated that SMAD2 in BMDMs was phosphorylated within 30 min and 60 min of treatment with 25 ng or 50 ng/ml activin A; these effects were abolished by the addition of 250 ng/ml FST288 (Supplementary Fig. S4A and B).

Whole transcriptome analysis of BMDMs treated with activin A in vitro. Experimental set-up for transcriptomic analysis in BMDMs. Cells were left untreated (CTRL) or stimulated with 50 ng/ml activin A (Act), 250 ng/ml FST288 (FST), or a combination of both (Act + FST) (A; n = 3). Venn diagram of up- (B) and downregulated (C) DEGs in BMDMs treated with Act, Act + FST, or FST versus CTRL cells. Heat map (D) showing expression of the top 50 DEGs in CTRL, Act, Act + FST, and FST groups (based on false discovery rate > 0.05 and minimal count number > 5). Heat maps showing significant DEGs enriched for the terms “Inflammation mediated by chemokine and cytokine signaling pathway” (E) and “Extracellular matrix organization” (F) from the Panther and Reactome databases, respectively in CTRL, Act, Act + FST, and FST groups of BMDMs, presented with Z-score normalization. Arrows indicate the investigated genes
Next, a whole transcriptomic analysis of BMDMs was performed after treatment with activin A, FST288, or a combination of both (Fig. 5A). Overall, the samples from untreated (CTRL), FST288-alone or FST288 + activin A-treated BMDMs showed similar gene expression patterns, which differed from those identified in activin A-treated BMDMs (Fig. 5B–D). Comparison of the activin A-stimulated BMDMs and the CTRL group revealed 936 upregulated and 799 downregulated DEGs (Fig. 5B and C). Further analysis of the top 50 DEGs identified by the term “inflammation mediated by chemokine and cytokine signaling pathway” from the Panther database demonstrated that several cytokine and chemokine signaling-related genes were regulated by activin A. For example, Ccr2 and Cxcr4 were upregulated, while Ccl2 was downregulated following activin A treatment (Fig. 5E), pointing to dual pro- and anti-inflammatory effects mediated by activin A. In addition, the selection of the top 50 significantly altered DEGs enriched for the term “extracellular matrix organization” from the Reactome database indicated that many upregulated genes, including Tgfbr1, Fn1, Mmp2, Mmp14, Pdgfa, and Itgav, were related to TGF-β signaling and ECM organization (Fig. 5F). Therefore, we focused on the selected genes (Ccr2, Cxcr4, Fn1, Mmp2, Mmp14, Pdgfa, Itgav, and Itgb3) in the subsequent experiments.
Activin A upregulates CCR2 in BMDMs and induces the migration of macrophages along a CCL2 gradient
The above RNAseq data analysis indicated that treating BMDMs with 50 ng/ml activin A-induced elevated Ccr2 expression. This was confirmed by Western blotting (Fig. 6A), while the addition of FST288 abolished this increase at both mRNA and protein levels (Figs. 5E and 6A). To investigate these findings at a functional level, we analyzed the ability of BMDMs (isolated from WT and Ccr2−/− mice in the absence/presence of activin A and/or FST288) to migrate along a CCL2 gradient. Given that CCL2 stimulates macrophage migration through binding to CCR2 [51], increasing the levels of CCR2 was anticipated to increase migratory capacity. Accordingly, the results of transwell migration assays revealed that activin A significantly increased migration by WT BMDMs, while this effect was abolished by the addition of FST288 (Fig. 6B). In contrast, activin A did not influence the migratory capability of Ccr2−/− BMDMs (Fig. 6B).

Levels of CCR2, fibronectin, and CXCR4 are elevated in BMDMs following exposure to activin A. Western blotting analysis and quantification of CCR2 protein in BMDMs 6 days after treatment with 25 ng/50 ng/ml activin A, 250 ng/ml FST288, or a combination of both (A); β-actin was used as a loading control (n = 6). Migration of BMDMs generated from WT and Ccr2−/− mice in the presence of 50 ng/ml activin A, 250 ng/ml FST288, or both was quantified by incubating within a 500 ng/ml CCL2 gradient in a transwell system (B); migrated cells in the lower chamber were quantified by flow cytometry (n = 3). Relative expression (RE) of Fn1 (C), Cxcr4 (H), and Cxcl12 (I) in BMDMs 6 days after treatment with 25 ng/50 ng/ml activin A, 250 ng/ml FST288, or a combination of both was determined by qRT-PCR analysis and normalization to Hprt (n = 8). Fibronectin (green), F4/80 (red), and DNA (Topro3; blue) immunofluorescence staining in BMDMs left untreated or treated with 50 ng/ml activin A, 250 ng/ml FST288, or a combination of both (D). After gating CD45+CD11c−Ly6C−F4/80+CD11b+ macrophages, the percentages of fibronectin+ (E) and CXCR4+ (F, G) macrophages in single-cell BMDM suspensions following treatment with 25 ng/50 ng/ml activin A, 250 ng/ml FST288, or a combination of both were measured by flow cytometry (n = 5). Values are mean ± SEM; statistical analyses used the Kruskal–Wallis test followed by Dunn’s multiple comparison test or one-way/two-way ANOVA followed by Bonferroni’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Activin A increases fibronectin and CXCR4 expression in BMDMs
In vivo analyses demonstrated that numbers of both fibronectin+ and CXCR4+ macrophages were elevated in fibrotic lesions during EAO (Fig. 3 and Supplementary Fig. S3). Moreover, RNAseq confirmed that fibronectin (Fn1) and Cxcr4 were upregulated upon activin A stimulation of BMDMs (Fig. 5E and F). Consistent with these findings, qRT-PCR and immunofluorescence showed that fibronectin mRNA and protein levels were increased in BMDMs after activin A treatment (Fig. 6C and D); these effects were inhibited by FST288. Activin A treatment also induced changes in the morphology of BMDMs from round, oval, or irregular to more elongated in shape (Fig. 6D); this was accompanied by a decrease in the mean fluorescence intensity of the macrophage-specific marker F4/80 (Supplementary Fig. S4C and D). In addition, as TNF stimulates activin A secretion by mouse SCs in vitro, culture medium from TNF-stimulated SCs (SCCM) was used to evaluate the effects on macrophages of activin A originating directly from testicular cells [24]. Analysis by qRT-PCR confirmed that TNF-stimulated SCCM induced a significant increase in Fn1 mRNA expression in BMDMs (Supplementary Fig. S5A), while non-TNF-SCCM had no effect. FST288 inhibited the increase in Fn1 mRNA, confirming that the effect of SCCM was due to activin A (Supplementary Fig. S5A). Moreover, flow cytometry showed that activin A treatment of BMDMs significantly increased the percentage of fibronectin+ macrophages (Fig. 6E), while the percentage of fibronectin+ monocytes was significantly decreased in a dose-dependent manner (Supplementary Fig. S4E).
