
Abstract
PIK3CA mutations are amongst the most prevalent somatic mutations in cancer and are associated with resistance to first-line treatment along with low survival rates in a variety of malignancies. There is evidence that patients carrying PIK3CA mutations may benefit from treatment with acetylsalicylic acid, commonly known as aspirin, particularly in the setting of colorectal cancer. In this regard, it has been clarified that Class IA Phosphatidylinositol 3-kinases (PI3K), whose catalytic subunit p110α is encoded by the PIK3CA gene, are involved in signal transduction that regulates cell cycle, cell growth, and metabolism and, if disturbed, induces carcinogenic effects. Although PI3K is associated with pro-inflammatory cyclooxygenase-2 (COX-2) expression and signaling, and COX-2 is among the best-studied targets of aspirin, the mechanisms behind this clinically relevant phenomenon are still unclear. Indeed, there is further evidence that the protective, anti-carcinogenic effect of aspirin in this setting may be mediated in a COX-independent manner. However, until now the understanding of aspirin’s prostaglandin-independent mode of action is poor. This review will provide an overview of the current literature on this topic and aims to analyze possible mechanisms and targets behind the aspirin sensitivity of PIK3CA-mutated cancers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
According to the GLOBOCAN 2020 study, cancer is a leading cause of death in industrialized countries and in most developing countries, accounting for nearly 10 million deaths worldwide [1]. For instance, according to recent estimates, every third death in Germany among people over the age of 45 is cancer-related. In addition, 1.52 million people suffering from cancer had to be treated in hospital in 2018 [2]. Colorectal cancer (CRC) is the third most common and the second most deadly cancer being responsible for approximately 10% of all cancer-related deaths worldwide [1, 3]. Known risk factors for the development of colorectal carcinoma include familial and genetic predisposition, chronic inflammatory bowel diseases as well as a diet low in fiber and vegetables, obesity, and lack of exercise [4].
For the pathogenesis of colorectal carcinoma, the so-called adenoma carcinoma-sequence is assumed. It is supposed that the majority of colorectal carcinomas develop in a multi-stage process (sequence) of genetic changes from initially benign tumors (villous and tubular adenomas) of the colon mucosa. The malignant transformation of the cells occurs gradually via mutations of various tumor suppressor and proto-oncogenes, such as APC (adenomatous polyposis coli), KRAS (kirsten rat sarcoma virus), and p53 (also known as TP53, tumor protein 53), and subsequently leads to uncontrolled proliferation of the enterocytes and promotes invasive growth and metastasis of the degenerated cells to regional and distant lymph nodes and organs, such as the liver and lungs [5, 6]. Thus, although CRC is associated with poor lifestyle choices, genetic mutations play a crucial role in morbidity and mortality [7]. However, since CRC, like all cancers, is a collective term for a variety of different cancer entities, there is no one-size-fits-all solution to treatment.
The PIK3CA gene encodes for the catalytic subunit p110α of class IA phosphatidylinositol 3-kinases (PI3K) [8]. In vivo and in vitro research has shown that mutations within this gene are associated with poor prognosis for cancer patients and resistance to standard treatments such as chemotherapy and monoclonal antibody therapy [9,10,11,12,13,14,15,16,17,18]. Mutations in the PI3K/Akt/mTOR (mechanistic target of rapamycin kinase)-pathway are amongst the most common across different types of cancer, including not only CRC but also lung, breast, and prostate cancer [19], which have collectively killed more than three million people worldwide in 2020 [1]. This signaling pathway is crucial in cell cycle regulation making it a promising target for anti-cancer drug therapy [20].
Aspirin is one of the most commonly used drugs worldwide. It is widely available and affordable for a large portion of the world’s population as an over-the-counter-drug. There are studies implying that regular intake of low-dose aspirin (acetylsalicylic acid, ASA; ≤ 150 mg per day) may have a chemopreventative and even curative effects on CRC [21,22,23,24,25,26,27]. Nonetheless, many health experts advise against prophylactic intake of aspirin due to possible side effects, such as gastrointestinal ulceration and bleeding as well as hemorrhagic stroke [28, 29]. In recent years, there has been increasing evidence that, in the context of CRC, solely patients with PIK3CA mutations benefit from the use of aspirin. Therefore, the PIK3CA mutation status has been suggested as a predictor for the efficacy of aspirin treatment in CRC [30,31,32,33,34]. However, the mechanisms behind this clinical phenomenon are yet unclear. In the following, we will discuss several signal transduction pathways whose deregulation may be mechanistically related to the effects of PIK3CA mutations in cancer development and progression, as well as to particularities of aspirin action in affected individuals.
The PI3K/Akt/mTOR-Pathway
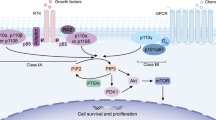
PI3K are a group of lipid kinases that regulate highly conserved signaling pathways involved in cell proliferation, adhesion, motility, apoptosis, and angiogenesis, thus influencing important hallmarks of cancer [8, 35, 36]. PI3K phosphorylate phosphoinositides at the D3-position of the inositol ring leading to the generation of second messengers. Three classes of PI3K are known [37]. Class I PI3K are heterodimers consisting of a regulatory subunit p85 and a catalytic subunit p110 [38]. The catalytic subunit p110α of class IA PI3K is made up of five domains: an adaptor binding domain (ABD), a Ras (rat sarcoma viral oncogene)-binding domain (RBD), a C2 domain, a helical domain and a kinase domain (Fig. 1a) [8]. The p85α subunit also consists of the following five domains: a Src homology 3 (SH3) domain, a breakpoint-cluster region homology (BH) domain flanked by two proline-rich regions, also known as Rho-GTPase binding (RhoGAP) domain [39], an N-terminal SH2 domain (nSH2), an inter-SH2 (iSH2) domain and a C-terminal SH2 domain (cSH2) (Fig. 1b). The nSH2 domain is responsible for the regulation of p110α and the iSH2 domain is required for tethering (Fig. 1c) [8, 40].

Structure of PI3Kα. a Regulatory subunit p85α (NM_181523.3) which is encoded by the PIK3R1 gene consists of five domains: a Src homology 3 (SH3) domain, a Rho-GTPase-activating protein (RhoGAP) domain, an N-terminal SH2 domain (nSH2; cyan), an inter-SH2 domain (iSH2; purple) and a C-terminal SH2 domain (cSH2). The niSH2 domain is depicted in image c, the crystal structures of the white domains are not yet available. b Catalytic subunit p110α (NM_181523.3), encoded by PIK3CA, also consists of five domains: an adaptor binding domain (ABD; blue), a Ras-binding domain (RBD; orange), a C2 domain (yellow), a helical domain (red) and a kinase domain (green). The most common PIK3CA mutations are E542K and E545K in the helical domain as well as H1047R in the kinase domain. c 3D-model of p110α in complex with the niSH2 domain of p85α generated using PyMOL software (4OVU, PDB [41]). The domains are colored according to images a and b
Class IA PI3K are mainly activated by growth factor receptor tyrosine kinases (RTK) or associated adaptor proteins [38, 42,43,44]. Ligand binding of growth factor receptors leads to autophosphorylation of RTK, which in turn leads to binding of the p85 SH2-domain to phospho-YXXM-motifs (pY; X indicating any amino acid) within the RTK. Upon this, the heterodimer dissociates and p110α are released from autoinhibition by p85α, revealing the catalytic site [45,46,47]. Following dissociation, p110α is recruited to the plasma membrane, a process mediated by GTPases of the Ras family [48]. At the membrane p110α phosphorylates its substrate phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3) under use of adenosine triphosphate (ATP). The second messenger PIP3 in turn recruits adaptor and effector proteins with a pleckstrin homology (PH) domain, including Akt (also known as protein kinase B, PKB) and phosphoinositide-dependent kinase 1 (PDK1), to the cellular membrane. PIP3 is later recycled to PIP2 by PTEN (phosphatase and tensin homologue, deleted on chromosome 10) [8]. Akt is activated by phosphorylation of T308 and S473 by PDK1 and mTORC2 (mechanistic target of rapamycin, complex 2), respectively [20, 49, 50]. The serine and threonine kinase Akt/PKB is a key player in the regulation of cell survival, proliferation, metabolism, and growth [51,52,53]. The PI3K/Akt/mTOR pathway is also connected to other pathways such as the Ras/Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK), NF-κB (Nuclear factor κ B), Notch, and APC/Wnt/β-catenin pathways which are also highly involved in carcinogenesis [50, 54,55,56,57,58,59,60,61,62]. The PI3K pathway as well as the most important connecting pathways is depicted in Fig. 2.

