
Abstract
During sepsis, the importance of alterations in cell metabolism is underappreciated. The cellular metabolism, which has a variable metabolic profile in different cells and disease stages, is largely responsible for the immune imbalance and organ failure associated with sepsis. Metabolic reprogramming, in which glycolysis replaces OXPHOS as the main energy-producing pathway, is both a requirement for immune cell activation and a cause of immunosuppression. Meanwhile, the metabolites produced by OXPHOS and glycolysis can act as signaling molecules to control the immune response during sepsis. Sepsis-induced "energy shortage" leads to stagnated cell function and even organ dysfunction. Metabolic reprogramming can alleviate the energy crisis to some extent, enhance host tolerance to maintain cell survival functions, and ultimately increase the adaptation of cells during sepsis. However, a switch from glycolysis to OXPHOS is essential for restoring cell function. This review summarized the crosstalk between metabolic reprogramming and immune cell activity as well as organ function during sepsis, discussed the benefits and drawbacks of metabolic reprogramming to show the contradictions of metabolic reprogramming during sepsis, and assessed the feasibility of treating sepsis through targeted metabolism. Using metabolic reprogramming to achieve metabolic homeostasis could be a viable therapy option for sepsis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sepsis is a serious life-threatening disease in the ICU, with at least 19 million patients reported worldwide each year [1, 2]. In general, a strong pro-inflammatory effect eliminates the pathogen in the early stages of inflammation, and the immune system progressively shifts to an anti-inflammatory state as the disease progresses, mostly to minimize inflammation and promote tissue healing. The balance between pro- and anti-inflammatory is an essential component of the normal functioning of the body's immune system. However, during sepsis, the disrupted pro- and anti-inflammatory balance typically results in inflammatory storms and immunosuppression, which lead to excessive cellular damage and inability to respond to secondary infections, ultimately causing fatal multi-organ dysfunction syndrome (MODS). MODS is the most serious complication of sepsis, but its pathogenesis is still poorly understood. Severe immunosuppression and MODS create a therapeutic dilemma in sepsis, despite decades of research by researchers, there have been no significant breakthroughs in treatment, which is still dominated by antibiotics and organ support. The dynamics of cellular energy metabolism, the basis of cellular activity during sepsis, is a role that cannot be ignored.
Metabolic reprogramming occurs in almost all types of cells during sepsis. Immune cells undergo tumor-like changes—the "Warburg effect" [3,4,5,6], where cells switch to glycolysis as their primary source of energy instead of oxidative phosphorylation (OXPHOS) under aerobic conditions, which is critical to immune cell activity during sepsis. The reprogramming that glycolysis replaces OXPHOS is also observed in organ cells, such as tubular epithelial cells (TECs) [7] and cardiomyocytes [8]. Singer et al. [9,10,11] have argued that sepsis-induced MODS may represent a defensive strategy for the host in response to metabolic dysregulation and insufficient energy, which links the vital and functional activities of organ cells to metabolic reprogramming. Furthermore, the relevance of metabolic reprogramming to sepsis was highlighted again by Raymond et al. [12] in 2013, who found that the metabolomes and proteomes of patients at the hospital who would ultimately die differed markedly from those of patients who would survive. Sepsis is a disease with a complicated metabolism, and the significance of metabolic reprogramming in sepsis requires more exploration. Identifying the crosstalk between metabolism and disease is essential for the diagnosis and treatment of sepsis. Moreover, as the concept that the oxygen delivered to the cells is sufficient is gradually recognized, "cytopathic hypoxia" caused by mitochondrial dysfunction or changes in metabolic enzyme activity may be a better explanation for metabolic reprogramming.
This review aims to describe alterations in oxidative metabolism and glycolysis in immune and organ cells during sepsis and to summarize their interrelation with cellular function. We emphasized the importance and complexity of metabolic reprogramming in sepsis, as well as suggested to maintain metabolic homeostasis may be a strategy for the future treatment of sepsis. At the same time, the key role of mitochondria as a metabolic organelle in sepsis metabolism regulation will also be underlined.
Glycolysis is a double-edged sword for the immune system during sepsis
Energy metabolism in cells is driven mainly by OXPHOS and glycolysis. Since the study in the 1950s demonstrated that lymphocyte activation was associated with an increase in glucose consumption [13], the key role of metabolism in the activation, proliferation, and differentiation of immune cells has been gradually identified [14] and evidenced by studies of tumors [15] and diabetes [16, 17]. Immune dysfunction is one of the most prominent clinical features of sepsis. Knowledge of the effects of metabolism on immune function will help to elucidate the imbalance between the pro-inflammatory and anti-inflammatory.
Metabolic reprogramming of immune cells during sepsis
Innate immune cells (monocytes, macrophages, granulocytes, natural killer cells, and dendritic cells), adaptive immune cells (lymphocytes), and certain vascular cells (endothelial cells and vascular smooth muscle cells) all play roles in the inflammatory response. The innate immune cells recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through surface receptors (Toll-like receptors (TLRs) and c-type lectin receptors (CLRs)), cytoplasmic receptors (nucleotide-binding oligomerization domain (Nod)-rich leucine repeat receptors (NLRs) and RIG-I-like receptors (RLRs)] [18] to release signaling and effector molecules to trigger inflammatory responses and kill invading microbes, as well as mobilize adaptive immune cells (T lymphocytes and B lymphocytes) to generate delayed but stronger and more specific immune responses. During the activation of immune cells, the energy metabolic pathway is reprogrammed from OXPHOS to glycolysis.
Although resting dendritic cells (DCs) are dependent on mitochondrial OXPHOS fuelled by the β-oxidation of lipids [19,20,21], metabolic reprogramming from OXPHOS to glycolysis triggered by TLR signaling is essential for DC maturation and associated biologic functions [21]. When glycolysis is blocked with the glycolytic inhibitor 2-DG [6], DCs activation is considerably reduced with the release of IL-6 and TNF substantially impaired [6, 21]. Similar to DCs, mature neutrophils’ glycolysis is associated with their metastatic activity [22] and the creation of neutrophil extracellular networks (NETs), which kill bacteria after metastasis to the target region [23]. Macrophages are induced into M1-or M2-type after being activated by LPS. The differentiation between M1- and M2-type macrophages plays the opposite role in sepsis. Porta et al. [24] and Pena's team [25] have illustrated that M1-type macrophages are responsible for secreting pro-inflammatory factors and promoting inflammation, and M2-type macrophages are responsible for reducing inflammation and repairing damaged tissues. In the resting state, macrophages create energy mostly by OXPHOS [26, 27], whereas M2-type macrophages primarily utilize fatty acid oxidation (FAO) to support the anti-inflammatory function and M1-type macrophages mainly use glycolysis not only for faster ATP production but also to obtain the biosynthetic raw material [28].
OXPHOS provides energy to immature T cells, whereas activated effector T lymphocytes switch to glycolysis to speed up ATP production [29,30,31]. Different T cell subsets, however, have different metabolic properties. For instance, effector T cells (Teffs) that dominate the pro-inflammatory response, such as Th1, Th2, and Th17 [29], mainly rely on glycolysis, whereas regulatory T-cells (Tregs) and memory T cells (Tmems) which mainly play an anti-inflammatory role mostly use fatty acids in their metabolism, and mTORC1 activation drives Teff cells differentiation while suppressing Treg cells [32,33,34]. However, some research has found that sped Treg cell proliferation by Ethyl Pyruvate has been connected to glycolysis rather than OXPHOS [31].
Metabolic reprogramming mainly involves suppressed mitochondrial oxidative function and an increased glycolytic capacity. LPS causes increased glycolysis while inhibiting mitochondrial respiration by suppressing nitrosylation of cytochrome C oxidase and complex enzyme I in the electron transport chain [27, 35]. The mTOR-HIF-1α pathway is also activated in macrophages by LPS, which prompts increased expression of HIF-1α downstream glycolytic enzymes like glucose transporter protein 1 (GLUT1), fructose-2,6-bisphosphatase 3 (PFKFB3), hexokinase 2 (HK2), pyruvate kinase M2 (PKM2), and lactate dehydrogenase (LDH) to elevate glycolytic capacity [36]. AMPK promotes β-oxidation of fatty acids by regulating proliferator-activated receptor γ (PPAR-γ) and carnitine palmitoyl transferase 1 (CPT1), but in sepsis, the downregulated AMPK pathway promotes the glycolytic capacity of macrophages [37, 38] and directly affects the secretion of inflammatory and anti-inflammatory factors like IL-1β and IL-6.