Similarly, activin A treatment caused a fivefold increase in Cxcr4 mRNA expression in cultured BMDMs; increased percentages of CXCR4+ macrophages (approximately 22-fold) and monocytes (approximately fourfold) were also observed (Fig. 6F–H and Supplementary Fig. S4F). In contrast, the mRNA expression of the CXCR4 ligand Cxcl12 was reduced in activin A-treated BMDMs (Fig. 6I). Moreover, although TNF-stimulated SCCM did not induce increased Cxcr4 expression in BMDMs, the inclusion of FST288 resulted in reduced levels of Cxcr4 mRNA (Supplementary Fig. S5B). These data clearly demonstrate the pro-fibrotic function of activin A in BMDMs.
The expression of MMPs that are dysregulated during EAO is affected by activin A in BMDM culture
As shown above, increased Inhba expression correlated with elevated levels of Mmp and Timp1 mRNAs in EAO testes in vivo (Fig. 4C–F). Moreover, RNAseq indicated that expression of Mmp2 and Mmp14 was increased in activin A-stimulated BMDMs (Fig. 5F). Therefore, we investigated the effects of activin A stimulation and Ccr2 absence on MMPs in vitro and in vivo. Analysis by qRT-PCR showed that activin A significantly upregulated the expression of Mmp2 and Mmp14 in BMDMs, while Mmp9 and Timp1 mRNAs were downregulated (Fig. 7A); these effects were blocked by FST288. Treatment with TNF-stimulated SCCM also led to a 50-fold increase in Mmp2 expression (Supplementary Fig. S5C). Notably, treatment of BMDMs with TNF-stimulated SCCM also upregulated the expression of Mmp14 (46-fold), Mmp9 (16-fold), and Timp1 (1.5-fold); however, these increases were not abolished by FST288, pointing to regulation by SC-derived factors other than activin A (Supplementary Fig. S5D–F). In agreement with the qRT-PCR results, gelatin zymography showed that activin A-induced significant upregulation of MMP2 and downregulation of MMP9 enzymatic activity (Fig. 7B and C). However, in contrast, induction of EAO resulted in increased expression of Mmp9 and Timp1 in addition to Mmp2 and Mmp14 in WT testes (Fig. 7D–G). Moreover, the EAO-induced increases in Mmp2 and Mmp14 mRNA were significantly reduced in Ccr2−/− testes (Fig. 7D and E). Although Mmp9 (Fig. 7F) was also downregulated, this decrease was not statistically significant due to the variable levels of Mmp9 expression in Ccr2−/− EAO testes. Our results also indicated that Timp1 expression was not influenced by Ccr2 deficiency (Fig. 7G). Interestingly, Spearman correlation analysis of mRNA data showed that Adgre1 (the gene encoding F4/80) was significantly positively correlated with Mmp2, Mmp14, Mmp9, and Timp1 in WT and Ccr2−/− EAO testes indicating a connection between macrophages and MMP expression (Fig. 7H and Supplementary Fig. S6A–C).

Modulation of MMPs and TIMP1 by activin A in BMDMs in vitro and EAO testes in vivo. Relative expression (RE) of Mmp2, Mmp14, Mmp9, and Timp1 in BMDMs 6 days after treatment with 25 ng/50 ng/ml activin A, 250 ng/ml FST288, or a combination of both was determined by qRT-PCR analysis and normalization to Hprt (A; n = 8). Representative gelatin zymography gel of BMDMs 7 days after treatment with 50 ng/ml activin A, 250 ng/ml FST288, or a combination of both (B); serum-free RPMI-1640 medium was used as a control. Six independent gelatin-zymography replicates were performed and the relative enzymatic activities of MMP2 and MMP9 were calculated by quantifying band intensity (C). RE of Mmp2 (D), Mmp14 (E), Mmp9 (F), and Timp1 (G) in untreated, adjuvant control, and EAO testes from WT and Ccr2−/− mice was determined by qRT-PCR and normalization to 18S rRNA and Hprt (n = 7–13). The correlation between Adgre1 and Mmp14 RE was determined in WT and Ccr2−/− EAO testes (H; n = 21). Values are mean ± SEM. Statistical analyses used the Kruskal–Wallis test followed by Bonferroni’s multiple comparison test or one-way/two-way ANOVA followed by Tukey’s multiple comparison test; correlation significance was analyzed using Spearman’s coefficient. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Integrin subunits and PDGFs expression are dysregulated in activin A-treated BMDMs and WT EAO testes
Integrins (receptors for ECM proteins) and PDGFs produced by immune cells contribute to ECM deposition and fibrosis development [52, 53]. Our RNAseq results suggested that activin A treatment regulated the expression of Pdgfa and integrin subunits Itgb3 and Itgav (Fig. 5F). Therefore, we went on to investigate the expression of integrin and PDGF genes in activin A-treated BMDMs and EAO testes by qRT-PCR. In BMDMs, activin A-induced significant dose-dependent upregulation of Itgav, Itgb5, Pdgfa, Pdgfb, and Pdgfrb expression, whereas Itgb3 mRNA was downregulated (Supplementary Fig. S7A and B). In WT EAO testes, expression of Itgav, Itgb3, Pdgfb, and Pdgfrb (Supplementary Fig. S7C, D and G, H) was also significantly increased, while upregulation of these genes was inhibited in the absence of Ccr2. Furthermore, expression of Itgav and Pdgfrb was positively associated with Adgre1 levels in EAO testis (Supplementary Fig. S6D and I). These two genes showed similar changes in vitro and in vivo, indicating that activin A may regulate the production of the αv integrin subunit and PDGFRβ in macrophages, facilitating the development of testicular fibrosis. However, the levels of Itgb5 and Pdgfa mRNAs were unchanged during EAO, and Itgb5 was expressed at lower levels in Ccr2−/− EAO testes, compared with WT EAO testes (Supplementary Fig. S7E and F).