Simplified depiction of the PI3K/Akt/mTOR-pathway. PI3K is activated by RTK or related adaptor proteins. Upon activation, it dissociates and the catalytic subunit p110α is recruited to the cell membrane, a process in which Ras family GTPases assist. At the membrane, it phosphorylates PIP2 to the second-messenger PIP3, which then activates a complex network of signaling pathways via Akt/PKB and PDK1. The key pathways influencing cell growth, proliferation, and survival, as well as metabolism, gene expression, angiogenesis, and metastasis are displayed. The PI3K/Akt/mTOR-pathway is also connected to other signaling pathways, such as the Ras/Raf/MEK/ERK-, Notch-, and APC/Wnt/β-catenin-pathway. The most important signaling effectors are highlighted in color. Solid lines indicate direct interaction whilst dashed lines indicate indirect interaction downstream. Transcription factors are marked by oval encircling [49, 50, 53, 58,59,60,61,62, 65,66,67,68,69,70,71,72]
Disturbances in the PI3K/Akt/mTOR pathway play a central role in cancer, the most common deregulations being a loss of PTEN, amplification of the PIK3CA or AKT gene locus and PIK3CA mutations [63,64,65]. This signaling pathway has, therefore, been suggested as a promising target for anti-tumor agents [20, 66].
PIK3CA mutations in Cancer: results from clinical and translational studies
The PIK3CA gene encodes for the catalytic subunit p110α of class IA PI3K. According to The Cancer Genome Atlas and the US National Cancer Institute, PIK3CA is the second most mutated gene across several major cancer types investigated, resulting in a mutational frequency of over 12% (Fig. 3) [19, 73]. Mutation frequency data vary among studies, with the highest values observed for hepatocellular carcinoma (36%) [74], CRC (32%) [75], breast cancer (25–44%) [76,77,78,79,80], head and neck squamous cell cancers (19%) [81], and ovarian cancer (12%) [82].

Distribution of the most frequently mutated genes in cancer. According to the US National Cancer Institute PIK3CA mutations are the second most common mutation in cancer (12.2% of cases), following TP53 mutations [19]
Three so-called hotspots of mutation have been identified, accounting for more than 75% of all PIK3CA sequence alterations [75]: two on exon 9 (E542K and E545K) in the helical domain at the interface of the p85/p110α binding site and one on exon 20 (H1047R) in the kinase domain (see Fig. 1) [40, 83]. These gain-of-function-mutations lead to an increase in PI3K activity and are oncogenic in vivo. [84,85,86] Molecular analyses have shown that the mechanism by which the gain of activity is achieved depends on the position of the mutation [46]. All three hotspot mutations are single-nucleotide substitutions that result in amino acid sequence changes. Mutations of the helical domain (e.g. E545K and E542K) lead to an instability of the nSH2-helical domain inhibitory contact [46, 87, 88]. E545K, for example, induces this by opposite amino acid charge compared to the wildtype causing a conformational change at the nSH2-helical interface [87]. Hence, helical domain-mutated p110α is independent of RTK activation; however, Ras-GTP activation is still necessary. Kinase domain mutations, such as H1047R, however, do not require activation by Ras-GTP, but are dependent on activation via p85 [46, 89]. Kinase domain mutation H1047R also leads to a conformational change, which increases membrane binding of PI3K and, thus, the turnover of the membrane-bound substrate PIP2 [88, 90] In vitro experiments have shown that helical and kinase domain mutations act independently of each other and synergistically, if combined [46].
PI3K/Akt signaling plays a major role in the development of CRC and other cancers [91]. PIK3CA mutations lead to increased proliferation, reduced apoptosis, and tumor invasion [84]. Yet, the predictive value of PIK3CA mutations for clinical outcome is controversial [92]. Many researchers argue in favor of using it as a prognostic marker. For instance, activation of the PI3K/Akt pathway is linked to poor prognosis in CRC [11, 13, 17] and multiple studies report patients with PIK3CA mutations to have an overall higher rate of recurrence and metastasis, as well as lower survival rates [11,12,13, 93]. Moreover, this correlation was not only seen in CRC, but also in other types of cancer, most notably breast cancer [92].
The greatest obstacles in cancer treatment are therapy resistance and tumor recurrence. Common treatments include surgery, pharmacotherapy, and radiation. Standard first-line pharmacological treatments against CRC are chemotherapy regimens like FOLFOX (folinic acid, 5-fluorouracil (5-FU), and oxaliplatin) or FOLFIRI (folinic acid, 5-FU, and irinotecan) in combination with monoclonal antibodies against growth factors or RTK such as VEGF-A (vascular endothelial growth factor A), e.g. bevacizumab, and EGFR (epidermal growth factor receptor), e.g. cetuximab or panitumumab [92]. PIK3CA mutations often coincide with therapy resistance [9]. There are multiple reports that PIK3CA mutations lead to resistance to targeted treatments against RTK [14, 15, 18]. This was the case in a study conducted by Perrone and colleagues. After sequencing samples derived from patients who did not respond to cetuximab treatment, they found that therapy resistance often coincided with KRAS mutation or deregulated PI3K signaling by mutations/loss of PIK3CA or PTEN [14]. A larger study by Sartore-Bianchi et al. yielded comparable results [18]. After conducting in vitro experiments with PIK3CA-mutated CRC cell lines, Jhawer et al. propose PIK3CA mutational status as a predictive biomarker regarding the efficacy of anti-EGFR therapy [15]. Patients with PIK3CA-mutated CRC have also shown resistance to chemotherapy regimens like FOLFOX or FOLFIRI as well as radiotherapy [16, 94]. Wang et al. found PIK3CA mutations to be responsible for the insensitivity of CRC stem cells towards first-line chemotherapy regimens [16]. In light of this evidence, inhibitors of the PI3K/Akt/mTOR-pathway as means of targeted treatment came into focus [65]. Indeed, administration of Akt and mTOR inhibitors in combination with anti-EGFR treatment to PIK3CA-mutated cell lines derived from CRC patients has shown promising results, at least in vitro [95]. Morii et al. also found that the Akt inhibitor perifosine can overcome chemoresistance of PIK3CA-mutated CRC cell lines to oxaliplatin and 5-FU [96]. As a result of PIK3CA-related drug development, the first selective inhibitor of PI3K p110α, alpelisib, was approved by the US Food and Drug Administration (FDA) in 2019 as treatment for PIK3CA-mutated hormone receptor positive/human epidermal growth factor receptor 2 negative (HR + /HER2-) breast cancer in combination with the estrogen receptor antagonist fulvestrant in post-menopausal women and men. The approval was based on the SOLAR 1 study, in which progression-free survival at a median follow-up of 20 months nearly doubled (11 vs. 5.7 months) for patients harboring PIK3CA mutations receiving the drug compared to the placebo control group. In contrast, patients without PIK3CA-mutated cancer did not benefit from alpelisib treatment [97, 98]. In addition, clinical trials have been set up to study whether patients suffering from metastatic colorectal cancer (mCRC) will benefit from alpelisib treatment. For instance, preliminary results from 102 patients enrolled in a combined phase 1b/randomised phase 2 clinical trial (NCT01719380) investigating the safety and efficacy of the BRAF inhibitor encorafenib and the monoclonal EGFR antibody cetuximab with or without alpelisib in patients suffering from advanced BRAF-mutant mCRC indicated that these patients may benefit from therapeutic intervention with alpelisib. Analysis of progression-free survival comparing the triplet to the doublet after 73 events showed a hazard ratio (95% confidence interval) of 0.69 (0.43–1.11; P = 0.064) with an overall response rate of 27% (16–41%) and 22% (12–36%), respectively [99]. A 2021 preclinical in vitro trial conducted by Aslam et al. showed that PIK3CA-mutated colon carcinoma cell lines are especially sensitive to simultaneous treatment with alpelisib and cyclin-dependent kinase 4 and 6 inhibitor ribociclib compared to wild-type cells [100]. Currently, the ALCAP clinical trial is underway to evaluate the benefits of combined alpelisib and capecitabine, a 5-FU prodrug, treatment in patients with PIK3CA-mutated mCRC (NCT04753203) [101]. Also, there is an ongoing study examining the efficacy of PI3K inhibitor MEN1611 in combination with cetuximab, also concerning PIK3CA-mutated mCRC (C-PRECISE-01, NCT04495621) [102].
Pharmacology of aspirin (acetylsalicylic acid)
History of aspirin
Aspirin (acetylsalicylic acid, ASA) is one of the most widely distributed drugs worldwide. As a non-opioid analgesic, it is widely used to treat pain, fever, and also inflammation. Indeed, for centuries salicylates found in plants, such as the willow or meadowsweet, have been used as remedies against the ailments mentioned above [103, 104]. ASA was first synthesized by Felix Hoffmann and Arthur Eichengrün in 1897 and subsequently distributed by Bayer under the name “Aspirin”. Being the first non-steroidal anti-inflammatory drug (NSAID), it quickly became popular as a “wonder drug”. Its popularity has, albeit, somewhat declined since due to common side-effects, such as gastrointestinal ulceration and bleeding, and the availability of other NSAIDs and acetaminophen (paracetamol) [103]. Although, first intended as a prodrug to avoid the gastric irritation often caused by salicylic acid, the antithrombotic properties of aspirin were discovered in the late 1960s and, thus, widespread use as an antiplatelet agent for the prevention and treatment of thromboembolic complications in the setting of cardiovascular disease has since expanded the prescribing scope even further [103, 105, 106]. Accordingly, the WHO has listed aspirin as an essential medicine since 1977 [105].