Enhanced glycolysis contributes to immune cell activity
The "Warburg effect" is favorable because of its ability to provide not only ATP but also metabolites that contribute to the activities of the immune cells [39]. The increased expression of GLUT1 during sepsis promotes the competitive uptake of glucose [40, 41], a source of carbon that can be used for biosynthetic in addition to being a source of energy [42]. When GLUT1 expression is depressed, glucose uptake and glycolysis are significantly inhibited, along with a significant decrease in Teff's vital and functional activities [29, 40]. On the other hand, the increased glucose uptake and glycolytic flux, as well as the high rate of ATP production by glycolysis, allow ATP from glycolysis to meet the energy requirements of activated immune cells, even when OXPHOS is inhibited. The glucose 6-phosphate (G-6-P) produced in the first phase of glycolysis can be imported into the pentose phosphate pathway (PPP) to nucleotide synthesis, while the NADPH produced is a cofactor necessary for a variety of metabolic processes, such as lipid synthesis [43]. Dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (G-3-P) from glycolysis are the raw materials for lipid synthesis and amino acid synthesis, respectively [44]. In addition, citrate is converted to cytoplasmic acetyl coenzyme A in the tricarboxylic acid (TCA) cycle, which is used to make cholesterol and fatty acids for lipid synthesis [45]. Glycolysis offers the biological raw materials that immune cells require for proliferation and synthesis.
Glycolysis provides enough energy and biomolecules to activate the immune system, allowing for a more efficient and quick immune response. In this way, limiting glycolysis appears to lessen the inflammatory storm [46, 47]. Hannah R et al. [48] have determined in bovine heart mitochondria that metformin directly inhibits complex I activity in a non-competitive manner and ATP synthase activity, which in turn leads to a decrease in OXPHOS and activation of AMPK in cells [49, 50], which support metformin to reduce macrophage NLRP3 inflammasome in diabetes [51, 52]. However, in 2021, Hongxu Xian et al. [53] found that independent of AMPK or NF-κB pathways, metformin blocked LPS-induced ATP-dependent synthesis of the NLRP3 ligand mtDNA and ultimately affected interleukin (IL)-1β production, while metformin may also affect inflammasome non-dependent IL-6 secretion via JNK and p38-MAPK activation, thereby attenuating LPS and SARS-CoV-2-induced ARDS. Similarly in LPS-stimulated BMDMs, metformin, without AMPK activation, decreased LPS-induced production of the pro-IL-1β at both the mRNA and protein levels and boosted LPS induction of the anti-inflammatory cytokine IL-10 [54]. ROS also has been reported involved in LPS-induced IL-1β mRNA production [55]; both metformin and rotenone influence IL-1β production by affecting ROS production of mitochondrial complex I [56]. mTOR is a central regulator of cellular metabolism, and targeting mTOR with rapamycin or derivatives thereof will generally activate downstream molecules such as HIF-1α [57] and GLUT1 [58] to promote glucose uptake and glycolysis [57]. Further, LPS-induced pro-IL-1β and subsequent ATP-induced IL-1β release were cut off by the rapamycin [59]. In CD8+ T cells, blockade of mTOR by rapamycin also promoted the differentiation of Tmem cells [60, 61], which may ultimately provide us with a novel therapeutic target for altering the course of immune disorders and inflammation.
An excessive level of glycolysis causes immunosuppression
The glycolysis supports the rapid activation of immune cells to release large amounts of pro-inflammatory factors and even cause "inflammatory storms," which are extremely prone to excessive cellular damage. Most patients can survive "inflammatory storms" but get worse due to immunosuppression. Immunosuppression is often manifested by reduced expression of genes encoding pro-inflammatory cytokines and chemokines that recruit T cells (e.g. TNF-α, IL-6, CCL2) but increased expression of anti-inflammatory cytokines (e.g. IL-4, IL-10) [62, 63], which means that the host is unable to respond effectively to the "secondary infection." This transition from "inflammatory storms" to hypo-inflammation is accompanied by a shift in the substrate utilization of immune cells from glucose to fatty acid oxidation. The human sepsis blood leukocytes have shown increased fatty acid transporters (eg.CD36) and CPT-1 levels during the immunosuppressed stage [64]. Divya Vats et al. [65] have shown that IL-4-mediated activation of STAT6 in macrophages induces uptake and oxidation of fatty acids as well as biogenesis, and PGC-1β knockdown decreases FAO while increasing the pro-inflammatory effector properties of macrophages. SIRT1 and SIRT6 link metabolism with the early and late stages of acute inflammation responses by supporting the switch from glycolysis to FAO [64]. In addition to FAO promoting the anti-inflammatory phenotype in immune cells, lactate produced by glycolysis during the pro-inflammatory phase also exerts a potent immunosuppressive effect.
Lactate's role in the clinical treatment of sepsis is undeniable, but it is far from being a simple metabolite, and more studies are identifying lactate as an active signaling molecule [66, 67], such as Lactate-GPR81 [68, 69] and Lactate-GPR132 signaling axis [70]. Particularly the functions of dynamic regulation and balance to histone lactation modifications and inflammation-related gene expression [71,72,73] give lactate a new identity in the regulation of immune function.
Zhang et al. [74] found that elevated intracellular lactate inhibits cytoplasmic RLRs-mediated activation of IFNs on M1-type cells. Also during macrophage cytosolic emesis, lactate increases the expression of anti-inflammatory genes Tgfb and IL-10 as well as M2-type genes Vegfa, Mgl1, and CD206 in nearby immune cells via paracrine secretion, which in turn promotes an anti-inflammatory milieu [75]. High levels of lactate also support the expression of the M2-type macrophage homeostatic genes Mrc1 and Arg1 to promote the M2-type macrophage development but suppress M1-type macrophages [76]. Similarly, lactate inhibits the inflammatory response and promotes the polarization of M2-type macrophages through GPR81-dependent antagonism of TLR4-mediated signaling pathways and consequently attenuated LPS-induced NF-κB activation [68, 69, 77,78,79]. High levels of lactate inhibit the differentiation of monocytes into DCs and hamper DCs formation and accumulation [80, 81].
When lactate is transported outside the cell via the monocarboxylate transporter protein MCT4, it modulates migration and cytokine production of immune cells in various ways and induces a "stop migration" signal mediated by the lactate transporters SLC5A12 (CD4 T cells) and SLC16A1 (CD8 T cells) [82]. These inhibitions of T-cells function and apoptosis are coupled with decreased expression of several glycolytic enzymes and glucose flux, such as downregulation of PFKFB3 leading to G-6-P toward the PPP [28, 83]. Lactate efflux causes acidification of the extracellular environment, which induces an incompetent state of CD8+ T cells with reduced cytolytic activity and cytokine secretion when the pH is between 6.0 and 6.5, and also causes immune cell death, whereas proton pump inhibitor treatment effectively restores T cell function [28]. MCT1/MCT4, lactate transporters into and out of cells, have emerged as new targets for tumor therapy [84,85,86], while there were few studies in sepsis. The effect of MCT1 or MCT4 on immunosuppression in sepsis is worthy of more study.
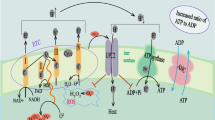
Metabolic reprogramming of immune cells to glycolysis during sepsis is an essential component of the initiation of the host defense response. Whether glycolysis is a passive choice due to the inhibition of OXPHOS or an active selection of immune cells, its role in the pro-inflammatory reaction is crucial. At the same time, the lactate produced by glycolysis feedback promotes the anti-inflammatory response, which is helpful to maintain immune homeostasis. However high-level lactate will cause immune suppression by promoting immune cell death or inactivation, which disrupts the immune homeostasis of the body. Figure 1 shows the crosstalk between metabolic reprogramming and the activities of immune cells. Cellular self-regulation is very clever and fine, and controlling immune cell metabolism at the right time is a crucial strategy for maintaining immunological homeostasis. Once a wide range of immune cells has been activated, how to modulate the degree of glycolysis at the right time to restrict the inflammatory response while avoiding the immunosuppression induced by excessive lactate is what future research should focus on.