Discussion
In this study, we aimed to investigate the role of the activin A/CCL2-CCR2/macrophage axis in the development of testicular fibrosis. We hypothesized that activin A may be involved in the regulation of inflammatory and fibrotic responses during EAO by regulating the potential pro-fibrotic properties of newly-recruited CCR2+ macrophages. To examine these possibilities, we compared the progression of EAO in WT and Ccr2−/− mice as well as in mice overexpressing follistatin to reduce activin bioactivity. Our results demonstrated that CCR2 and activin A, acting through macrophages, are crucial for the development of testicular inflammation and fibrosis in EAO. In short, testicular inflammation and fibrosis were reduced in the absence of Ccr2, as indicated by lower numbers of immune cells expressing ECM proteins (fibronectin, collagen I) and decreased production of collagen, MMPs, as well as inflammatory mediators (indicated by changes in Il-10, Ccl2, and Inhba expression). Furthermore, activin A acted as an inducer of CCR2 and fibrosis-related mediators (fibronectin, CXCR4, MMP2, and PDGF) in macrophages. Inhibition of activin A in vivo by overexpression of FST during EAO decreased testicular fibrosis and expression of collagen I by TMs.
EAO is a well-established mouse model of autoimmune-based male infertility featuring marked testicular deposition of ECM proteins (including collagens and fibronectin), increased numbers of TMs, and elevated levels of inflammatory mediators such as TNF, CCL2, and activin A. These pathological alterations combine to cause tissue destruction, inhibition of spermatogenesis, and subsequent infertility [8, 13, 21, 24]. Overall, our study revealed CCR2 and activin A to be important players in the progression of fibrosis in testicular inflammation. Furthermore, our findings underlined the pro-fibrotic role of monocyte-derived macrophages in the development of testicular fibrosis.
CCR2 is a receptor for CCL2, an inflammatory chemokine that mediates the chemotaxis of leukocytes to injury sites [54]. In this study, the absence of CCR2 during testicular inflammation not only preserved the tissue from the deposition of ECM and production of activin A, IL-10, and CCL2 but also protected the testis from damage by reducing the accumulation of immune cells. This is in accordance with previous reports demonstrating reduced inflammatory and fibrotic responses in different organs in the absence of CCR2 signaling [14, 28, 30, 55,56,57]. Here, we showed that in contrast to WT animals, Ccr2−/− mice exhibited normal levels of Il-1a, Il-10, Ccl2, and Inhba mRNA in the testes following EAO induction, pointing to the essential contribution of CCR2+ cells to the induction of the inflammatory response.
Since the mechanisms leading to the fibrotic response and the origin of ECM proteins during the progression of testicular inflammation were unknown, we wanted to address these questions. ECM-producing cells originate from a variety of sources, including bone marrow-derived circulating cells known as fibrocytes [15, 58]. Fibrocytes are characterized by the co-expression of hematopoietic markers such as CD45, CD34, or CD11b and mesenchymal markers such as collagen or vimentin [59]. These cells mainly migrate to injury sites by chemotaxis via chemokine ligand/receptor pathways, including CCL2/CCR2 or CXCL12/CXCR4 [59, 60]. Fibrocytes can also originate from a subpopulation of monocytes via monocyte-to-fibroblast transition [61,62,63]. Our results indicate that during the severe stage of EAO, immune cells (particularly macrophages) act as important sources of fibronectin and collagen I production. Since the CD34 marker was not included in our analysis, we cannot definitely exclude the possibility that a proportion of the ECM-expressing CD45+ cells may have been fibrocytes. The significant reduction in the accumulation of CD45+ cells and TMs expressing fibronectin, collagen I, and CXCR4 observed in EAO Ccr2−/− testis indicates the importance of CCR2 and CXCR4 signaling in testicular ECM expression. In addition, the very high levels of Cxcl12 and Cxcr4 mRNA detected in WT EAO testis were significantly reduced in the absence of Ccr2, underlining the importance of newly-recruited CCR2+ cells as a source of these molecules during the testicular fibrotic response. Similarly, the expression of fibronectin in TMs under pathological conditions in the human testis (impaired spermatogenesis and collagen accumulation) points to the possible involvement of macrophages in fibrotic remodeling. These data are in agreement with earlier reports demonstrating CCR2-dependent inhibition of BMDM and fibrocyte infiltration in kidney injury, colon fibrosis, and colitis [28, 30, 64,65,66]. Moreover, CXCR4+ macrophages have also been shown to act as a pro-fibrotic cell subpopulation in the kidney and lung [67,68,69]. Future studies are required to establish whether CCR2+CXCR4+ TMs are the key players involved in the EAO fibrotic response. However, currently, we cannot exclude the possibility that the production of Cxcl12 by CCR2+ cells may account for the reduced infiltration of CXCR4+ cells in Ccr2−/− mouse testes.