Safety of aspirin
Even though aspirin is considered a relatively safe drug, there are risks associated with its intake, especially if taken long-term [107]. The most common adverse effects of aspirin are an increased risk of bleeding and injuries to the gastroduodenal mucosa [28, 29, 107, 108]. While fatal bleeding incidents are rare, life-threatening complications, such as intracranial and gastrointestinal hemorrhage, do occur and should be considered [28, 29, 109]. Gastrointestinal side-effects are the most frequent; these range from dyspepsia to gastrointestinal bleeding and peptic ulcers [28, 29, 110,111,112]. Particular caution should be heeded when prescribing long-term aspirin use to the elderly (≥ 75 years), a group that also has an increased risk of developing malignant colorectal tumors [4, 108]. Since most trials regarding the safety of aspirin were conducted in middle-aged patients, risk assessment for the elderly is difficult [28, 108]. The ASPREE trial (Aspirin in Reducing Events in the Elderly), conducted from 2010 to 2014 in the USA and Australia, found that participants (ages 70 and older; ≥ 65 years for Hispanic and black participants in the US) taking 100 mg of aspirin daily actually had higher all-cause mortality than those receiving placebo. Most deaths in the aspirin group were attributed to cancer, mainly of the gastrointestinal tract (including CRC) [113]. Therefore, health experts only recommend prophylactic long-term use of low-dose aspirin (min. 10 years; ≤ 150 mg) for patients aged 50–69 with a life expectancy of at least 10 years and an increased risk of developing CRC or cardiovascular disease [28, 29].
Pharmacokinetics of aspirin
Following oral ingestion, aspirin is absorbed from the gastrointestinal tract in its unhydrolyzed form by passive diffusion [107, 114, 115]. Aspirin is absorbed from the gastrointestinal tract, with the main areas of absorption being the small intestine and, to a lesser extent, the stomach [115,116,117]. However, the quantity of non-absorbed aspirin reaching the colon and rectum is unclear, raising the question of whether colorectal cancer or epithelial cells are exposed to aspirin that passes through the colonic lumen or exclusively to aspirin that circulates in the bloodstream after absorption in more proximal portions of the intestine.
After absorption, it is cleaved by esterases into an acetyl moiety and its primary metabolite salicylic acid by cells of the gut mucosa and (primarily) the liver following first-order kinetics [107, 114, 115, 118, 119]. Free acetate yielded by hydrolysis can enter the tricarboxylic acid (TCA) cycle [114]. Plasma levels of unhydrolyzed aspirin rise quickly after absorption and peak after about 15–20 min. Thereafter, they decrease rapidly, while the salicylate levels increase [120]. According to Needs et al. the peroral bioavailability of aspirin is approximately 70%, indicating that about 70% of a perorally administered aspirin dose enters the systemic circulation unhydrolyzed [115]. However, the absorption rate depends on the kind of formulation; aqueous solutions or fast-dissolving formulations lead to quicker absorption from the gastrointestinal tract and higher plasma levels of non-metabolized aspirin [107, 121]. In the blood stream, aspirin, as well as salicylate, is bound to proteins, mainly albumin, and distributed throughout the body [107, 114, 122, 123].
Mechanism of action of aspirin
Aspirin’s main mechanism of action is the acetylation of proteins, which is entirely non-specific [124, 125]. Examples include the acetylation of human serum albumin, resulting in altered protein function due to conformational changes [124, 126]. Additionally, aspirin has shown to acetylate tumor suppressor protein p53 [19, 124, 127]. There are also accounts of aspirin-acetylating histones, for instance lysine residues such as K56 and K122 on histone H3 [128, 129]. In this context, Wang et al. identified a total of 523 proteins to be targets of aspirin acetylation in HCT116 CRC cells, amongst them mTOR and others affected by the PI3K pathway, such as eIF2 and eIF4A1. In further experiments they demonstrated that aspirin does indeed suppress mTOR function as evidenced by a reduction in phosphorylation (Ser235/236) of the mTORC1 target S6 in HCT116 cells and mouse embryonic fibroblasts [130]. Nevertheless, the causality between specific acetylation of mTOR and mTOR inactivation needs to be verified in future experimental analyses by exchange mutations of the detected acetylated amino acid residues.
The most well-known effect of aspirin, to date, is its influence on the prostanoid system which was discovered by John Vane in 1971 [131]. Prostanoids are a class of lipid mediators that can be divided into prostaglandins and thromboxanes. They derive from the unsaturated fatty acid arachidonic acid which is released from the plasma membrane by phospholipases A2 [132, 133]. Cyclooxygenases (COX), also known as prostaglandin G/H synthases (PGHS) or prostaglandin-endoperoxide synthases (PTGS), catalyze the transformation of arachidonic acid into prostaglandin H2 (PGH2) which is a precursor prostanoid [132,133,134]. COX exist in two isoforms, COX-1 and COX-2. Commonly, COX-1 is seen as a constitutive form responsible for the maintenance of the gastroduodenal mucosa and tissue homeostasis in general, whereas COX-2 has been identified as an inducible form in most cell types which is mainly expressed in response to inflammation [132], although this static role allocation has been questioned [135]. Aspirin covalently acetylates COX-1 at S530 and COX-2 at S516, thereby irreversibly inhibiting the production of PGH2 [107, 136]. The irreversibility of COX inhibition distinguishes aspirin from other “traditional” NSAIDs, such as ibuprofen, which reversibly inhibit the activity of COX-1 and COX-2 through competitive antagonism with arachidonic acid at the active site of the enzymes [107, 137, 138]. The duration of aspirin’s effect is, therefore, also not limited by its half-life but by the turnover rate of the target protein and, in the case of therapeutic use of aspirin as an antiplatelet agent, by the platelet lifespan [107]. Although aspirin has the potential to inhibit both COX isoforms, it is more selective towards COX-1, with IC50 values at isolated enzymes of 5 µg/mL (COX-1) and 210 µg/mL (COX-2), respectively [139, 140]. In contrast, most NSAIDs are predominantly nonselective in inhibiting COX-1 and COX-2, although agents such as meloxicam, nimesulide, and diclofenac have been reported to possess a 18- to 29-fold greater potency towards COX-2 in vitro [138, 141]. In addition, selective COX-2 inhibitors act preferentially on COX-2 and, therefore, appear to differ from nonselective COX inhibitors with regard to the spectrum of side effects. As such, they are less likely to cause gastric and duodenal ulceration, whereas they increase the incidence of thromboembolic events and renovascular hypertension, the latter two side effects being most likely due to a reduction in the bioavailability of vascular endothelial (vasculoprotective) prostacyclin with, however, preserved thromboxane A2 (TxA2)-dependent platelet aggregation and vasoconstriction [142, 143]. Indeed, COX-2-selective inhibitors rofecoxib and valdecoxib have been withdrawn from the market due to an increased risk for the occurrence of cardiovascular events, including myocardial infarction [143].
PGH2 formed by COX is then further processed into a variety of prostaglandins and thromboxane by specific synthases [132, 133]. The most relevant being prostaglandin E2 (PGE2), D2, F2α, prostacyclin (PGI2) and TxA2. While both COX isoforms lead to the biosynthesis of prostaglandins, TxA2 synthesis predominantly depends on COX-1-derived precursors [132, 133, 144]. This dependence of TxA2 formation on COX-1 activity is due to COX isoform-preferential coupling of synthases. COX-1 predominantly interacts with thromboxane A synthase 1 (TBXA1S) and with PGF and cytosolic PGE synthases, whereas COX-2 primarily couples to prostacyclin synthase and microsomal PGE synthases, the latter two being induced by cytokines and tumor promotors [133]. In line with this concept, the COX-2 selective inhibitor celecoxib in doses of up to 800 mg did not inhibit TxA2-induced platelet aggregation in humans, while reducing systemic prostacyclin biosynthesis as indicated by a reduction in urinary excretion of the prostacyclin metabolite 2,3-dinor 6-keto-PGF1α in these individuals [142]. Prostanoids act locally and mediate their effects via G-protein coupled receptors (GPCR) [132, 133]. They are ubiquitously expressed throughout the body, where they fulfill a wide variety of functions [134]. For example, PGE2 is involved in the response to inflammation and nociception [133, 134]. Moreover, prostaglandins such as PGE2 and PGI2 contribute to renal blood flow and function and, in the case of PGE2, also to gastroduodenal mucosa protection [145]. Prostanoids also play an important role in the regulation of the cardiovascular system, most notably TxA2 and PGI2. TxA2 induces vasoconstriction, platelet aggregation, and endothelial dysfunction, whereas PGI2 exerts opposite effects which is why it is considered a protective functional antagonist of TxA2 [146].