The crosstalk between metabolic reprogramming and immune cells activities. Immune cells rely mainly on OXPHOS for ATP production in the resting state. During sepsis, activated immune cells rely on increased glycolysis for ATP and various biosynthetic materials, such as Ru-5-P, DHAP, and G-3-P, to promote immune cell differentiation and proliferation. Macrophages differentiate into M1-type macrophages that promote inflammation and anti-inflammatory macrophages are differentiated into M2. T cells differentiate into pro-inflammatory effector T cells and anti-inflammatory regulatory T cells. Pro-inflammatory cells secrete large amounts of inflammatory factors (IL-6, IL-1β, and TNF-α) causing a host inflammatory response, at which time the balance between OXPHOS and glycolysis can still be maintained. However, when glycolysis is further enhanced, the balance is upset and lactate suppresses pro-inflammatory response and promotes anti-inflammatory response through various mechanisms, ultimately resulting in immunosuppression. GLUT1 glucose transporter protein 1, PFKFB3 fructose-2,6-bisphosphatase 3, HK2 hexokinase 2, PKM2 pyruvate kinase M2, LDH lactate dehydrogenase, DHAP dihydroxyacetone phosphate, G-3-P glyceraldehyde 3-phosphate, 3-P-G 3-phosphoglycerate, Glu-6-P glucose 6-phosphate, Fru-1,6-P 1,6-Fructose diphosphate, PPP pentose phosphate pathway, MCT4 monocarboxylate transporter 4, M0 Macrophage, Th1,Th2 effector T cells, Tregs regulatory T cells, ARG1 arginase 1, MGL1 Macrophage galactose-type lectin 1
Metabolic reprogramming is a cell adaptability mechanism but also causes MODS
After Hotchkiss et al. [87] in 1999 demonstrated that cell death cannot explain the development of organ dysfunction during sepsis, Takasu et al. [88] also discovered that cardiomyocytes and kidney cells from sepsis-dead patients did not show necrosis or apoptosis in 2013, which means that sepsis-induced MODS may have other underlying mechanisms. A recent study found that mitochondrial DNA was broken and mitochondrial homeostasis-related genes were down-regulated in kidney biopsy samples from dead SAKI patients [89], and more and more studies have linked mitochondrial dysfunction to sepsis. A bold prediction appears plausible—energy shortage linked with impaired mitochondrial function in sepsis may be a crucial component contributing to MODS.
Tolerance: a new way to understand MODS
In 2008, a defense present in plants that reduces the negative effects of infection was also found in animals-tolerance [90, 91]. The discovery has opened up new areas of research into pathogen–host relationships and provided new perspectives for the understanding of sepsis-induced MODS. Even in adverse conditions, tolerance offers cells the opportunity to survive even at the expense of their functional activity, i.e., "where there is life, there is hope."
In the long evolutionary process of life, limited resources are utilized for the body's life preservation, growth, and reproduction, and wise energy allocation is necessary. A host can evolve two types of defense mechanisms to increase its fitness when challenged with a pathogen resistance, which drives the immune to clear the pathogens, and tolerance, which reduces infection impact on health in other ways. The requirement of a large amount of energy in the resistance process during sepsis leads to energy competition in the host, especially in the case of decreased ATP due to mitochondrial damage or hypoxia [92, 93]. The host needs to make an energy trade-off between resistance (mobilizing immune cells) and tolerance (maintaining organ function) and ultimately prioritizes the needs of immune cells [94]. Kirthana et al. [95] demonstrated that activation of immunity by LPS finally triggered energy conservation, and the energy trade-off between immunity and homeothermy triggers entry into a hypometabolic–hypothermic state that enhances tissue tolerance. Kirthana et al. [95] also thought tissue tolerance is the mechanism of hibernation. Hypothermia–hypometabolism have been shown to enhance survival in acute sickness in both animal and human studies [96, 97]. Sepsis-induced MODS is characterized by minimal cell death, reduced cellular oxygen consumption, and normal/elevated tissue oxygen levels [9]. In addition, in survivors, organs usually return to function within days to weeks [98]. So Singer et al. [10] hold that MODS may be a hibernation phenomenon due to adaptive shutdown aimed at reducing cellular energy requirements, with a trade-off between organ function and cellular viability. Miguel et al. [99] have also proposed that tolerance is a defense strategy to maintain the health of the body by limiting organ damage.
Driving immunity to clear pathogens is an energy-intensive process, which means that less energy is available to organ cells; a reasonable energy distribution is also required between the vital and functional activities of the cells to ensure cells’ survival. When turtle liver cells were exposed to hypoxia, researchers noticed that cellular energy and oxygen consumption dropped, but that critical activities including intracellular ion balance were intact [100]. Cell function was likewise stopped in a model of induced hibernation in rat hepatocytes, but Na+/K+ ATPase activity was maintained, and hepatocytes restored oxygen consumption and ATP generation to normal oxidation after reoxygenation capability [101].
Tolerance reflects the flexibility of cells to balance their survival and function, and energy allocation is an important manifestation of tolerance. The correlation between tolerance, energy trade-offs, and MODS highlights the central role of energy in sepsis and also implies an impact of metabolic reprogramming on organ cell function.
The metabolic reprogramming is a key indicator of tolerance.
The overarching principles and mechanisms that control the expression of tissue tolerance programs remain largely unclear, but there is no doubt that energy is an important factor in tolerance. We all know that infected animals' metabolism skews toward FAO for ATP, accompanied by increased levels of free fatty acids in the blood. A study by Khan et al. [102] showed that the level of cellular lipid metabolites deviates from a "safe range" when comparing plasma lipid metabolism profiles between septic and non-septic patients and that the level of lipid metabolites is related to sepsis mortality. FAO is primarily controlled by PPAR-α (encoded by the NR1C1 gene), and several studies have reported decreased PPAR-α levels in the organs and whole blood during sepsis. The rapid decline of hepatic PPAR-α levels causes excess free fatty acids, and PPAR-α agonist pemafibrate protects against lipotoxicity and tissue damage by improving hepatic PPAR-α function in sepsis [103]. Takuma et al. [104] demonstrated that mice lacking PPAR-α had poor renal functions and that diminished PPAR-α signaling increased the incidence of AKI. Drosatos et al. [105] also found that JNK inhibition prevented LPS-mediated cardiac dysfunction by increasing PPAR-α expression to improve FAO.
Although the sepsis-induced “starvation response" promotes FAO to be considered the primary energy supplier, there is still a suppression of FAO during sepsis, and the oxygen and the normal function of mitochondria for ATP are difficult to achieve during sepsis, so FAO is not the best way to enhance tolerance. Glycolysis, not required mitochondria and oxygen, becomes the main candidate for increased tolerance during sepsis. Recent studies suggest that metabolic adaptations to bacterial infections are a critical determinant of tissue tolerance [94, 106]. As previously indicated, the "Warburg effect" is used in the process of immune cell activation to maintain energy requirements and support biosynthetic chemicals, as well as to create a memory for a specific insult and modulate the response to future insults (a process known as trained immunity) [57]. Katherine et al. [106] found that glycolysis manipulation alters the disease tolerance of mice suffering from malaria, and supplemental glucose improves survival by promoting glycolysis.
Glycolysis during sepsis is the main pathway that determines the allocation of energy to organ cells, and changes in glycolysis reflect the activation and strength of cellular tolerance. It is reasonable to assume that the reprogramming from OXPHOS to glycolysis is a protective mechanism that maintains the cellular survival state by enhancing cellular tolerance and reducing cellular damage. During sepsis, both glucose uptake and the expression of glycolysis-related enzymes were significantly altered in organ cells. Sepsis elevates glucose uptake and the expression of GLUT1 protein 1.7-fold in the skeletal muscle of septic rats to enhance glycolysis [107]. In LPS-induced pulmonary fibrosis, LPS causes lung fibroblast aerobic glycolysis through the activation of the PI3K-Akt-mTOR/PFKFB3 pathway [108]. LPS also activates key molecules that regulate metabolisms such as HIF-1α [109] and AMPK [110]. These reprogramming of OXPHOS to glycolysis may be a pre-determined defense mechanism.
Metabolic reprogramming may also be the result of mitochondrial dysfunction or hypoxia. However, it is undeniable that glycolysis alleviates the energy crisis and promotes the tolerance of cells in harsh environments. The shift to glycolysis of cardiomyocytes to survive ischemia and hypoxia is thought to be an energy compensatory mechanism [111], and Singer et al. [10]also suggested that sepsis-induced low cardiac output is a "hibernation"-like phenomenon exhibited by the myocardium in the presence of energy deficit. Cells re-prioritize energy expenditure to maintain vital activities at the expense of organ function in low-energy situations, and the decrease in cellular function may be a protective mechanism facing inadequate energy production. At this point, the more energy produced by glycolysis, the stronger tolerance, and the more survival organ cells.