Our results also showed that expression of Mmp2, Mmp14, Mmp9, and Timp1 was increased in WT EAO testes, indicating activation of the ECM machinery. The gelatinases MMP2 and MMP9 mainly cleave collagen IV, an important component of the basement membrane, making it easier for leukocytes to transmigrate [70, 71]. Activation of MMP2 requires the involvement of MMP14 [72]. Aggregated ECM proteins require degradation by MMPs to prevent fibrosis; on the other hand, TIMP1 inhibits the degradative function of MMP proteins, thereby exacerbating the fibrotic process. However, elevated levels of MMPs could also contribute to immune cell migration or activate PTCs /fibroblasts to produce more ECM proteins, thereby promoting tissue fibrosis [73, 74]. Thus, the process of testicular fibrosis appears to depend on the dynamic balance between MMP and TIMP activities. In this study, loss of Ccr2 abrogated the elevated expression of Mmp2 and Mmp14 occurring in EAO testes. Interestingly, the expression of these genes positively correlated with levels of Adgre1 mRNA, indicating that macrophages are involved in MMP and TIMP1 production and thereby in the promotion of testicular fibrosis at the inflammation stage.
The changes in integrins and PDGF gene expression observed in EAO may also contribute to the fibrotic phenotype, as these groups of molecules are known as important promoters of fibrosis [52, 53, 75, 76]. The increased levels of Itgav/Itgb3 (encoding integrin αvβ3) and Pdgfb/Pdgfrb mRNAs detected during EAO (dependent on Ccr2 expression) were likely due to the infiltration of macrophages, as Itgav and Pdgfrb correlated positively with Adgre1 expression. Furthermore, PDGF acts as a strong chemoattractant for fibrocytes in pulmonary fibrosis, while pharmacological blockade of the PDGF/PDGFR axis is considered a promising treatment option [77].
Taken together, these in vivo data indicate that CCR2 contributes to the development of testicular fibrosis during inflammation by mediating leukocyte infiltration, cytokine release, and ECM protein production by immune cells, particularly TMs. In addition, our results suggest that CCR2 + TMs may produce multiple factors, including the CXCL12/CXCR4 axis, MMPs, αv integrin, PDGFB, and PDGFRβ, which potentially act as downstream signaling molecules to regulate immune processes during testicular inflammation (summarized in Fig. 8).

Model of the role of activin A, CCL2/CCR2, and macrophages in fibrotic remodeling during EAO. Increased expression of TNF during testicular inflammation induces SCs to release higher levels of CCL2 and activin A. In response to elevated levels of CCL2, macrophages expressing CCR2 are attracted to the site of injury and infiltrate the inflamed testes. Activin A stimulates CCR2 expression on macrophages and drives them to a pro-fibrotic phenotype by inducing the expression of CXCR4, collagen I, fibronectin, MMPs, integrins and PDGFs. Together, these processes facilitate the development of fibrosis in EAO testes. In EAO mice, inflammatory response and fibrotic remodeling can be inhibited by the knockout of Ccr2, blocking macrophage infiltration, or by the use of FST, an activin A antagonist. (GC dying germ cells, SC Sertoli cells, PTC peritubular cells, Mφ macrophages)
In addition to CCR2, activin A is another important fibrotic regulator [6, 23]. In the current study, activin A stimulated CCR2 expression (at the mRNA and protein level) in macrophages and reduced Ccl2 levels. Similarly opposing effects of activin A on Ccr2 and Ccl2 expression have been demonstrated in human monocyte-derived macrophages [78]. Our findings indicated that activin A can increase the responsiveness of inflammatory BMDMs to CCL2 by enhancing the expression of CCR2. Furthermore, according to our results, activin A also induced the expression of CXCR4 in macrophages. As CXCR4 is highly expressed by macrophages during testicular inflammation, reduced infiltration by monocytes/macrophages in Ccr2−/− mice during EAO may explain why the level of testicular Cxcr4 mRNA did not increase in CCR2-depleted mice following induction of EAO (see Fig. 8).
Previously, we showed that activin A-induced the expression of fibrosis-specific genes in mouse PTCs and NIH 3T3 fibroblasts. Furthermore, expression of the activin dimer subunit was increased, not only in inflamed mouse testes but also in human testes with impaired spermatogenesis and focal leukocytic infiltrates [13, 24]. Notably, the severity of the induced inflammation and damage was directly proportional to the level of activin A [13, 24]. These findings, together with the effects of Ccr2 deficiency during EAO (including reduced infiltration of macrophages, lower levels of ECM deposition and Inhba) implicate activin A as a key regulator of testicular inflammation and fibrosis, acting through TMs. In vivo inhibition of activin A by overexpression of follistatin clearly demonstrated the role of activin A in the development of testicular inflammation; for example, suppression of the accumulation of TM-derived collagen I by follistatin indicated that activin A affected the progression of testicular fibrosis by regulating pro-fibrotic properties of macrophages. Moreover, the expression of Inhba correlated positively with testicular Mmp and Timp1 expression during EAO. Therefore, elevated levels of MMPs and TIMP1 in EAO testes were associated with both macrophage infiltration and activin A expression.