However, not only formation of the mentioned prostaglandins and thromboxane A2 depend on the activity of COX enzymes, but also COX have been shown to be involved, for instance, in dihomoprostaglandin formation from adrenic acid [147] or in the (mostly COX-2-dependent) formation of the F2-isoprostane and thromboxane A2 receptor agonist 8-iso-PGF2α (8-iso-prostaglandin F2α) [144, 148]. Uninhibited, COX also generate a small amount of the arachidonic acid derivates 11-(R)-hydroxyeicosatetraenoic acid (11-HETE), as well as a racemic mixture of 15-(S)- and (R)-hydroxyeicosatetraenoic acid (15-HETE), the majority being the (S)-enantiomer [137, 149]. Unlike other NSAIDs, aspirin has been reported to qualitatively alter the enzymatic substrate specificity and activity of COX-2. Indeed, aspirin-acetylated COX-2 is involved in the formation of aspirin-triggered specialized proresolving mediators, including aspirin-triggered lipoxins and resolvins, which appear to play a role in the anti-inflammatory and anti-cancer effects of the drug [150, 151]. In this context, acetylation of COX-2 does not completely inhibit its function [137, 149]. Conformational changes due to the acetylation by aspirin lead to a shift in the stereochemistry of the product, resulting in a higher ratio of 15-(R)-HETE formation compared to 15-(S)-HETE (3:1) [137, 149, 152]. Whereas 15-(S)-HETE has been shown to be involved in the process of inflammation, angiogenesis, and the pathogenesis of cancer, especially CRC [153,154,155,156,157,158,159], 15-(R)-HETE can be converted to, i.e. 15 epi-lipoxin A4 or B4 by leukocyte 5-lipoxygenase in a transcellular process during the interaction of COX-2-expressing endothelial or epithelial cells with polymorphonuclear leukocytes [151]. In a similar transcellular fashion, aspirin-induced acetylation of COX-2 initiates the formation of so-called aspirin-triggered resolvins from docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) [151]. Resolvins are categorized as either D-series (derived from DHA) or E-series (derived from EPA) and aspirin-triggered epimers have been identified for each family, although compared with the pharmacodynamics of other NSAIDs, a unique activity of aspirin-acetylated COX-2 appears to be its participation in the formation of aspirin-triggered resolvins of the D-series [160]. In this regard, both resolvins and lipoxins promote the resolution of inflammation by stimulating phagocytosis of cellular debris and counteracting the release of pro-inflammatory cytokines without being immunosuppressive [151]. In addition, they have been reported to counteract tumor growth in animal models [161, 162]. Compared with native resolvins, aspirin-triggered forms (R-epimers) appear to resist rapid inactivation by oxidoreductases more effectively and, therefore, have longer half-lives, and as shown, for example, for aspirin-triggered resolvin D1, they appear to have greater efficacy in mediating anti-inflammatory effects, at least in murine models of inflammation [163]. Interestingly, both aspirin-triggered lipoxins and resolvins have been attributed significant anticancer effects in preclinical studies by promoting phagocytosis of apoptotic and necrotic tumor cells by macrophages and counteracting macrophage-induced secretion of pro-inflammatory cytokines [164], suggesting that this pharmacodynamic property of aspirin may be clinically relevant with respect to the anti-inflammatory and anticancer effects of the drug. Nevertheless, it is unclear to what extent these aspirin-triggered lipid mediators play a role in the aspirin sensitivity of PIK3CA-mutated CRC. Yet, it is conceivable that activating PIK3CA mutations may lead to (i) increased expression of COX-2 in cancer or cancer-associated endothelial cells, (ii) to increased interaction between polymorphonuclear leukocytes and PIK3CA-mutated colorectal cancer cells, or (iii) to interaction-induced increased expression of 5-lipoxygenase in polymorphonuclear leukocytes, thereby increasing the efficacy of aspirin-triggered local formation of lipid mediators with potential anticancer activity.
Apart from the acetyl moiety, salicylic acid also interacts with signaling pathways independently, making aspirin one drug with two pharmacologically active compounds [107, 114]. So far, it is not clear if the anti-cancer properties exhibited by aspirin are mediated by acetylation or its salicylate moiety. The mechanism of action of salicylic acid is more complex and, thus, not yet well understood. Salicylate is also known for its anti-inflammatory effect. One possible explanation is its interference with COX-2 expression. There are multiple reports of salicylate inhibiting COX-2 mRNA transcription [165,166,167]. Xu et al. found that COX-2 mRNA transcription inhibition was mediated by interleukin-1β (IL-1β), phorbol 12-myristate 13-acetate and lipopolysaccharide (LPS) [165]. This is consistent with a 2002 study published by Cieslik and colleagues. They stated that salicylate suppresses both COX-2 as well as inducible nitric oxide synthase (iNOS) expression by reducing the binding CCAAT/Enhancer-binding protein-β to the promoter, which is stimulated by LPS and the cytokine interferon-γ (IFN-γ) [166]. Later, Chae et al. reported that salicylate inhibits phosphorylation of IκBα (NFKB inhibitor alpha) and subsequent degradation mediated by the cytokine TNFα (tumor necrosis factor α) via the ERK1/2-MAPK-pathway. The degradation of IκBα is necessary to activate NF-κB downstream and induce COX-2 expression [167]. According to Wang and Brecher’s 1999 study, salicylate inhibits the expression of iNOS by means of ERK, MAPK, IFN-γ, and TNFα [168]. Kiss et al. also found that salicylate decreases NF-κB activity via inhibition of IκB (IκB protein) [169]. As a downstream target of Akt NF-κB is substantially involved in the oncogenic activity of the PI3K pathway (Fig. 2) [69, 170,171,172]. According to Hawley et al. salicylic acid also activates metabolic regulator adenosine monophosphate–activated protein kinase (AMPK) [173]. Taken together, these data suggest that the salicylate moiety of aspirin may significantly counteract the PIK3CA mutation-triggered overactivity of several relevant effectors of the PIK3 pathway, e.g., by inhibiting NF-κB-dependent signal transduction and by downregulating COX-2 expression, thus possibly contributing to the beneficial effects of aspirin on PIK3CA-mutant CRC.
Aspirin sensitivity of PIK3CA-mutated CRC
There is substantial evidence that aspirin is effective in the prevention and treatment of CRC. For example, in a randomized controlled trial involving over 1,000 patients, Baron et al. found that low-dose aspirin (81 mg) had a chemopreventive effect on colorectal adenoma [25]. Also, in a long-term cohort study performed in Sweden over a period of more than 20 years, aspirin use was associated with a 35% reduced risk of CRC [174]. In a meta-analysis that included seven trials each on aspirin therapy after the diagnosis of CRC and seven trials on aspirin use before the diagnosis of CRC, Peiwei Li and colleagues concluded that aspirin is effective for the treatment of CRC after diagnosis but not for primary prevention in people at high risk of developing CRC [27]. Interestingly, an overall survival benefit associated with aspirin use after diagnosis was observed in both colon and rectal cancers, although this survival benefit of aspirin use after diagnosis appeared to be limited to patients with COX-2-positive as well as PIK3CA-mutant tumors. In agreement with this study, Bastiaannet et al. also found aspirin to lower the risk of mortality in CRC patients post-diagnosis [175]. Also, in gastrointestinal tract cancers, prolonged patient survival has been observed with low-dose aspirin [176]. Similar observations have been made in non-gastrointestinal malignancies, such as head and neck, breast and lung cancer [177,178,179,180,181,182]. Thus, these findings together suggest that aspirin may exhibit broad anti-cancerogenic effects which are not limited to CRC.
Different landmark studies suggest that the effect of aspirin on cancer is limited to patients with PIK3CA-mutated tumors [30,31,32,33]. This was the case in an observational study of over 900 CRC patients published in 2012 by Liao et al. in which aspirin use significantly improved survival in patients with PIK3CA-mutant CRC, whereas survival in patients with wild-type PIK3CA CRC was not improved [30]. Zumwalt et al. compared the effect of aspirin on various colon carcinoma cell lines and found that aspirin treatment of PIK3CA-mutant colon cancer cells leads to a downregulation of cell-cycle-related genes and reduced tumor growth in mouse xenografts [33]. Gu et al. confirmed these findings, observing that aspirin induces apoptosis and leads to G0/G1 cell-cycle arrest only in colon carcinoma cell lines carrying PIK3CA-mutations [32]. Nonetheless, the exact mechanism behind the aspirin sensitivity of PIK3CA-mutated tumors remains unclear. In the following, we present the current knowledge on this topic and discuss possible mechanisms by which aspirin may inhibit tumorigenesis related to PI3K signaling.