Switching back from glycolysis to OXPHOS is essential for MODS
Although switching to glycolysis enhances the tolerance to ensure cell survival for a short period and effectively avoids re-injury to mitochondria and cells by ROS, as well as downstream adverse reactions caused by ROS are suppressed, long-term glycolysis is damaging to organ function cells. Exogenous lactate caused an increase in mitochondrial damage in HK2 cells after 6 h, and cell survival was severely reduced after 24 h [112]. Long-term glycolysis has been reported to cause permanent renal shrinkage and fibrosis in TECs, increasing the probability of transitioning from AKI to chronic kidney disease [7]. By quickly reverting OXPHOS to an energy supply mode, the prognosis of sepsis can be greatly improved. For example, Shikonin, a powerful PKM2 inhibitor, reduces serum lactate and HMGB1 levels to protect mice from sepsis [113]. The activation of SIRT1, an important facilitator protein of OXPHOS, can enhance the survival of sepsis mice, according to Opal et al. [114] and Vidula et al. [115]. Treatment with the glycolysis inhibitor 2-DG enhanced heart function and survival outcomes in a mouse model of septic cardiomyopathy [8]. Although glycolysis may help cells survive in the early stages of sepsis, it is clear that a return to OXPHOS metabolism is necessary, especially for those relying on OXPHOS.
The strength of tolerance can alter the outcome of cells in adverse situations. Lives evolve under survival pressure, and timely trade-offs are a crucial approach to adapting to the rule of superiority and inferiority. Cell evolution has resulted in a collaboration between OXPHOS and glycolysis for energy supply. When OXPHOS is disrupted, the small amount of ATP produced by glycolysis is mainly used to sustain life activities, and the restoration of OXPHOS is necessary for the recovery of cellular function. Tolerance offers us a unique perspective on MODS. As shown in Fig. 2, metabolic reprogramming promotes tissue tolerance to protect organ cells during sepsis.

Metabolic reprogramming promotes tissue tolerance to increase organ cell adaptation during sepsis. Under normal conditions, cells obtain ATP from OXPHOS and glycolysis. Resting Immune cells have fewer energy requirements and therefore most of the energy is allocated to organ cells to support functional activities. In sepsis, ATP is preferentially allocated to immune cells to support immune activation for clearing the pathogens, which results in less ATP for organ cells. To reduce energy consumption, organ cells shut down functional activities and use ATP as much as possible to maintain cell life activities, which leads to stagnation in organ cell function even MODS. This is an adaptation mechanism of the host during sepsis. When OXPHOS is suppressed, enhanced glycolysis rapidly supplies ATP to maintain cellular survival and encourages stronger tissue tolerance to adapt to sepsis more easily. It also provides an opportunity for cells to restore OXPHOS to promote recovery of cellular function. GLUT1 glucose transporter protein 1, PFKFB3 fructose-2,6-bisphosphatase 3, HK2 hexokinase 2, PKM2 pyruvate kinase M2, LDH lactate dehydrogenase, Glu-6-P glucose 6-phosphate, Fru-6-P Fructose 6-phosphate, MODS multi-organ dysfunction syndrome
Mitochondria are essential to the metabolism of sepsis
It is vital to re-establish the function of OXPHOS in immune cells as well as organ cells. The operation of OXPHOS requires a healthy inner mitochondrial membrane and flawless mitochondrial activity, so the contribution of mitochondria to metabolic reprograming during sepsis cannot be ignored.
Factors affecting mitochondrial function in sepsis
Mitochondrial function is affected during sepsis mainly through several aspects, as shown in Table 1. First, sufficient oxygen is a prerequisite for mitochondrial ATP synthesis. Although the idea that tissue hypoxia is caused by insufficient oxygen delivery during sepsis is gradually being questioned, the mismatch between macrocirculation and microcirculation [116, 117], abnormal volume distribution, and decreased cardiac function [118] during sepsis may all contribute to insufficient tissue oxygen delivery. Even if the various enzyme activity involved in oxygen oxidation are tolerable, ATP cannot be effectively generated under low oxygen levels. When ATP generation falls below a certain level, the apoptotic and necrotic pathway is triggered [119, 120]. Pyruvate dehydrogenase complex (PDC) is the major metabolic enzyme for mitochondrial oxidative energy production, hence, a decrease in its activity will have an immediate influence on mitochondrial energy production. Additionally, PDC is also a bifurcation point between OXPHOS and glycolysis, and its role in metabolic reprogramming cannot be ignored. During sepsis, PDC activity is regulated by HIF-1a [121], glucocorticoid receptor (GR) [122], and peroxisome proliferator-activated receptor (PPAR-a) [123, 124] pathways, which have direct impacts on OXPHOS capability. Meanwhile, the large amounts of NO [125, 126], H2S [127], ROS [128], and other reactive substances that are produced during inflammation can inhibit the mitochondria function by suppressing ETC complexes and the TCA cycle. Mitochondrial homeostasis (biogenesis, fusion, autophagy) plays a decisive role in the regulation of cellular energy metabolism and signaling pathways. Genes related to mitochondrial biosyntheses, such as peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1), mitochondrial transcription factor (Tfam), and nuclear respiratory factor-1 (NRF-1), are also explicitly and significantly repressed [129], whereas increased PGC-1 contributes to organ function recovery [130]. FUNDC1 interacts with LC3 to regulate mitochondrial homeostasis [131], and Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy [132]. AMPK as the key player in metabolism focuses on the regulation of various aspects of mitochondrial homeostasis [133] and promotes the interaction between mitochondrial fission and autophagy through the regulation of Drp1, Mff, PINK, and Parkin [134]. Uncoupling protein 2 (UCP2) is thought to dissipate the proton gradient across the inner membrane and uncouple the respiratory chain from ATP generation [135]. During sepsis, calcium/calmodulin-dependent protein kinase(CaMK)-IV shifts the balance toward mitochondrial fission and away from fusion by directly phosphorylating fission protein Drp1 and reducing the expression of the fusion proteins Mfn1/2 and OPA1 [136]. CaMK-IV also is a direct PINK1 kinase and regulator of Parkin expression to control mitophagy [136]. Also, stress response during the early stages of sepsis promotes massive hormone secretion [137,138,139,140], which to some extent affects mitochondrial function. Thyroid hormones can increase mitochondrial biosynthesis [141, 142] and regulate mitochondrial autophagy [143]. Insulin resistance in type 2 diabetes is closely related to mitochondrial function [144, 145]. Kitada et al. [146] found that SIRT1 also can improve insulin resistance by promoting fatty acid oxidation and mitochondrial biogenesis via deacetylation of PGC-1α and PPAR-α activation in skeletal muscle.
Treatment of sepsis requires proper mitochondrial function
Mitochondria have long been thought to have functions in the progression of the disease by regulating cell metabolism and influencing ATP production. It is also responsible for a wide range of cellular functions, such as cellular calcium homeostasis and programmed cell death.
Mitochondrial free radicals and ROS can disrupt cell signaling, while the mitochondria themselves are the main targets of ROS damage. UCP2 is a mitochondrial membrane protein that reduces mitochondrial ROS generation by triggering proton leak and thereby lowers mitochondrial ROS production [135]. In a study of septic cardiomyopathy, inhibiting UCP2 enhanced ROS, mitochondrial swelling, and cardiac damage [147], whereas overexpressing UCP2 increased caspase3 activity and Bax protein accumulation, and attenuated cardiomyocyte apoptosis [148]. Upregulation of SIRT3-mediated inhibition of oxidative stress preserved mitochondrial function and induced autophagy in small intestinal epithelial cells, which partially alleviated sepsis-induced small intestinal injury [149]. Suppressed PGC-1α by LPS triggers the reprogramming of cardiac energy metabolism, which is associated with decreased ventricular function in septic cardiomyopathy [150]. At the same time, the mitochondrial homeostasis, including the dynamic process of mitochondrial fusion/fission (Mfn2, OPA1, Drp1), mitochondrial biogenesis (NRF-1, PGC-1α, Tfam), and mitochondrial mitophagy (Parkin, PINK1), protects the lung [136] and kidneys [136] from ongoing oxidative injury [151].
Mitochondria also play a role in modulating immune cell function [152]. TLRs enhanced by mitochondrial ROS allow macrophages to receive and conduct signals more sensitively, improving immune cells' ability to clear harmful bacteria [153]. Secondly, mitochondria-specific protein, mitochondrial antiviral signaling protein (MAVS), accumulates on the surface of mitochondria to serve as a signaling platform to participate in the anti-RNA virus RLR pathway [74, 154]. At the same time, mitochondrial DAMPs, such as mitochondrial DNA and n-formyl peptides (n-fp) [155], are released to the cytosol where they could be sensed by various PRRs to activate the immune response.