Activin A treatment influenced the transcriptome of BMDMs in vitro and increased the expression of Fn1, Ccr2, Cxcr4, Mmp2, Mmp14, Itgav, Pdgfa, Pdgfb, and Pdgfrb genes as well as the levels of fibronectin, CCR2, CXCR4, and MMP2 proteins. Moreover, activin A enhanced the migratory ability of macrophages in response to CCL2 stimulation. Interestingly, in addition to increased expression of fibrosis-related genes, activin A-induced transformation to spindle-shaped morphology and diminished F4/80 fluorescence in BMDMs, indicating possible conversion to a fibroblastic phenotype in vitro. Similarly, activin A reportedly reprogrammed pre-tumorigenic macrophages to a tumor-associated phenotype, promoted conversion of CD4+CD25− naïve T cells into regulatory T cells, and induced a pro-fibrotic transcriptome in fibroblasts, thereby transforming them into myofibroblasts [79,80,81]. Furthermore, macrophages from the synovium of active rheumatoid arthritis patients, which exhibited pro-inflammatory, macrophage-polarizing properties, were inhibited by the addition of an anti-activin A neutralizing antibody [82].
Combining these results, we postulate that activin A promotes the progression of testicular fibrosis by inducing the production of fibrosis-related mediators in macrophages. Furthermore, although activin A treatment reduced the expression of Mmp9, Timp1, and Itgb3 in BMDMs, other factors, such as TNF, may increase the levels of MMP9 and TIMP1 in macrophages or other testicular cells, thereby promoting their expression in EAO testes [83]. In addition, cells such as fibroblasts can produce large amounts of β3 integrin in response to inflammatory stimuli, which could underlie the high levels of Itgb3 mRNA observed in EAO testes [84].
Critically, it should be noted that utilizing BMDMs matured in the presence of M-CSF as a surrogate for TMs may not exactly replicate all functional aspects of the TMs. However, M-CSF is a critical regulator of the maturation and tissue-specific functions of TMs [85]. As TMs make up a very small proportion of the normal mouse testis cell population, in vitro experimentation on isolated TMs would require an unreasonably large number of mice. In accordance with 3R principles, TMs were replaced by BMDMs, drastically reducing the number of animals required.
Taken together, our in vivo and in vitro results illustrate that activin A promotes the differentiation of macrophages to a pro-fibrotic phenotype through the induction of a variety of inflammatory mediators and receptor molecules (summarized in Fig. 8). Activin A-stimulated BMDMs are potential fibroblast precursors, which likely facilitate tissue fibrosis. In addition, by inducing CCR2 expression, activin A potentially affects the recruitment of monocytes/macrophages to injury sites. Although our data indicate that CCR2, activin A, and macrophages are responsible for the testicular fibrotic response, the precise immune mechanisms that regulate this process require further investigation. Future studies are needed to address the direct influence of activin A using targeted depletion of activin A receptors (in monocytes/macrophages) to analyze their role in inflammatory responses and fibrotic remodeling.
In conclusion, our data indicate that CCR2 and activin A regulate the development of fibrosis during testicular inflammation and underline the crucial pro-fibrotic function of macrophages in inflammation-associated fibrotic remodeling in EAO. Future therapeutic targeting of CCR2 and/or activin A may offer a possible strategy to address testicular fibrosis.
Data availability
The data presented in this study are available in the article and supplementary material. Transcriptome data are available online at the GEO repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE210004).
Abbreviations
- αSMA:
-
α-Smooth muscle actin
- BMDM:
-
Bone marrow-derived macrophage
- CCL2:
-
C–C motif chemokine ligand 2
- CCR2:
-
C–C motif chemokine receptor type 2
- DEGs:
-
Differentially expressed genes
- EAO:
-
Experimental autoimmune orchitis
- ECM:
-
Extracellular matrix
- FST:
-
Follistatin
- IL:
-
Interleukin
- MMP:
-
Matrix metalloproteinase
- PDGF:
-
Platelet-derived growth factor
- PTC:
-
Peritubular cell
- SC:
-
Sertoli cell
- SCCM:
-
Sertoli cell-conditioned medium
- TGF-β:
-
Transforming growth factor-β
- TIMP:
-
Tissue inhibitor of metalloproteinase
- TMs:
-
Testicular macrophages
- TNF:
-
Tumor necrosis factor
- WT:
-
Wild type
References
Agarwal A, Baskaran S, Parekh N et al (2021) Male infertility. Lancet 397:319–333. https://doi.org/10.1016/S0140-6736(20)32667-2
Bryan ER, Kim J, Beagley KW, Carey AJ (2020) Testicular inflammation and infertility: Could chlamydial infections be contributing? Am J Reprod Immunol 84:e13286. https://doi.org/10.1111/aji.13286
Vanderborght M, Wyns C (2018) Fertility and infertility: definition and epidemiology. Clin Biochem 62:2–10. https://doi.org/10.1016/j.clinbiochem.2018.03.012
Schuppe HC, Pilatz A, Hossain H et al (2017) Urogenital infection as a risk factor for male infertility. Dtsch Arztebl Int 114:339–346. https://doi.org/10.3238/arztebl.2017.0339
Jacobo P, Guazzone VA, Theas MS, Lustig L (2011) Testicular autoimmunity. Autoimmun Rev 10:201–204. https://doi.org/10.1016/j.autrev.2010.09.026