COX-2-related aspirin-sensitivity of cancer cells
The most obvious answer would be that this phenomenon is related to inhibition of COX enzymes, as they are by far the best researched targets of aspirin [183]. The COX-2/PGE2 signaling axis contributes to most hallmarks of cancer, promoting cancer-associated angiogenesis as well as proliferation, survival, migration, and invasion of cancer cells [184]. Also, COX-2 is induced by inflammation, a process strongly linked to the development of cancer, CRC in particular [132, 185, 186]. Colorectal tumors often display elevated COX-2 expression levels as seen in both patient-derived tissues and animal models [187,188,189]. Importantly, overexpression of COX-2 is associated with poor survival of CRC patients [190], whereas selective COX-2 inhibition in colon cancer cells has shown to suppress tumor growth and to induce apoptosis as well as cell-cycle arrest [191, 192]. With regard to the clinical relevance of COX-2 in the pathogenesis of CRC, Veettil and colleagues published a systematic review in 2019 concluding that the benefit of CRC chemoprevention with COX-2-inhibitor celecoxib outweighed the risk of cardiovascular side effects after reviewing three randomized controlled trials and three post-trial studies which compared the incidence of recurrence of colorectal adenomas in patients given celecoxib at varying doses or placebo [193]. In line with these findings, Valverde et al. found that combined celecoxib and cetuximab treatment inhibited growth and induced apoptosis in non-KRAS-mutated CRC cells in vitro and in mouse xenografts via impairment of the EGFR/RAS/β-catenin/FOXM1 signaling axis [194]. Also, other NSAIDs like sulindac have proven to induce apoptosis in colon cancer cells, thereby pointing to a class effect of COX inhibitors in CRC treatment or prevention that may be attributable to the ability of these drugs to inhibit COX-2 function [188, 195]. There is evidence from non-CRC cancers indicating that COX-2 signaling is related to PI3K signaling. For instance, Uddin et al. observed that COX-2 inhibition via aspirin, a selective COX-2 inhibitor (NS398), and gene silencing by siRNA led to a reduction of Akt phosphorylation, which resulted in cell cycle inhibition and apoptosis of ovarian cancer xenografts in mice [196]. Further, in a study published by Tury and colleagues, the COX-2-selective inhibitor celecoxib was effective against PIK3CA-mutated patient-derived breast cancer xenografts, but not against PIK3CA-wildtype xenografts [197]. In CRC, Liao et al. found aspirin was most effective on tumors with both PIK3CA mutation and COX-2 expression, but the sample size in this study was too small to draw reliable conclusions [30]. Indeed, PI3K has been identified as a downstream target of COX-2/PGE2 but may also be involved in the regulation of COX-2 expression in the context of a positive feedback loop [198,199,200,201,202,203,204,205]. In this regard, PGE2-driven EP4 activation and a subsequent EP4-related activation of the GRK/β-arrestin/Src/PI3K/GSK3 pathway has been proposed, leading, for example, to nuclear translocation of β-catenin and β-catenin-dependent gene expression [206]. Moreover, an EP4-selective agonist activated the PI3K/ERK pathway in colon carcinoma cells possibly via EGFR transactivation and thereby rescued proliferation suppressed by indomethacin or COX-2 inhibitors [203]. In this context, PGE2 was shown to induce EGFR transactivation and subsequent PI3K signaling via EP4, β-arrestin and c-Src in colon cancer cells [202] In addition, PGE2 via EP2 and EP4 has also shown to modify the activity of other growth factor receptors, such as insulin-like growth factor 1 receptor (IGFR) [207]. Interestingly, COX-2 mRNA expression and PGE2 synthesis may also be regulated by PI3K via insulin-like growth factors (IGF) 1 and 2, as seen in in vitro experiments with Caco-2 colon carcinoma cells. Here, both the inhibition of PI3K and antagonism of the receptor suppressed COX-2 mRNA transcription [200]. Interestingly, and as addressed in the section on aspirin pharmacodynamics, aspirin can alter the enzymatic activity of COX-2 through acetylation such that it generates precursors of aspirin-triggered forms (R-epimers) of lipoxins and resolvins that arise from these precursor molecules in transcellular processes of COX-2-expressing epithelial or cancer cells and polymorphonuclear leukocytes [151]. These lipid mediators have been shown to elicit both anti-inflammatory and anti-cancer effects in preclinical models, the latter presumably by stimulating phagocytosis of tumor cell debris by macrophages and by inhibiting macrophage-induced inflammation [151]. Although the exact background of the generation of aspirin-triggered lipoxins and resolvins in PIK3CA-mutated and COX-2-overexpressing CRC remains unclear to date, it can, therefore, be postulated that aspirin mediates enhanced generation of these lipid mediators in the presence of increased COX-2 expression and activity, which in turn may contribute to the antitumor effects of the drug.
However, it must also be noted that there are clinical data that contradict the relevance of COX-2 as an important target of aspirin in PIK3CA-mutated CRC. For instance, in a study by Gray et al., aspirin intake led to increased patient survival in colon cancers with increased COX-2 expression, but the PIK3CA mutational status determined was not related to this effect [208]. Moreover and interestingly, in a trial by Enric Domingo and colleagues published in 2013, patients with PIK3CA-mutated CRC benefited from aspirin, albeit no improvement was seen in the patients being treated with the COX-2-inhibitor rofecoxib [34]. In this context, it must be noted that in most studies PIK3CA mutations were not differentiated as to whether they were located in the helical or in the kinase domain. This could explain in part contradictory study outcomes. For example, as described above, the COX-2/PGE2/EP4 signaling axis is able to increase PI3K activity via RTK (trans)activation, an effect that is unlikely to have a relevant impact on downstream signaling events with PIK3CA mutations in the helical domain because of uncoupling of these mutants from RTK signaling. Off note, this is also the reason why PIK3CA mutations in the helical domain are discussed as a cause of resistance to, for example, EGFR inhibitor therapy and are being investigated in clinical trials (C-PRECISE-01, NCT04495621) as a target in such CRC entities [102]. Nonetheless, aspirin could also elicit anti-tumor effects in these cancer entities via downregulation of COX-2 mRNA expression (potentially via the salicylate moiety of the drug) and a shift in COX-2 enzymatic activity (acetylation by aspirin). Thus, the results presented in this section collectively suggest that COX-2 is a relevant target for aspirin sensitivity of PIK3CA-mutated CRC, as both PIK3CA activity and COX-2 expression and activity may be amplified as part of a positive feedback loop. Contributing to the clinical phenomenon of aspirin sensitivity of PIK3CA-mutated CRC could also be increased formation of aspirin-triggered lipoxins and resolvins in COX-2-overexpressing tumors, which may also provide a promising explanation for different clinical outcomes of aspirin- and selective COX-2 inhibitor-treated patients suffering from PIK3CA-mutated CRC. However, given the conflicting clinical and experimental results in this context, further research is needed to clarify the exact mechanistic interconnections.
COX-1- and TXA2-related mechanisms
Aspirin is an NSAID with clinically relevant preference for COX-1 and it has the potential to inhibit COX-2 at higher doses [139, 140, 183]. In this context, it seems plausible that the effect of aspirin on PIK3CA-mutated CRC is mediated preferentially by COX-1 or TxA2, the latter being a prostanoid whose generation depends predominantly on COX-1-mediated PGH2 biosynthesis [132, 133]. Angiogenesis, as well as platelet aggregation play an important role in tumor growth and metastasis, both are influenced by TxA2 [209,210,211,212,213]. Thus, these processes could be inhibited by aspirin’s anti-platelet properties and in particular through a reduction in platelet TxA2 synthesis, which appears to be essential to prepare metastatic intravascular niches at least in preclinical cancer models in vivo [214]. The inhibition of platelet-activated metastasis by aspirin could be mediated by inhibition of Akt, as suggested by a preclinical in vivo study on breast cancer cells [215]. Here, platelets were shown to promote metastasis of these cells through activation of the Akt signaling pathway and aspirin treatment of platelets resulted in inhibition of Akt and subsequent inhibition of pro-metastatic IL-8 production. Also, Li et al. proposed that PI3K is critical for platelet secretion as they were able to stimulate PI3K-dependent phosphorylation of Akt after activation of the thromboxane A2 receptor using the TxA2 mimetic U46619 [216]. COX-1 activity (and subsequent TxA2 formation) may also be involved in the development of CRC as experiments regarding azoxymethane-induced CRC in rats suggest [217, 218]. In line with these findings, Wu et al. observed that the selective COX-1 inhibitor SC-560 induced cell-cycle arrest and macroautophagy in colon cancer cells [219]. A study by Sakai et al. reported that TxA2 levels were upregulated in human colorectal carcinomas and CRC cell lines due to overexpression of TBXA1S, resulting in increased cell proliferation [220]. In addition, COX-1 can ease symptoms of colitis by upregulating β-arrestin via PGE2 and PI3K/Akt, indicating that the activity of these signaling effectors upstream may also be influenced by COX-1 in the colonic epithelium [221]. Taken together, COX-1 signaling in cancer and its inducible mechanisms have long been overlooked. Therefore, to fully understand the development of CRC and the beneficial effects of aspirin in PIK3CA-mutated CRC in this context, further research in this area is essential. However, based on current knowledge, COX-1 appears to act similarly to COX-2 upstream of PI3K and therefore the effect of aspirin on COX-1 activity is unlikely to explain the particular effects of the drug on PIK3CA-mutated CRC.