The specific regulatory effects of mitochondrial metabolism on the innate immune pathway are influenced by metabolites in the metabolic pathway. TCA is blocked by downregulation of isocitrate dehydrogenase and overexpression of immune-responsive gene 1 protein (IRG1) [156, 157], resulting in the conversion of accumulated citrate to itaconate [158, 159]. Itaconate can highly activate macrophages [160, 161] but also inhibit complex II (also known as succinate dehydrogenase), which prevents succinate from being oxidized [160]. Furthermore, succinate oxidation governs the inflammatory phenotype of the macrophage, as indicated by increased inflammatory gene expression and decreased anti-inflammatory gene expression [162, 163].
Conclusion
OXPHOS is the metabolic pathway of most cells in the physiological state, but during sepsis, the metabolism of cells is reprogrammed to glycolysis. Cells use both mechanisms to enhance their defenses in adverse situations. Crosstalk between immune activity and metabolic reprogramming during sepsis shows that switching to glycolysis increases immune activation, but that excessive glycolysis paradoxically causes immunosuppression. As the most key factor affecting tissue tolerance during sepsis, glycolysis can transiently maintain cellular vital activity, prolong survival, protect organs from ATP deficiency-induced death in sepsis, and in some way reduce oxidative damage to mitochondria and cells by OXPHOS. But, for achieving recovery of cellular functional activity in organs, the well-timed restoration of OXPHOS is the most necessary process. Whereas the basis of repairing OXPHOS is to ensure that mitochondrial structure and function are maintained, mitochondrial protection deserves more attention in the treatment of sepsis.
However, facing metabolic differences among different cell types and different severity of the disease, the most important question we need to consider is how to find the best time to use these reprogramming mechanisms. The same metabolic modality may lead to opposite results, e.g. glycolysis is beneficial for immune cell activation but detrimental in TECs [164]. Early restoration of OXPHOS appears to be the best option, but OXPHOS is also harmful to the fragile mitochondria and cells during sepsis. The metabolic flexibility determines the efficient operation of the metabolic reprogramming, which favors cells' ability to adapt to adverse conditions. The fixable metabolic patterns due to loss of metabolic flexibility exacerbate cell dysfunction. To take full advantage of the crosstalk between metabolic reprogramming and cell function, we also need to find the key regulators that modulate the metabolic flexibility of the cell. However, we presently don't recognize enough about septic metabolism and will face significant challenges in the future. Mitochondria, as the core organelles of cellular metabolism, could be a breakthrough to exploit metabolic reprogramming.
Data availability
Not applicable.
References
Schrijver IT, Théroude C, Roger T (2019) Myeloid-derived suppressor cells in sepsis. Front Immunol 10:327. https://doi.org/10.3389/fimmu.2019.00327
Salomao R, Ferreira BL, Salomao MC et al (2019) Sepsis: evolving concepts and challenges. Braz J Med Biol Res 52(4):e8595. https://doi.org/10.1590/1414-431X20198595
Warburg O (1956) On the origin of cancer cells. Science 123(3191):309–314. https://doi.org/10.1126/science.123.3191.309
He Y, Wang N, Shen Y et al (2014) Inhibition of high glucose-induced apoptosis by uncoupling protein 2 in human umbilical vein endothelial cells. Int J Mol Med 33(5):1275–1281. https://doi.org/10.3892/ijmm.2014.1676
Soto-Heredero G, Gómez De Las Heras MM, Gabandé-Rodríguez E et al (2020) Glycolysis—a key player in the inflammatory response. Febs J 287(16):3350–3369. https://doi.org/10.1111/febs.15327
Everts B, Amiel E, Huang SC et al (2014) TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat Immunol 15(4):323–332. https://doi.org/10.1038/ni.2833
Lan R, Geng H, Singha PK et al (2016) Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 27(11):3356–3367. https://doi.org/10.1681/asn.2015020177
Zheng Z, Ma H, Zhang X et al (2017) Enhanced glycolytic metabolism contributes to cardiac dysfunction in polymicrobial sepsis. J Infect Dis 215(9):1396–1406. https://doi.org/10.1093/infdis/jix138
Singer M, De Santis V, Vitale D et al (2004) Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet 364(9433):545–548. https://doi.org/10.1016/s0140-6736(04)16815-3
Stanzani G, Tidswell R, Singer M (2020) Do critical care patients hibernate? Theoretical support for less is more. Intensive Care Med 46(3):495–497. https://doi.org/10.1007/s00134-019-05813-9
Singer M (2008) Cellular dysfunction in sepsis. Clin Chest Med 29(4):655–660. https://doi.org/10.1016/j.ccm.2008.06.003 (viii–ix)
Langley RJ, Tsalik EL, Van Velkinburgh JC et al (2013) An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci Transl Med 5(195):195ra95. https://doi.org/10.1126/scitranslmed.3005893
Oren R, Farnham AE, Saito K et al (1963) Metabolic patterns in three types of phagocytizing cells. J Cell Biol 17(3):487–501. https://doi.org/10.1083/jcb.17.3.487
Loftus RM, Finlay DK (2016) Immunometabolism: cellular metabolism turns immune regulator. J Biol Chem 291(1):1–10. https://doi.org/10.1074/jbc.R115.693903
Kaymak I, Williams KS, Cantor JR et al (2021) Immunometabolic interplay in the tumor microenvironment. Cancer Cell 39(1):28–37. https://doi.org/10.1016/j.ccell.2020.09.004
Wu H, Ballantyne CM (2020) Metabolic inflammation and insulin resistance in obesity. Circ Res 126(11):1549–1564. https://doi.org/10.1161/circresaha.119.315896
Esser N, Legrand-Poels S, Piette J et al (2014) Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract 105(2):141–150. https://doi.org/10.1016/j.diabres.2014.04.006
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820. https://doi.org/10.1016/j.cell.2010.01.022
Zaccagnino P, Saltarella M, Maiorano S et al (2012) An active mitochondrial biogenesis occurs during dendritic cell differentiation. Int J Biochem Cell Biol 44(11):1962–1969. https://doi.org/10.1016/j.biocel.2012.07.024
Ryu SW, Han EC, Yoon J et al (2015) The mitochondrial fusion-related proteins Mfn2 and OPA1 are transcriptionally induced during differentiation of bone marrow progenitors to immature dendritic cells. Mol Cells 38(1):89–94. https://doi.org/10.14348/molcells.2015.2285
Krawczyk CM, Holowka T, Sun J et al (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115(23):4742–4749. https://doi.org/10.1182/blood-2009-10-249540
Tan C, Gu J, Chen H et al (2020) Inhibition of aerobic glycolysis promotes neutrophil to influx to the infectious site via CXCR2 in sepsis. Shock 53(1):114–123. https://doi.org/10.1097/shk.0000000000001334
Awasthi D, Nagarkoti S, Sadaf S et al (2019) Glycolysis dependent lactate formation in neutrophils: a metabolic link between NOX-dependent and independent NETosis. Biochim Biophys Acta Mol Basis Dis 1865(12):165542. https://doi.org/10.1016/j.bbadis.2019.165542
Porta C, Rimoldi M, Raes G et al (2009) Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A 106(35):14978–14983. https://doi.org/10.1073/pnas.0809784106
Pena OM, Pistolic J, Raj D et al (2011) Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol 186(12):7243–7254. https://doi.org/10.4049/jimmunol.1001952
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11(11):723–737. https://doi.org/10.1038/nri3073
Shapouri-Moghaddam A, Mohammadian S, Vazini H et al (2018) Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233(9):6425–6440. https://doi.org/10.1002/jcp.26429
Certo M, Tsai CH, Pucino V et al (2021) Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol 21(3):151–161. https://doi.org/10.1038/s41577-020-0406-2
Michalek RD, Gerriets VA, Jacobs SR et al (2011) Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186(6):3299–3303. https://doi.org/10.4049/jimmunol.1003613
Gerriets VA, Kishton RJ, Nichols AG et al (2015) Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest 125(1):194–207. https://doi.org/10.1172/jci76012
Koprivica I, Gajić D, Pejnović N et al (2020) Ethyl pyruvate promotes proliferation of regulatory T cells by increasing glycolysis. Molecules. https://doi.org/10.3390/molecules25184112