Lustig L, Guazzone VA, Theas MS et al (2020) Pathomechanisms of autoimmune based testicular inflammation. Front Immunol 11:583135. https://doi.org/10.3389/fimmu.2020.583135
Naito M, Terayama H, Hirai S et al (2012) Experimental autoimmune orchitis as a model of immunological male infertility. Med Mol Morphol 45:185–189. https://doi.org/10.1007/s00795-012-0587-2
Fijak M, Pilatz A, Hedger MP et al (2018) Infectious, inflammatory and “autoimmune” male factor infertility: How do rodent models inform clinical practice? Hum Reprod Update 24:416–441. https://doi.org/10.1093/humupd/dmy009
Guazzone VA, Rival C, Denduchis B, Lustig L (2003) Monocyte chemoattractant protein-1 (MCP-1/CCL2) in experimental autoimmune orchitis. J Reprod Immunol 60:143–157. https://doi.org/10.1016/j.jri.2003.08.001
Bhushan S, Theas MS, Guazzone VA et al (2020) Immune cell subtypes and their function in the testis. Front Immunol 11:583304. https://doi.org/10.3389/fimmu.2020.583304
Meinhardt A, Dejucq-Rainsford N, Bhushan S (2022) Testicular macrophages: development and function in health and disease. Trends Immunol 43:51–62. https://doi.org/10.1016/j.it.2021.11.003
Rival C, Theas M, Suescun M et al (2008) Functional and phenotypic characteristics of testicular macrophages in experimental autoimmune orchitis. J Pathol 215:108–117. https://doi.org/10.1002/path
Nicolas N, Michel V, Bhushan S et al (2017) Testicular activin and follistatin levels are elevated during the course of experimental autoimmune epididymo-orchitis in mice. Sci Rep 7:42391. https://doi.org/10.1038/srep42391
Wang M, Yang Y, Cansever D et al (2021) Two populations of self-maintaining monocyte-independent macrophages exist in adult epididymis and testis. Proc Natl Acad Sci USA 118:e2013686117. https://doi.org/10.1073/PNAS.2013686117
Mack M, Yanagita M (2015) Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int 87:297–307. https://doi.org/10.1038/ki.2014.287
Wynn TA, Vannella KM (2016) Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44:450–462. https://doi.org/10.1016/j.immuni.2016.02.015
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737. https://doi.org/10.1038/nri3073
Arpino V, Brock M, Gill SE (2015) The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol 44–46:247–254. https://doi.org/10.1016/j.matbio.2015.03.005
Wijayarathna R, de Kretser DM (2016) Activins in reproductive biology and beyond. Hum Reprod Update 22:342–357. https://doi.org/10.1093/humupd/dmv058
Hedger MP, Winnall WR (2012) Regulation of activin and inhibin in the adult testis and the evidence for functional roles in spermatogenesis and immunoregulation. Mol Cell Endocrinol 359:30–42. https://doi.org/10.1016/j.mce.2011.09.031
Nicolas N, Muir JA, Hayward S et al (2017) Induction of experimental autoimmune orchitis in mice: responses to elevated circulating levels of the activin-binding protein, follistatin. Reproduction 154:293–305. https://doi.org/10.1530/REP-17-0010
Morianos I, Papadopoulou G, Semitekolou M, Xanthou G (2019) Activin-A in the regulation of immunity in health and disease. J Autoimmun 104:102314. https://doi.org/10.1016/j.jaut.2019.102314
de Kretser DM, O’Hehir RE, Hardy CL, Hedger MP (2012) The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol Cell Endocrinol 359:101–106. https://doi.org/10.1016/j.mce.2011.10.009
Kauerhof AC, Nicolas N, Bhushan S et al (2019) Investigation of activin A in inflammatory responses of the testis and its role in the development of testicular fibrosis. Hum Reprod 34:1536–1550. https://doi.org/10.1093/humrep/dez109
Jiang L, Qi Y, Kong X et al (2021) Activin A as a novel chemokine induces migration of L929 fibroblasts by ERK signaling in microfluidic devices. Front Cell Dev Biol 9:660316. https://doi.org/10.3389/fcell.2021.660316
Hedger MP, de Kretser DM (2013) The activins and their binding protein, follistatin-Diagnostic and therapeutic targets in inflammatory disease and fibrosis. Cytokine Growth Factor Rev 24:285–295. https://doi.org/10.1016/j.cytogfr.2013.03.003
Fantuzzi L, Tagliamonte M, Gauzzi MC, Lopalco L (2019) Dual CCR5/CCR2 targeting: opportunities for the cure of complex disorders. Cell Mol Life Sci 76:4869–4886. https://doi.org/10.1007/s00018-019-03255-6
Xia Y, Entman ML, Wang Y (2013) CCR2 regulates the uptake of bone marrow-derived fibroblasts in renal fibrosis. PLoS One 8:e77493. https://doi.org/10.1371/journal.pone.0077493
Hasegawa M, Sato S (2008) The roles of chemokines in leukocyte recruitment and fibrosis in systemic sclerosis. Front Biosci-Landmark 13:3637–3647. https://doi.org/10.2741/2955
Kuroda N, Masuya M, Tawara I et al (2019) Infiltrating CCR2+ monocytes and their progenies, fibrocytes, contribute to colon fibrosis by inhibiting collagen degradation through the production of TIMP-1. Sci Rep 9:8568. https://doi.org/10.1038/s41598-019-45012-6
Lei T, Moos S, Klug J et al (2018) Galectin-1 enhances TNFα-induced inflammatory responses in Sertoli cells through activation of MAPK signalling. Sci Rep 8:3741. https://doi.org/10.1038/s41598-018-22135-w
Boring L, Gosling J, Chensue SW et al (1997) Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Investig 100:2552–2561. https://doi.org/10.1172/JCI119798
Kohno S, Munoz JA, Williams TM et al (1983) Immunopathology of murine experimental allergic orchitis. J Immunol 130:2675–2682