NF-κB signaling
Another possibility for the aspirin-sensitivity of PIK3CA-mutated CRC could be explained by prostaglandin-independent mechanisms. The NF-κB pathway is involved in the response to inflammation and infection, but also cancer development. A study by Martha Slattery and colleagues in 2018 showed that the NF-κB signaling pathway is dysregulated in CRC, as are p53 and Wnt/β-catenin signaling [172, 222]. NF-κB and β-catenin are involved in angiogenesis and metastasis in CRC [170]. Interestingly, both aspirin and salicylate were identified as specific inhibitors of IKK-β (IκB kinase-beta) activity in vitro and in vivo, an effect that depended on direct binding of the drugs to IKK-β to interfere with ATP binding [223]. Indeed, Jinbo Fu and colleagues demonstrated that aspirin suppressed chemoresistance to 5-FU in 5-FU-resistant CRC cell lines SW620 and SW480 by abrogating 5-FU-induced NF-κB activation, thereby enhancing the antitumor activity of 5-FU [224]. Also, salicylate treatment has been suggested to reduce chemotherapy resistance by inhibiting NF-κB activity [225] and it may inhibit Mucin-1 (MUC1)-mediated tumor migration and invasion by inhibiting Akt [226]. Thus, these results suggest that aspirin-mediated inhibition of tumor pathogenesis may also involve modulation of atypical signaling pathways not previously associated with aspirin pharmacodynamics. NF-κB signaling is not only related to PI3K/Akt, but also the Wnt/β-catenin pathway, and their connection is necessary to fully understand PI3K signaling in CRC and aspirin sensitivity of PIK3CA-mutant CRC entities.
Wnt and β-catenin-dependent signaling
The canonical Wnt/β-catenin pathway is one of the most crucial pathways in the development of CRC [227, 228]. It drives cell cycle progression and maintains the undifferentiated state of intestinal stem cells [227]. Pathway activation starts with a glycoprotein of the Wnt family extracellularly binding to the N-terminal domain of a G-protein coupled receptor of the Frizzled family. This activates Dishevelled (DVL), which is then translocated to the plasma membrane. In turn, DVL recruits the destruction complex consisting of APC, glycogen synthase kinase (GSK) 3β, Axin and casein kinase 1, inactivating it by removal from the cytosol [229]. In the cytosol, the destruction complex marks the protein β-catenin for ubiquitination and subsequent degradation by the proteasome [228]. However, unhindered by the complex, β-catenin relocates to the nucleus where it enables the transcription of various proteins responsible for cell differentiation, proliferation, and migration, for example, the proto-oncogene c-myc [230]. Wnt/β-catenin signaling is important for the homeostasis of the intestinal epithelium [231]. In complex with E-cadherin, β-catenin forms cell–cell junctions regulating epithelial cell adhesion and polarity. Deregulation may lead to epithelial-mesenchymal transition (EMT) [232]. EMT refers to the process of transition of epithelial cells into cells with mesenchymal properties, in which cells lose cell polarity and cell–cell adhesion to adopt a migratory and invasive phenotype, often considered the first step towards metastasis [233].
It is believed that the majority of CRCs arise via the adenoma carcinoma-sequence, a series of mutational events leading to the gradual transformation of benign polyps into malignant carcinomas [5, 234]. Alterations in the Wnt/β-catenin pathway, especially APC mutations, play a central role in this process [5, 235]. The Wnt/β-catenin and PI3K pathways are closely connected. One example is the connection via GSK3β, a substrate of Akt and member of the β-catenin destruction complex [58, 235]. It is phosphorylated by Akt at S9 leading to its inactivation [236]. Albeit, there is no evidence that phosphorylation of GSK3β by Akt directly influences the activity of the Wnt pathway [237, 238]. Further, Zeng et al. could show that tuberous sclerosis (TSC) 2 deletion and subsequent mTORC1 activation inhibits Wnt signaling by regulating Frizzled protein levels in a DVL-dependent way [239]. This is an interesting finding in the context of PIK3CA-mutated CRC because mTORC1 is a downstream target of PI3K-Akt by inhibiting TSC1/2 (see Fig. 2) and because there is evidence for aspirin-mediated direct acetylation and inactivation of mTOR and the mTORC1 complex [130, 240]. Also, a 2007 publication by Fang et al. suggests that Akt directly phosphorylates β-catenin at S552 resulting in dissociation of cell junctions and accumulation of β-catenin in the cytosol and the nucleus of human epidermoid carcinoma cells [241]. In accordance, Steffen Ormanns and colleagues later found that β-catenin transcriptional activity depended on PI3K activity, presumably due to Akt phosphorylation. In this context, PI3K inhibition did not affect the subcellular localization of β-catenin but impaired the binding of β-catenin to Wnt target gene promoters and decreased the expression of Wnt target genes [242]. Apart from this, it also should be noted that both pathways are also connected to NF-κB signaling (see Fig. 2 and previous section) [69, 243, 244].
Interestingly, there is evidence that sensitivity of cancer cells to aspirin is mediated by Wnt and β-catenin signaling. In an in vitro approach to investigate the influence of aspirin on Wnt/β-catenin signaling in CRC, Bos and colleagues demonstrated that aspirin treatment induced a decreased expression of Wnt target genes and increased phosphorylation of β-catenin in DLD-1 and SW480 colon carcinoma cell lines. Phosphorylation of β-catenin most likely led to its ubiquitination, as cytosolic β-catenin subsequently decreased. They, therefore, assumed that this effect is mediated by protein phosphatase 2A (PP2A), a negative regulator of Akt [245, 246]. PP2A is critically involved in cell–cell adhesion and EMT by stabilizing the β-catenin/E-cadherin complex. Interestingly, the findings of Bos et al. are consistent with two studies published in recent years. In 2019 Jin and Wuo observed that aspirin inhibits Wnt-mediated EMT in SW480 colon carcinoma cells [247]. In addition, a paper published in 2021 by Dunbar et al. showed that aspirin could reverse a Wnt-induced stem-like phenotype in intestinal organoids derived from mouse and human organoids lacking APC. In addition, motility and invasion of colon cancer cells were reduced, effects mediated via the induction of Dickkopf-1, a Wnt-antagonist, through aspirin treatment [248]. Taken together, these data suggest that beneficial effects of aspirin on CRC pathogenesis may be mediated, at least in part, by inhibition of Wnt/β-catenin signaling. However, it is unclear to what extent the inhibitory effects of aspirin might be of particular relevance in PIK3CA-mutated CRC in this context. Additional future studies are therefore needed to elucidate this issue.
Cancer stem cells
As discussed in the previous section, Wnt/β-catenin signaling is among the most important factors in CRC carcinogenesis. In addition, it plays an important role in intestinal homeostasis and cancer stem cell (CSC) development, which will be discussed in the following.
The epithelium of the intestine is continually replenished with a turnover rate of approximately 3–4 days [249]. New cells derive from so-called crypts of Lieberkühn which line the intestinal wall [249,250,251]. These crypts harbor pluripotent intestinal stem cells (ISCs) responsible for renewal of the intestinal mucosa and its homeostasis [249, 251], a process highly regulated by the Wnt/β-catenin pathway [231]. It is conceivable that colorectal CSCs derive from ISCs in a series of mutational events leading to a loss of proliferative control mechanisms [251, 252]. Importantly, CSCs could then represent key players in metastasis, therapy resistance, and CRC recurrence [252, 253]. Most cancer therapies, such as radiation and chemotherapeutic drugs, target replication mechanisms of highly proliferating cells, such as cancer cells [252, 253]. CSCs are usually in a quiescent state, thus, evading those therapies [252, 253]. Even after seemingly successful chemo- or radiotherapy, surviving CSCs may lead to tumor recurrence and metastasis [252, 253]. Specifically targeting CSCs is difficult and the objective of current research. The PI3K/Akt/mTOR and related pathways, such as the Notch and Wnt/β-catenin pathways, have come into focus of this research [253]. Chen et al. observed that the dual PI3K/mTOR inhibitor BEZ235 suppresses the proliferation of CSCs [254]. Wang et al. also linked PI3K/Akt signaling to stem cell-like properties and 5-FU resistance in CRC [255]. They suggested that the observed effect is mediated by metastasis‑associated colon cancer 1 (MACC1), which has also been proposed as a biomarker predicting metastasis, poor prognosis, and therapy resistance [255, 256]. Similar observations have also been made in other cancers: in an in vitro model of therapy resistant ovarian cancer, Thakur and Ray found that NF-κB activates TNFα and PIK3CA via a feedback loop leading to the maintenance of stem cell-like characteristics [257]. According to Zhou et al., activation of the PI3K/Akt/mTOR pathway is responsible for stemness in gastric carcinoma cells [258]. In this context, several studies confirm that NANOG, a transcription factor essential for the maintenance of embryonic and CSCs, is regulated by the PI3K/Akt pathway [255, 259,260,261,262]. In CRC, high levels of NANOG are associated with vascularization and more aggressive behavior in general [263, 264]. Indeed, it is possible that CSCs are the target of aspirin’s anti-cancer properties. Chen et al. reported that aspirin (but no other NSAIDs) directly interacts with p300 in the nucleus, promotes H3K9 acetylation, activates FasL expression, and induces apoptosis in patient-derived colorectal CSCs, whereas these effects of aspirin were not observed in non-CSCs most likely because of H3K9 hypermethylation [265]. Interestingly, salicylate and its derivates have also shown to interfere with p300 [266, 267]. Previously, p300 had been reported to be a target of Akt phosphorylation [268]. Wang and colleagues demonstrated that aspirin reduced colorectal xenograft tumor growth in nude mice, an effect that was associated with a reduction in stemness-related transcription factors, such as c-Myc, OCT4, and NANOG. Interestingly, suppression of NANOG was also able to block the anti-tumorigenic effect of aspirin, suggesting that NANOG is an important downstream target of aspirin action in CRC [269]. In esophageal squamous cell carcinoma, aspirin was able to overcome cisplatin resistance in CSCs by inhibiting the phosphorylation of Akt, suggesting an involvement of PI3K. Aspirin most likely acetylated histones leading to altered gene expression, in this case remodeling of chromatin in the region encoding the pro-apoptotic Bcl-2-like protein 11 (BIM) [270]. Both Khoo and Saha suggest that aspirin may overcome chemotherapy resistance in preclinical models of CSCs via interleukin 6 (IL6), a pro-inflammatory cytokine which acts downstream of NF-κB and is regulated by COX-dependent as well as COX-independent mechanisms [271, 272]. Khoo et al. believe the effect to be due to COX-2 inhibition, whereas Saha et al. speculate aspirin to interfere with the IL6-NF-κB-feedback loop [271, 272]. Taken together, the effect of aspirin on CSCs and the involvement of PI3K in their regulation may provide an explanation for how aspirin treatment can overcome treatment resistance and counteract metastasis in PIK3CA-mutated CRC. Nevertheless, studies on the influence of aspirin or its salicylate moiety on CSCs carrying activating somatic mutations of PIK3CA are lacking. This important and interesting field of research is therefore still in its infancy and further research on this topic is to be expected in the near future.