Baixauli F, Acín-Pérez R, Villarroya-Beltrí C et al (2015) Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab 22(3):485–498. https://doi.org/10.1016/j.cmet.2015.07.020
Desdín-Micó G, Soto-Heredero G, Mittelbrunn M (2018) Mitochondrial activity in T cells. Mitochondrion 41:51–57. https://doi.org/10.1016/j.mito.2017.10.006
Jung J, Zeng H, Horng T (2019) Metabolism as a guiding force for immunity. Nat Cell Biol 21(1):85–93. https://doi.org/10.1038/s41556-018-0217-x
Orecchioni M, Ghosheh Y, Pramod AB et al (2019) Macrophage polarization: different gene signatures in M1(LPS+) vs classically and M2(LPS-) vs alternatively activated macrophages. Front Immunol 10:1084. https://doi.org/10.3389/fimmu.2019.01084
Deng H, Wu L, Liu M et al (2020) Bone marrow mesenchymal stem cell-derived exosomes attenuate LPS-induced ARDS by modulating macrophage polarization through inhibiting glycolysis in macrophages. Shock 54(6):828–843. https://doi.org/10.1097/shk.0000000000001549
Huang J, Liu K, Zhu S et al (2018) AMPK regulates immunometabolism in sepsis. Brain Behav Immun 72:89–100. https://doi.org/10.1016/j.bbi.2017.11.003
Wang Y, Xu Y, Zhang P et al (2018) Smiglaside A ameliorates LPS-induced acute lung injury by modulating macrophage polarization via AMPK-PPARγ pathway. Biochem Pharmacol 156:385–395. https://doi.org/10.1016/j.bcp.2018.09.002
Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27(1):441–464. https://doi.org/10.1146/annurev-cellbio-092910-154237
Macintyre AN, Gerriets VA, Nichols AG et al (2014) The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20(1):61–72. https://doi.org/10.1016/j.cmet.2014.05.004
Freemerman AJ, Johnson AR, Sacks GN et al (2014) Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem 289(11):7884–7896. https://doi.org/10.1074/jbc.M113.522037
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033. https://doi.org/10.1126/science.1160809
Donnelly RP, Finlay DK (2015) Glucose, glycolysis and lymphocyte responses. Mol Immunol 68(2 Pt C):513–519. https://doi.org/10.1016/j.molimm.2015.07.034
Kim J-A, Yeom Y (2017) Metabolic signaling to epigenetic alterations in cancer. Biomol Ther. https://doi.org/10.4062/biomolther.2017.185
Batista-Gonzalez A, Vidal R, Criollo A et al (2019) New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol 10:2993. https://doi.org/10.3389/fimmu.2019.02993
Pålsson-Mcdermott EM, O’neill LAJ (2020) Targeting immunometabolism as an anti-inflammatory strategy. Cell Res 30(4):300–314. https://doi.org/10.1038/s41422-020-0291-z
Gauthier T, Chen W (2022) Modulation of macrophage immunometabolism: a new approach to fight infections. Front Immunol 13:780839. https://doi.org/10.3389/fimmu.2022.780839
Bridges HR, Jones AJY, Pollak MN et al (2014) Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 462(3):475–487. https://doi.org/10.1042/BJ20140620
Hawley SA, Ross FA, Chevtzoff C et al (2010) Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11(6):554–565. https://doi.org/10.1016/j.cmet.2010.04.001
Ouyang J, Parakhia RA, Ochs RS (2011) Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem 286(1):1–11. https://doi.org/10.1074/jbc.M110.121806
Yang F, Qin Y, Wang Y et al (2019) Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int J Biol Sci 15(5):1010–1019. https://doi.org/10.7150/ijbs.29680
Lee HM, Kim JJ, Kim HJ et al (2013) Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62(1):194–204. https://doi.org/10.2337/db12-0420
Xian H, Liu Y, Rundberg Nilsson A et al (2021) Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 54(7):1463-1477.e11. https://doi.org/10.1016/j.immuni.2021.05.004
Kelly B, Tannahill GM, Murphy MP et al (2015) Metformin inhibits the production of reactive oxygen species from NADH: ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J Biol Chem 290(33):20348–20359. https://doi.org/10.1074/jbc.M115.662114
Bauernfeind F, Bartok E, Rieger A et al (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187(2):613–617. https://doi.org/10.4049/jimmunol.1100613
Batandier C, Guigas B, Detaille D et al (2006) The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr 38(1):33–42. https://doi.org/10.1007/s10863-006-9003-8
Cheng SC, Quintin J, Cramer RA et al (2014) mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345(6204):1250684. https://doi.org/10.1126/science.1250684
Kasprzak A (2021) Insulin-like growth factor 1 (IGF-1) signaling in glucose metabolism in colorectal cancer. Int J Mol Sci. https://doi.org/10.3390/ijms22126434
Torp MK, Yang K, Ranheim T et al (2019) Mammalian target of rapamycin (mTOR) and the proteasome attenuates IL-1β expression in primary mouse cardiac fibroblasts. Front Immunol 10:1285. https://doi.org/10.3389/fimmu.2019.01285
Delgoffe GM, Kole TP, Zheng Y et al (2009) The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30(6):832–844. https://doi.org/10.1016/j.immuni.2009.04.014
Sukumar M, Liu J, Ji Y et al (2013) Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123(10):4479–4488. https://doi.org/10.1172/jci69589
Zhang Q, Hu Y, Zhang J et al (2019) iTRAQ-based proteomic analysis of endotoxin tolerance induced by lipopolysaccharide. Mol Med Rep 20(1):584–592. https://doi.org/10.3892/mmr.2019.10264
Morris M, Li L (2012) Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch Immunol Ther Exp (Warsz) 60(1):13–18. https://doi.org/10.1007/s00005-011-0155-9
Liu TF, Vachharajani VT, Yoza BK et al (2012) NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem 287(31):25758–25769. https://doi.org/10.1074/jbc.M112.362343
Vats D, Mukundan L, Odegaard JI et al (2006) Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4(1):13–24. https://doi.org/10.1016/j.cmet.2006.05.011
Brooks GA (2020) The tortuous path of lactate shuttle discovery: from cinders and boards to the lab and ICU. J Sport Health Sci 9(5):446–460. https://doi.org/10.1016/j.jshs.2020.02.006
Manoharan I, Prasad PD, Thangaraju M et al (2021) Lactate-dependent regulation of immune responses by dendritic cells and macrophages. Front Immunol 12:691134. https://doi.org/10.3389/fimmu.2021.691134
Errea A, Cayet D, Marchetti P et al (2016) Lactate inhibits the pro-inflammatory response and metabolic reprogramming in murine macrophages in a GPR81-independent manner. PLoS ONE 11(11):e0163694. https://doi.org/10.1371/journal.pone.0163694
Hoque R, Farooq A, Ghani A et al (2014) Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 146(7):1763–1774. https://doi.org/10.1053/j.gastro.2014.03.014
Chen P, Zuo H, Xiong H et al (2017) Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A 114(3):580–585. https://doi.org/10.1073/pnas.1614035114
Zhang D, Tang Z, Huang H et al (2019) Metabolic regulation of gene expression by histone lactylation. Nature 574(7779):575–580. https://doi.org/10.1038/s41586-019-1678-1
Irizarry-Caro RA, Mcdaniel MM, Overcast GR et al (2020) TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc Natl Acad Sci U S A 117(48):30628–30638. https://doi.org/10.1073/pnas.2009778117
Liberti MV, Locasale JW (2020) Histone lactylation: a new role for glucose metabolism. Trends Biochem Sci 45(3):179–182. https://doi.org/10.1016/j.tibs.2019.12.004
Zhang W, Wang G, Xu ZG et al (2019) Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell 178(1):176-189.e15. https://doi.org/10.1016/j.cell.2019.05.003
Morioka S, Perry JSA, Raymond MH et al (2018) Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 563(7733):714–718. https://doi.org/10.1038/s41586-018-0735-5
Liu N, Luo J, Kuang D et al (2019) Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α-mediated tumor progression. J Clin Invest 129(2):631–646. https://doi.org/10.1172/jci123027
Cai TQ, Ren N, Jin L et al (2008) Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem Biophys Res Commun 377(3):987–991. https://doi.org/10.1016/j.bbrc.2008.10.088
Yang K, Xu J, Fan M et al (2020) Lactate suppresses macrophage pro-inflammatory response to LPS stimulation by inhibition of YAP and NF-κB activation via GPR81-mediated signaling. Front Immunol 11:587913. https://doi.org/10.3389/fimmu.2020.587913
Dietl K, Renner K, Dettmer K et al (2010) Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol 184(3):1200–1209. https://doi.org/10.4049/jimmunol.0902584