Hauptman D, Perić MH, Marić T et al (2021) Leydig cells in patients with non-obstructive azoospermia: Do they really proliferate? Life (Basel) 11:1266. https://doi.org/10.3390/life11111266
Ježek D (2013) Atlas on the human testis: normal morphology and pathology. Springer, London
Kiernan J (2008) Histological and histochemical methods: theory and practice, 4th ed. Scion Publishing, Bloxham, UK
Sidis Y, Mukherjee A, Keutmann H et al (2006) Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 147:3586–3597. https://doi.org/10.1210/en.2006-0089
Bailey JD, Shaw A, McNeill E et al (2020) Isolation and culture of murine bone marrow-derived macrophages for nitric oxide and redox biology. Nitric Oxide 100–101:17–29. https://doi.org/10.1016/j.niox.2020.04.005
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Andrews S (2010) FASTQC. A quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Dobin A, Davis CA, Schlesinger F et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Liao Y, Smyth GK, Shi W (2014) FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. https://doi.org/10.1093/bioinformatics/btt656
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
UniProt Consortium (2014) Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 42:191–198. https://doi.org/10.1093/nar/gkt1140
Leeman M, McKay J, Murray G (2002) Matrix metalloproteinase 13 activity is associated with poor prognosis in colorectal cancer. J Clin Pathol 55:758–762. https://doi.org/10.1136/jcp.55.10.758
Mehrad B, Burdick MD, Strieter RM (2009) Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int J Biochem Cell Biol 41:1708–1718. https://doi.org/10.1016/j.biocel.2009.02.020
Mantovani A, Sica A, Sozzani S et al (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686. https://doi.org/10.1016/j.it.2004.09.015
Xu J, Mora A, Shim H et al (2007) Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol 37:291–299. https://doi.org/10.1165/rcmb.2006-0187OC
Robert S, Gicquel T, Victoni T et al (2016) Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci Rep 36:e00360. https://doi.org/10.1042/BSR20160107
Dzenko KA, Song L, Ge S et al (2005) CCR2 expression by brain microvascular endothelial cells is critical for macrophage transendothelial migration in response to CCL2. Microvasc Res 70:53–64. https://doi.org/10.1016/j.mvr.2005.04.005
Margadant C, Sonnenberg A (2010) Integrin-TGF-Β crosstalk in fibrosis, cancer and wound healing. EMBO Rep 11:97–105. https://doi.org/10.1038/embor.2009.276
Klinkhammer BM, Floege J, Boor P (2018) PDGF in organ fibrosis. Mol Aspects Med 62:44–62. https://doi.org/10.1016/j.mam.2017.11.008
Gschwandtner M, Derler R, Midwood KS (2019) More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol 10:2759. https://doi.org/10.3389/fimmu.2019.02759
Liang J, Jung Y, Tighe RM et al (2012) A macrophage subpopulation recruited by CC chemokine ligand-2 clears apoptotic cells in noninfectious lung injury. Am J Physiol Lung Cell Mol Physiol 302:933–941. https://doi.org/10.1152/ajplung.00256.2011
Braga TT, Correa-Costa M, Silva RC et al (2018) CCR2 contributes to the recruitment of monocytes and leads to kidney inflammation and fibrosis development. Inflammopharmacology 26:403–411. https://doi.org/10.1007/s10787-017-0317-4
Raghu H, Lepus CM, Wang Q et al (2017) CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann Rheum Dis 76:914–922. https://doi.org/10.1136/annrheumdis-2016-210426
Hinz B, Phan SH, Thannickal VJ et al (2007) The myofibroblast: one function, multiple origins. Am J Pathol 170:1807–1816. https://doi.org/10.2353/ajpath.2007.070112
Reilkoff RA, Bucala R, Herzog EL (2011) Fibrocytes: emerging effector cells in chronic inflammation. Nat Rev Immunol 11:427–435. https://doi.org/10.1038/nri2990
Mack M (2018) Inflammation and fibrosis. Matrix Biol 68–69:106–121. https://doi.org/10.1016/j.matbio.2017.11.010
Dong Y, Yang M, Zhang J et al (2016) Depletion of CD8 + T cells exacerbates CD4 + T cell-induced monocyte-to-fibroblast transition in renal fibrosis. J Immunol 196:1874–1881. https://doi.org/10.4049/jimmunol.1501232
Niedermeier M, Reich B, Gomez MR et al (2009) CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci USA 106:17892–17897. https://doi.org/10.1073/pnas.0906070106
Yang J, Lin SC, Chen G et al (2013) Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J Am Soc Nephrol 24:1644–1659. https://doi.org/10.1681/ASN.2013030217
Do C, Drel V, Tan C et al (2019) Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Investig Dermatol 139:2134-2143.e2. https://doi.org/10.1016/j.jid.2019.03.1145
Wilkening A, Krappe J, Mühe AM et al (2020) C-C chemokine receptor type 2 mediates glomerular injury and interstitial fibrosis in focal segmental glomerulosclerosis. Nephrol Dial Transplant 35:227–239. https://doi.org/10.1093/ndt/gfy380
Hachiya K, Masuya M, Kuroda N et al (2021) Irbesartan, an angiotensin II type 1 receptor blocker, inhibits colitis-associated tumourigenesis by blocking the MCP-1/CCR2 pathway. Sci Rep 11:19943. https://doi.org/10.1038/s41598-021-99412-8
Yuan A, Lee Y, Choi U et al (2015) Chemokine receptor Cxcr4 contributes to kidney fibrosis via multiple effectors. Am J Physiol Renal Physiol 308:F459–F472. https://doi.org/10.1152/ajprenal.00146.2014