Noncoding RNA
Even though a relevant portion of the genome is transcribed, only 1–2% encode for protein and are translated by the ribosome. Nevertheless, for decades, non-coding RNA (ncRNA) was mainly considered as "transcriptional junk" with no biologically relevant function [273, 274]. With accumulating evidence of involvement of ncRNAs in signal transduction, this perception has rapidly changed [275]. The types of ncRNA to be focused on in this review are microRNA (miRNA), long noncoding RNA (lncRNA), and circular RNA (circRNA). These regulatory RNAs have been observed to play a role in the development of CRC and have, therefore, been proposed as therapeutic targets and diagnostic markers [275,276,277,278]. Although it has been proven that altered ncRNA expression is a central element in the formation of cancerous entities, the underlying regulatory mechanisms are complex and not well explored [275, 277, 279]. Indeed, according to Anastasiadou et al., ncRNAs act as parts of vast and intricate networks [275].
miRNAs are short noncoding RNA strands with an average length of 22 bases, deriving mainly from intronic sequences [280]. Their most prominent function is post-transcriptional gene regulation by base-pairing to the 3’ untranslated region (3’UTR) of messenger RNAs (mRNAs), thus, hindering translation or inducing degradation of their target mRNAs [281]. Most miRNAs can bind to multiple mRNAs, also their expression and actions may differ in a cell-type specific way [280]. In the context of cancer pathogenesis, miRNA expression has been shown to play a crucial role in the regulation of cell cycle progression, growth, and metabolism, and, thereby, carcinogenesis. For instance, deregulated miRNAs can promote tumorigenesis by both inhibiting the transcription of tumor suppressor genes as well as failing to do so in the case of oncogenes [282].
LncRNAs are defined as transcripts with a length of > 200 nucleotides [282]. Apart from not being translated, lncRNAs are believed to be processed in the same manner as mRNAs, including transcription by RNA polymerase II as well as post-transcriptional modification, such as 5’ capping, polyadenylation, and splicing [283]. LncRNAs interact with DNA, mRNA, miRNA, and proteins in a multitude of ways. For instance, they can regulate gene expression by blocking access of transcription factors, polymerases, or miRNA to DNA or RNA, or they can also act in the opposite manner, by guiding transcription factors towards DNA or by modifying chromatin remodeling and accessibility [284].
CircRNAs, as the name indicates, are not linear strands of nucleotides but closed loops without 5’ or 3’ ends [285]. CircRNA is less explored than miRNA and lncRNA, most of what is known has been discovered within the past decade [286]. It has been suggested that circRNA descends from protein-coding regions of the genome and is generated from pre-mRNA in a process called “backsplicing” [285] and some circRNAs may even be translated [287]. So far, it has been discovered that circRNA can act as a molecular sponge for miRNA by presenting complementary binding sites akin to those of mRNA targets [288].
There is proven crosstalk between the PI3K pathway and ncRNA networks [289]. For example, the CRNDE lncRNA transcript is activated via the PI3K/Akt/mTOR and Raf/MAPK pathway by insulin/IGF in colorectal carcinoma cells, resulting in altered metabolism and the induction of the Warburg effect, which describes metabolic changes by which cancer cells switch to anaerobic glycolysis even in the presence of oxygen and fully functioning mitochondria [290]. A paper by Khan and Law further reported that the lncRNA RAMS-11 is significantly overexpressed in various CRC cell lines, a phenomenon that correlates with increased proliferation and metastasis. Moroever, downregulation of RAMS-11 resulted in increased apoptosis by inhibition of Akt/mTOR signaling via AMPK [291]. Multiple studies associate the expression of oncogenic lncRNA HOTAIR with PI3K signaling [290, 292, 293]. HOTAIR may act as a molecular sponge for miRNAs such as miR-34a and mir-206, which are known to interact with the PI3K pathway [294, 295]. In a similar fashion, miR-34a also binds to lncRNA SNHG7 enabling translation of GALNT7 [296]. Li and colleagues were able to show that this competition leads to an increase in PI3K/Akt/mTOR activity and related cell proliferation in CRC cells [296]. MiR-206 inhibits Akt and its downstream target GSK3β via c-Met, resulting in reduced proliferation, migration, and invasion of CRC cells [297]. Overexpression of miR-590-3p desensitizes colon carcinoma cells to radiation by promoting PI3K and Akt phosphorylation [298]. Most notably, El-Daly et al. discovered that miR-370 acts as a tumor suppressor by inhibiting PIK3CA and EGFR mRNA expression through 3’UTR base-pairing in an in vitro and in vivo model of CRC [299]. CircRNAs have shown to influence PI3K/Akt signaling, as well. For instance, circ-0001313 is overexpressed in CRC cells where it induces cell proliferation and inhibition of apoptosis by sponging AKT2 mRNA-directed miR-510-5p [300]. Circ_0008285 acts as a tumor suppressor by enabling PTEN translation, although, however, it appears to be downregulated in CRC cells [301]. Just recently, Chong and colleagues demonstrated that downregulation of circLHFPL2 most likely promotes sustained activation of the PI3K/Akt pathway by acting as a sponge for miR-556-5p and miR-1322 in CRC cells to modulate PTEN expression [302]. These results demonstrate that there are numerous reciprocal interactions between PI3K or PI3K-dependent signal transduction and ncRNA which have been shown to influence the phenotype of CRC entities. However, systematic studies on the impact of activating PIK3CA mutations on the expression of ncRNAs in CRC remain surprisingly scarce.
Results from experimental studies indicate that aspirin exerts anti-carcinogenic properties via ncRNA. Examples include a study by Lan et al., who found aspirin to negatively regulate miR-21 expression through inhibition of TCF4, a transcription factor of the Wnt/β-catenin signaling pathway, in patient-derived colon cancer epithelium [303]. Further, Guo et al. could show the involvement of aspirin in the transcription of lncRNA OLA1P2 in cultured cancer cell lines as well as mouse xenografts. Aspirin, they observed, enabled expression of the transcription factor FOXD3 via demethylation of the FOXD3 promotor, leading to transcription of OLA1P2. In turn, OLA1P2 blocked the nuclear import of phosphorylated STAT3 [304]. STAT3 is a phosphorylation target of mTOR [305]. In a 2021 study, aspirin treatment of colon carcinoma cell lines lead to inhibition of lncRNAs NEAT1 and LOC152578 resulting in reduced cell growth and metastasis [306]. Although ncRNAs have now come into focus with regard to aspirin-mediated inhibitory effects on CRC pathogenesis, systematic studies on the role of ncRNAs in aspirin sensitivity of PIK3CA-mutated CRC are scarce to date. Nevertheless, ncRNA may play a significant role in mediating the aspirin-induced effects associated with PIK3CA-mutated CRC. Therefore, it is expected that future research projects will soon shed light on this hitherto poorly studied issue.