Puig-Kröger A, Pello OM, Muñiz-Pello O et al (2003) Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: effect of lactate and glucose-degradation products. J Leukoc Biol 73(4):482–492. https://doi.org/10.1189/jlb.0902451
Gottfried E, Kunz-Schughart LA, Ebner S et al (2006) Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107(5):2013–2021. https://doi.org/10.1182/blood-2005-05-1795
Haas R, Smith J, Rocher-Ros V et al (2015) Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol 13(7):e1002202. https://doi.org/10.1371/journal.pbio.1002202
Yang Z, Fujii H, Mohan SV et al (2013) Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med 210(10):2119–2134. https://doi.org/10.1084/jem.20130252
Weiss HJ, Angiari S (2020) Metabolite transporters as regulators of immunity. Metabolites. https://doi.org/10.3390/metabo10100418
Sun X, Wang M, Wang M et al (2020) Role of proton-coupled monocarboxylate transporters in cancer: from metabolic crosstalk to therapeutic potential. Front Cell Dev Biol 8:651. https://doi.org/10.3389/fcell.2020.00651
Fiaschi T, Marini A, Giannoni E et al (2012) Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 72(19):5130–5140. https://doi.org/10.1158/0008-5472.Can-12-1949
Hotchkiss RS, Swanson PE, Freeman BD et al (1999) Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 27(7):1230–1251. https://doi.org/10.1097/00003246-199907000-00002
Takasu O, Gaut JP, Watanabe E et al (2013) Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med 187(5):509–517. https://doi.org/10.1164/rccm.201211-1983OC
Van Der Slikke EC, Star BS, Van Meurs M et al (2021) Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-AKI. Crit Care 25(1):36. https://doi.org/10.1186/s13054-020-03424-1
Schafer JF (1971) Tolerance to plant disease. Annu Rev Phytopathol 9(1):235–252. https://doi.org/10.1146/annurev.py.09.090171.001315
Schneider DS, Ayres JS (2008) Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol 8(11):889–895. https://doi.org/10.1038/nri2432
Mcdade TW (2005) Life history, maintenance, and the early origins of immune function. Am J Hum Biol 17(1):81–94. https://doi.org/10.1002/ajhb.20095
Rauw WM (2012) Immune response from a resource allocation perspective. Front Genet 3:267. https://doi.org/10.3389/fgene.2012.00267
Ayres JS, Schneider DS (2012) Tolerance of infections. Annu Rev Immunol 30(1):271–294. https://doi.org/10.1146/annurev-immunol-020711-075030
Ganeshan K, Nikkanen J, Man K et al (2019) Energetic trade-offs and hypometabolic states promote disease tolerance. Cell 177(2):399-413.e12. https://doi.org/10.1016/j.cell.2019.01.050
Bernard SA, Gray TW, Buist MD et al (2002) Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 346(8):557–563. https://doi.org/10.1056/NEJMoa003289
Marion DW, Penrod LE, Kelsey SF et al (1997) Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med 336(8):540–546. https://doi.org/10.1056/nejm199702203360803
Crouser ED (2004) Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion 4(5):729–741. https://doi.org/10.1016/j.mito.2004.07.023
Soares MP, Gozzelino R, Weis S (2014) Tissue damage control in disease tolerance. Trends Immunol 35(10):483–494. https://doi.org/10.1016/j.it.2014.08.001
Buck LT, Hochachka PW, Schön A et al (1993) Microcalorimetric measurement of reversible metabolic suppression induced by anoxia in isolated hepatocytes. Am J Physiol 265(5 Pt 2):R1014–R1019. https://doi.org/10.1152/ajpregu.1993.265.5.R1014
Subramanian RM, Chandel N, Budinger GR et al (2007) Hypoxic conformance of metabolism in primary rat hepatocytes: a model of hepatic hibernation. Hepatology 45(2):455–464. https://doi.org/10.1002/hep.21462
Khaliq W, Großmann P, Neugebauer S et al (2020) Lipid metabolic signatures deviate in sepsis survivors compared to non-survivors. Comput Struct Biotechnol J 18:3678–3691. https://doi.org/10.1016/j.csbj.2020.11.009
Van Wyngene L, Vanderhaeghen T, Timmermans S et al (2020) Hepatic PPARα function and lipid metabolic pathways are dysregulated in polymicrobial sepsis. EMBO Mol Med 12(2):e11319. https://doi.org/10.15252/emmm.201911319
Iwaki T, Bennion BG, Stenson EK et al (2019) PPARα contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol Rep 7(10):e14078. https://doi.org/10.14814/phy2.14078
Drosatos K, Drosatos-Tampakaki Z, Khan R et al (2011) Inhibition of c-Jun-N-terminal kinase increases cardiac peroxisome proliferator-activated receptor alpha expression and fatty acid oxidation and prevents lipopolysaccharide-induced heart dysfunction. J Biol Chem 286(42):36331–36339. https://doi.org/10.1074/jbc.M111.272146
Cumnock K, Gupta AS, Lissner M et al (2018) Host energy source is important for disease tolerance to malaria. Curr Biol 28(10):1635-1642.e3. https://doi.org/10.1016/j.cub.2018.04.009
Vary TC, Drnevich D, Jurasinski C et al (1995) Mechanisms regulating skeletal muscle glucose metabolism in sepsis. Shock 3(6):403–410
Hu X, Xu Q, Wan H et al (2020) PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab Invest 100(6):801–811. https://doi.org/10.1038/s41374-020-0404-9
Yeh CH, Cho W, So EC et al (2011) Propofol inhibits lipopolysaccharide-induced lung epithelial cell injury by reducing hypoxia-inducible factor-1alpha expression. Br J Anaesth 106(4):590–599. https://doi.org/10.1093/bja/aer005
Tan C, Gu J, Li T et al (2021) Inhibition of aerobic glycolysis alleviates sepsis-induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK-regulated autophagy. Int J Mol Med. https://doi.org/10.3892/ijmm.2021.4852
Kawaguchi S, Okada M (2021) Cardiac metabolism in sepsis. Metabolites. https://doi.org/10.3390/metabo11120846
Ji R, Chen W, Wang Y et al (2021) The Warburg effect promotes mitochondrial injury regulated by uncoupling protein-2 in septic acute kidney injury. Shock 55(5):640–648. https://doi.org/10.1097/shk.0000000000001576
Yang L, Xie M, Yang M et al (2014) PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun 5:4436. https://doi.org/10.1038/ncomms5436
Opal SM, Ellis JL, Suri V et al (2016) Pharmacological SIRT1 activation improves mortality and markedly alters transcriptional profiles that accompany experimental sepsis. Shock 45(4):411–418
Vachharajani VT, Liu T, Brown CM et al (2014) SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol 96(5):785–796. https://doi.org/10.1189/jlb.3MA0114-034RR
De Backer D, Ricottilli F, Ospina-Tascón GA (2021) Septic shock: a microcirculation disease. Curr Opin Anaesthesiol 34(2):85–91. https://doi.org/10.1097/aco.0000000000000957
Inkinen N, Pettilä V, Lakkisto P et al (2019) Association of endothelial and glycocalyx injury biomarkers with fluid administration, development of acute kidney injury, and 90-day mortality: data from the FINNAKI observational study. Ann Intensive Care 9(1):103. https://doi.org/10.1186/s13613-019-0575-y
Lin Y, Xu Y, Zhang Z (2020) Sepsis-induced myocardial dysfunction (SIMD): the pathophysiological mechanisms and therapeutic strategies targeting mitochondria. Inflammation 43(4):1184–1200. https://doi.org/10.1007/s10753-020-01233-w
Bartolák-Suki E, Imsirovic J, Nishibori Y et al (2017) Regulation of mitochondrial structure and dynamics by the cytoskeleton and mechanical factors. Int J Mol Sci. https://doi.org/10.3390/ijms18081812
Hanus J, Zhang H, Wang Z et al (2013) Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis 4(12):e965. https://doi.org/10.1038/cddis.2013.478
Dasgupta A, Wu D, Tian L et al (2020) Mitochondria in the pulmonary vasculature in health and disease: oxygen-sensing, metabolism, and dynamics. Compr Physiol 10(2):713–765. https://doi.org/10.1002/cphy.c190027
Connaughton S, Chowdhury F, Attia RR et al (2010) Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol Cell Endocrinol 315(1–2):159–167. https://doi.org/10.1016/j.mce.2009.08.011
Palomer X, Salvadó L, Barroso E et al (2013) An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol 168(4):3160–3172. https://doi.org/10.1016/j.ijcard.2013.07.150
Nakamura MT, Yudell BE, Loor JJ (2014) Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res 53:124–144. https://doi.org/10.1016/j.plipres.2013.12.001
Brown GC (2007) Nitric oxide and mitochondria. Front Biosci 12:1024–1033. https://doi.org/10.2741/2122