Dupin I, Allard B, Ozier A et al (2016) Blood fibrocytes are recruited during acute exacerbations of chronic obstructive pulmonary disease through a CXCR4-dependent pathway. J Allergy Clin Immunol 137:1036-1042.e7. https://doi.org/10.1016/j.jaci.2015.08.043
Chen Y, Pu Q, Ma Y et al (2021) Aging reprograms the hematopoietic-vascular niche to impede regeneration and promote fibrosis. Cell Metab 33:395–410. https://doi.org/10.1016/j.cmet.2020.11.019 (e4)
Khokha R, Murthy A, Weiss A (2013) Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol 13:649–665. https://doi.org/10.1038/nri3499
Kurzepa J, Mdro A, Czechowska G et al (2014) Role of MMP-2 and MMP-9 and their natural inhibitors in liver fibrosis, chronic pancreatitis and non-specific inflammatory bowel diseases. Hepatobiliary Pancreat Dis Int 13:570–579. https://doi.org/10.1016/S1499-3872(14)60261-7
Yosef G, Arkadash V, Papo N (2018) Targeting the MMP-14/MMP-2/integrin v3 axis with multispecific N-TIMP2– based antagonists for cancer therapy. J Biol Chem 293:13310–13326. https://doi.org/10.1074/jbc.RA118.004406
Kobayashi T, Kim HJ, Liu X et al (2014) Matrix metalloproteinase-9 activates TGF-β and stimulates fibroblast contraction of collagen gels. Am J Physiol Lung Cell Mol Physiol 306:1006–1015. https://doi.org/10.1152/ajplung.00015.2014
Song J, Wu C, Korpos E et al (2015) Focal MMP-2 and MMP-9 activity at the blood-brain barrier promotes chemokine-induced leukocyte migration. Cell Rep 10:1040–1054. https://doi.org/10.1016/j.celrep.2015.01.037
Camelo A, Dunmore R, Sleeman MA, Clarke DL (2014) The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front Pharmacol 4:173. https://doi.org/10.3389/fphar.2013.00173
Schnittert J, Bansal R, Storm G, Prakash J (2018) Integrins in wound healing, fibrosis and tumor stroma: high potential targets for therapeutics and drug delivery. Adv Drug Deliv Rev 129:37–53. https://doi.org/10.1016/j.addr.2018.01.020
Aono Y, Kishi M, Yokota Y et al (2014) Role of platelet-derived growth factor/platelet-derived growth factor receptor axis in the trafficking of circulating fibrocytes in pulmonary fibrosis. Am J Respir Cell Mol Biol 51:793–801. https://doi.org/10.1165/rcmb.2013-0455OC
Sierra-Filardi E, Nieto C, Domínguez-Soto Á et al (2014) CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol 192:3858–3867. https://doi.org/10.4049/jimmunol.1302821
Wietecha MS, Pensalfini M, Cangkrama M et al (2020) Activin-mediated alterations of the fibroblast transcriptome and matrisome control the biomechanical properties of skin wounds. Nat Commun 11:2604. https://doi.org/10.1038/s41467-020-16409-z
Antsiferova M, Piwko-Czuchra A, Cangkrama M et al (2017) Activin promotes skin carcinogenesis by attraction and reprogramming of macrophages. EMBO Mol Med 9:27–45. https://doi.org/10.15252/emmm.201606493
Cangkrama M, Wietecha M, Werner S (2020) Wound repair, scar formation, and cancer: converging on activin. Trends Mol Med 26:1107–1117. https://doi.org/10.1016/j.molmed.2020.07.009
Palacios BS, Estrada-Capetillo L, Izquierdo E et al (2015) Macrophages from the synovium of active rheumatoid arthritis exhibit an activin a-dependent pro-inflammatory profile. J Pathol 235:515–526. https://doi.org/10.1002/path.4466
Webster NL, Crowe SM (2006) Matrix metalloproteinases, their production by monocytes and macrophages and their potential role in HIV-related diseases. J Leukoc Biol 80:1052–1066. https://doi.org/10.1189/jlb.0306152
Balasubramanian S, Quinones L, Kasiganesan H et al (2012) β3 Integrin in cardiac fibroblast is critical for extracellular matrix accumulation during pressure overload hypertrophy in mouse. PLoS One 7:e45076. https://doi.org/10.1371/journal.pone.0045076
Cohen PE, Nishimura K, Zhu L, Pollard JW (1999) Macrophages: important accessory cells for reproductive function. J Leukoc Biol 66:765–772. https://doi.org/10.1002/jlb.66.5.765
Acknowledgements
We would like to thank Dr. Jessica Tamanini of Insight Editing London for language editing of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the International Research Training Group in “Molecular pathogenesis on male reproductive disorders”, a collaboration between Justus Liebig University (Giessen) and Monash University (Melbourne) (GRK1871/1-2) funded by the Deutsche Forschungsgemeinschaft and Monash University. The financial support of the China Scholarship Council to Wei Peng is also gratefully appreciated.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of the School of Medicine at the University of Zagreb (380-59-10106-20-111/171). Animal experiments were approved by the responsible local ethics committees on animal care (Regierungspraesidium Giessen GI 58/2014 — Nr. 735-GP and the Monash Medical Centre Animal Experimentation Committee). All experiments involving animals were carried out in strict accordance with the recommendations in the guide for the Care and Use of Laboratory Animals of the German law on animal welfare and the Australian Code for the Care and Use of Animals for Scientific Purposes.
Consent to participate
Informed consent was obtained from all men included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, W., Kepsch, A., Kracht, T.O. et al. Activin A and CCR2 regulate macrophage function in testicular fibrosis caused by experimental autoimmune orchitis. Cell. Mol. Life Sci. 79, 602 (2022). https://doi.org/10.1007/s00018-022-04632-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04632-4