Palmieri EM, Gonzalez-Cotto M, Baseler WA et al (2020) Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun. https://doi.org/10.1038/s41467-020-14433-7
Murphy B, Bhattacharya R, Mukherjee P (2019) Hydrogen sulfide signaling in mitochondria and disease. Faseb J 33(12):13098–13125. https://doi.org/10.1096/fj.201901304R
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94(3):909–950. https://doi.org/10.1152/physrev.00026.2013
Mao JY, Su LX, Li DK et al (2021) The effects of UCP2 on autophagy through the AMPK signaling pathway in septic cardiomyopathy and the underlying mechanism. Ann Transl Med 9(3):259. https://doi.org/10.21037/atm-20-4819
Tran M, Tam D, Bardia A et al (2011) PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121(10):4003–4014. https://doi.org/10.1172/jci58662
Zhang W, Siraj S, Zhang R et al (2017) Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 13(6):1080–1081. https://doi.org/10.1080/15548627.2017.1300224
Zhou H, Zhu P, Guo J et al (2017) Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol 13:498–507. https://doi.org/10.1016/j.redox.2017.07.007
Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19(2):121–135. https://doi.org/10.1038/nrm.2017.95
Seabright AP, Fine NHF, Barlow JP et al (2020) AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. Faseb J 34(5):6284–6301. https://doi.org/10.1096/fj.201903051R
Donadelli M, Dando I, Fiorini C et al (2014) UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol Life Sci 71(7):1171–1190. https://doi.org/10.1007/s00018-013-1407-0
Zhang X, Griepentrog JE, Zou B et al (2020) CaMKIV regulates mitochondrial dynamics during sepsis. Cell Calcium 92:102286. https://doi.org/10.1016/j.ceca.2020.102286
Zhou B, Liu J, Zeng L et al (2020) Extracellular SQSTM1 mediates bacterial septic death in mice through insulin receptor signalling. Nat Microbiol 5(12):1576–1587. https://doi.org/10.1038/s41564-020-00795-7
Hache G, Osuchowski M, Thiemermann C (2015) Does insulin protect the brain in mice and man with sepsis? Shock 44(3):287. https://doi.org/10.1097/shk.0000000000000423
Bloise FF, Santos AT, De Brito J et al (2020) Sepsis impairs thyroid hormone signaling and mitochondrial function in the mouse diaphragm. Thyroid 30(7):1079–1090. https://doi.org/10.1089/thy.2019.0124
Liu YC, Jiang TY, Chen ZS et al (2021) Thyroid hormone disorders: a predictor of mortality in patients with septic shock defined by Sepsis-3? Intern Emerg Med 16(4):967–973. https://doi.org/10.1007/s11739-020-02546-2
Yu G, Tzouvelekis A, Wang R et al (2018) Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med 24(1):39–49. https://doi.org/10.1038/nm.4447
Marín-García J (2010) Thyroid hormone and myocardial mitochondrial biogenesis. Vascul Pharmacol 52(3–4):120–130. https://doi.org/10.1016/j.vph.2009.10.008
Yau WW, Singh BK, Lesmana R et al (2019) Thyroid hormone (T(3)) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy 15(1):131–150. https://doi.org/10.1080/15548627.2018.1511263
Szendroedi J, Phielix E, Roden M (2011) The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 8(2):92–103. https://doi.org/10.1038/nrendo.2011.138
Tubbs E, Theurey P, Vial G et al (2014) Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63(10):3279–3294. https://doi.org/10.2337/db13-1751
Kitada M, Ogura Y, Monno I et al (2019) Sirtuins and type 2 diabetes: role in inflammation, oxidative stress, and mitochondrial function. Front Endocrinol 10:187–187. https://doi.org/10.3389/fendo.2019.00187
Zheng G, Lyu J, Liu S et al (2015) Silencing of uncoupling protein 2 by small interfering RNA aggravates mitochondrial dysfunction in cardiomyocytes under septic conditions. Int J Mol Med 35(6):1525–1536. https://doi.org/10.3892/ijmm.2015.2177
Huang J, Peng W, Zheng Y et al (2019) Upregulation of UCP2 expression protects against LPS-induced oxidative stress and apoptosis in cardiomyocytes. Oxid Med Cell Longev 2019:2758262. https://doi.org/10.1155/2019/2758262
Xu S, Li L, Wu J et al (2021) Melatonin attenuates sepsis-induced small-intestine injury by upregulating SIRT3-mediated oxidative-stress inhibition, mitochondrial protection, and autophagy induction. Front Immunol 12:625627. https://doi.org/10.3389/fimmu.2021.625627
Schilling J, Lai L, Sambandam N et al (2011) Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor γ coactivator-1 signaling. Circ Heart Fail 4(4):474–482. https://doi.org/10.1161/circheartfailure.110.959833
Shi J, Yu J, Zhang Y et al (2019) PI3K/Akt pathway-mediated HO-1 induction regulates mitochondrial quality control and attenuates endotoxin-induced acute lung injury. Lab Invest 99(12):1795–1809. https://doi.org/10.1038/s41374-019-0286-x
Banoth B, Cassel SL (2018) Mitochondria in innate immune signaling. Transl Res 202:52–68. https://doi.org/10.1016/j.trsl.2018.07.014
West AP, Brodsky IE, Rahner C et al (2011) TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472(7344):476–480. https://doi.org/10.1038/nature09973
Li S, Kuang M, Chen L et al (2021) The mitochondrial protein ERAL1 suppresses RNA virus infection by facilitating RIG-I-like receptor signaling. Cell Rep 34(3):108631. https://doi.org/10.1016/j.celrep.2020.108631
Balasubramanian K, Maeda A, Lee JS et al (2015) Dichotomous roles for externalized cardiolipin in extracellular signaling: promotion of phagocytosis and attenuation of innate immunity. Sci Signal 8(395):ra95. https://doi.org/10.1126/scisignal.aaa6179
Michelucci A, Cordes T, Ghelfi J et al (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A 110(19):7820–7825. https://doi.org/10.1073/pnas.1218599110
Tallam A, Perumal TM, Antony PM et al (2016) Gene regulatory network inference of immunoresponsive gene 1 (IRG1) identifies interferon regulatory factor 1 (IRF1) as its transcriptional regulator in mammalian macrophages. PLoS ONE 11(2):e0149050. https://doi.org/10.1371/journal.pone.0149050
Oneill LA (2015) A broken krebs cycle in macrophages. Immunity 42(3):393–394. https://doi.org/10.1016/j.immuni.2015.02.017
Strelko CL, Lu W, Dufort FJ et al (2011) Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc 133(41):16386–16389. https://doi.org/10.1021/ja2070889
Lampropoulou V, Sergushichev A, Bambouskova M et al (2016) Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 24(1):158–166. https://doi.org/10.1016/j.cmet.2016.06.004
Hooftman A, O’neill LAJ (2019) The immunomodulatory potential of the metabolite itaconate. Trends Immunol 40(8):687–698. https://doi.org/10.1016/j.it.2019.05.007
Mills EL, Kelly B, Logan A et al (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167(2):457-470.e13. https://doi.org/10.1016/j.cell.2016.08.064
Tannahill GM, Curtis AM, Adamik J et al (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496(7444):238–242. https://doi.org/10.1038/nature11986
Li Z, Lu S, Li X (2021) The role of metabolic reprogramming in tubular epithelial cells during the progression of acute kidney injury. Cell Mol Life Sci 78(15):5731–5741. https://doi.org/10.1007/s00018-021-03892-w
Acknowledgements
We thank Dr. Rongping Chen from Peking Union Medical College for her suggestions in writing the content of this manuscript.
Funding
The Beijing Municipal Science and Technology Commission provided financial assistance (No. Z201100005520038). This research grant was awarded to author Dawei Liu.
Author information
Authors and Affiliations
Contributions
JL, GZ, XW, and DL all contributed to the literature search, writing, and revising of the manuscript. JL, XW, and DL designed the frame of this manuscript. The first draft of the manuscript was written by JL. JL and GZ drew the Fig. 1 and Fig. 2. All authors contributed to this manuscript and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that there are no potential financial or other conflicts of interest in relation to this research and its publication.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors agree to publish this review.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, J., Zhou, G., Wang, X. et al. Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell. Mol. Life Sci. 79, 456 (2022). https://doi.org/10.1007/s00018-022-04490-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04490-0