Gut microbiota
The gastrointestinal microbiome plays an important role in inflammatory processes and the regulation of gut homeostasis, closely communicating with ISCs [307, 308]. Gut microbiota are believed to be involved in carcinogenesis [309]. While some infectious pathogens may drive cancer development, an imbalance in the microbial composition is often found in CRC patients compared to healthy controls [309,310,311]. Escherichia coli, Streptococcus gallolyticus/bovis, Bacteroides fragilis and Fusobacterium nucleatum, amongst others, have been linked to the development of CRC. [310, 312,313,314,315,316] In addition a pathogenic role of Enterococcus faecalis in this regard has been discussed but remains controversial [317]. One Factor which may contribute to dysbiosis is nutrition, especially a diet high in fat and low in fiber [309, 310, 313, 318]. There is evidence that the intake of aspirin affects the gut microbiome [319, 320]. In an observational study with healthy volunteers, Rogers and Aronoff noticed that the profile of the gut microbiome of NSAID users differed from that of non-users. In this study, the operational taxonomic units of bacterial species Prevotella, Bacteroides, Barnesiella, and the Ruminococcaceae family distinguished aspirin users from those abstaining from any kind of medication [320]. This is consistent with a later study by Prizment et al. who measured the relative abundance of multiple gut bacterial taxa before and after a six week treatment with 325 mg aspirin or placebo. They found relative increases in Akkermansia, Prevotella, and Ruminococcaceae as well as decreases in Parabacteroides, Bacteroides, and Dorea in the aspirin group compared to the placebo group [321]. In this context, the presence of Prevotella and Ruminococcaceae have been shown to negatively correlate with CRC, whilst the latter three have been positively correlated with the disease [322, 323]. In the case of Akkermansia there is conflicting evidence whether it is considered a beneficial microbe, especially with regard to inflammation [324, 325]. Zhao and colleagues observed aspirin treatment to induce a shift in microbiomal composition. Bacterial genera considered beneficial, such as Bifidobacterium and Lactobacillus, were enriched, while bacteria associated with CRC, like Alistipes finegoldii and B. fragilis, were reduced. In addition, they confirmed an inverse effect of aspirin treatment on tumor size and number in mouse xenografts, albeit only in animals treated with antibiotics. Importantly, some microbes, especially Lysinibacillus sphaericus, were found to degrade aspirin resulting in lower plasma levels [319]. Caitlin Brennan et al. found that aspirin and its metabolite salicylic acid alter the expression of several genes in the F. nucleatum strain Fn7-1 in culture, including genes encoding for chaperones, proteins related to the Wnt/β-catenin pathway, and ptgs2, the gene encoding for COX-2. At higher concentrations, aspirin also inhibited growth of the mentioned F. nucleatum strain Fn7-1. Further, they observed that aspirin reduced F. nucleatum-related growth of colorectal adenomas in mice. F. nucleatum strains isolated from human CRC tissues yielded consistent results. Additionally, aspirin treatment asserted significant antibacterial effects on B. fragilis and E. coli strains similar to those seen with F. nucleatum, although somewhat milder [326]. Interestingly, Han et al. report in a pre-print that infection of Caco-2 and HT-29 CRC cells lines with F. nucleatum leads to resistance against cetuximab in vitro and in mouse xenografts coinciding with an activation of the PI3K/Akt and JAK/STAT3 pathways in infected cells [327]. However, to our knowledge, a link between this and the sensitivity of gut bacteria towards aspirin has not yet been investigated, but it might be worth keeping in mind. Thus, to our knowledge, there is no evidence (but also no counterevidence) to date that aspirin sensitivity of PIK3CA-mutated CRC is related to the microbiome. Nevertheless, the colon microbial milieu plays an important role in intestinal epithelial homeostasis and carcinogenesis, so this must be taken into account when elucidating the specific pharmacodynamics of aspirin in PIK3CA-mutated CRC in future studies.
Tumor metabolic reprogramming
Changes in metabolism stand at the heart of tumorigenesis and are now considered a hallmark of cancer [328]. Cancer cells have a high demand for nutrients and energy to enable unhindered growth and proliferation, a need that cannot be met by the mechanisms utilized by “ordinary”, non-malignant cells [329]. Otto Warburg already observed metabolic reprogramming of cancer cells nearly a century ago. The Warburg effect states that tumor cells perform anaerobic glycolysis despite an adequate supply of oxygen [330]. This kind of metabolism might be favorable for highly proliferative cells by directing metabolites towards biomass production instead of generating ATP [331,332,333,334]. A heightened need for glutamine is also considered a hallmark of cancer metabolism [329]. In the cell, it can be converted to α-ketoglutarate to enter the TCA cycle and not only serves as an energy source, but also yields carbon and nitrogen for biosynthesis [329, 334, 335]. It is likely that metabolic shifts precede somatic mutations in CRC and promote the development of benign adenomas into carcinomas [334]. As metabolic changes are very common in cancer, they can be used for diagnosis and prognosis of disease. Imaging techniques like positron emission tomography (PET) scans are a frequently used option. CRC and other cancers can be identified through increased glucose metabolism, an indicator of the Warburg effect, by detecting positron emission of the glucose analog [18F]fluorodeoxyglucose after injection [336]. In addition, PET scan techniques detecting 11C-glutamine are currently being established [337]. Serum glutamine levels may also be used as diagnostic and prognostic marker. For instance, in a retrospective study of 123 newly diagnosed CRC patients Ling and colleagues found decreased glutamine levels to correlate with lower overall and progression-free survival [338].
The PI3K/Akt pathway plays a major role in the regulation of metabolism as well as metabolic reprogramming of cancer cells [61]. Phosphorylation of the glycolysis enzyme hexokinase 2 (HK2) by Akt results in increased proliferation, tumorigenesis, and metastasis of colon cancer mediated by NF-κB and hypoxia inducible factor 1α (HIF-1 α) both in vitro and in vivo in mouse xenograft tumors [339]. PI3K/Akt-mediated HK2 signaling has also shown to inhibit apoptosis in pediatric osteosarcoma [340]. In cervical carcinoma, PIK3CA mutations E545K and E542K lead to increased glucose metabolism and cell proliferation via AKT/GSK3β/β-catenin signaling in xenografts and in patient-derived samples [341]. Laboratory experiments as well as clinical trials have shown that PIK3CA-mutated CRC cells are especially dependent on glutamine for survival compared to PIK3CA wildtype cells [67, 342,343,344]. This might be related to the fact that the expression of glutamine transporter ASCT2 (also known as solute carrier family 1, member 5 (SLC1A5)) is regulated by PI3K-dependent mTOR signaling [345].
In recent years, glucose and glutamine metabolism have come into focus as potential mediators of aspirin’s effect on cancer. Further, there is substantial evidence that it targets the deregulated metabolism of cancer cells in a PI3K-dependent manner. For example, aspirin has shown to inhibit the enzyme glucose-6-phosphate dehydrogenase (G6PD) responsible for NADH production in the pentose phosphate pathway (PPP) by acetylating lysine residues in HCT116 and HT-29 CRC cells [346, 347]. G6PD is regulated by PI3K/Akt as a downstream target of mTORC1 [348,349,350]. Chen et al. found that PI3K activity decouples glycolysis and the TCA cycle, while promoting PPP through G6PD activation [347]. Further, aspirin downregulated glutamine and glucose levels as well as inflammation and tumor growth in a study of lung cancer in obese mice, effects that were associated with Akt phosphorylation and GLUT1 expression [351]. Experiments concerning a murine model of ulcerative colitis, a kind of inflammatory bowel disease, yielded that both glutamine and 5-aminosalicylic acid (5-ASA) were able to alleviate symptoms caused by oxidative stress-injury induced through inhibition of the PI3K/Akt signaling pathway [352]. According to a paper published by Hao et al. in 2016 PIK3CA mutations lead to an upregulation of glutamate pyruvate transaminase 2 (GPT2) in CRC cells via ATF4 (activating transcription factor 4)/PDK1/RSK2 (ribosomal S6 kinase 2) in an AKT-independent manner ultimately rendering the cells dependent on glutamine [67]. In 2020, Shogen Boku and colleagues could prove that administration of aspirin led to the same effect as glutamine deprivation in PIK3CA-mutated CRC cell lines. It did so by two mechanisms: G1-arrest was induced via the mTOR-pathway and glutaminolysis enzymes w-ere activated via ATF4 [68]. Taken together, one of the most effective methods of preventing tumor growth appears to be to deprive cancer cells of nutrients. The evidence for the effect of aspirin on deregulated metabolism in PIK3CA-mutated cancer is considerable. However, no clear metabolic targets have yet been identified in the aforementioned publications to explain the aspirin sensitivity of PIK3CA-mutant CRC, so additional research is needed here to elucidate the precise mechanistic background of this phenomenon.
Summary and conclusion
A vast number of cancer patients harbor PIK3CA mutations. Unfortunately, the prognosis of these patients is dismal in many cases, and the efficacy of standard treatments is often reduced. This leads to an urgent need of alternative treatments for these patients. Genetic profiling and subsequent targeted therapy may be a promising perspective for these individuals. PI3K and Akt inhibitors have shown much potential, aspirin, however, is low in cost and easily available. Nonetheless, the U.S. Preventive Services Task Force is cautious to universally recommend low-dose aspirin (< 100 mg/day) as preventative treatment against CRC due to side-effects, such as gastrointestinal bleeding and hemorrhagic stroke [28].
Moreover and importantly, the balance between harm and benefit is yet unclear. Thus, additional evidence is needed to elucidate the mechanisms behind the preventive and curative effects of aspirin with regard to colorectal carcinoma in order to identify patients who particularly benefit from aspirin therapy.
Although the effect of aspirin on the prostaglandin pathway is one of the best-researched drug mechanisms so far, it is still unclear how aspirin is linked to beneficial effects in individuals carrying somatic PIK3CA mutations. Aspirin is hydrolyzed in the body into acetyl and salicylic acid within a short time, making it two pharmacologically active compounds in one drug. The acetylation of molecules is nonspecific and is related to the concentration of aspirin at the site of action, which is particularly high in the gastrointestinal tract after peroral ingestion of the drug, although to our knowledge there are no available pharmacokinetic data on how high the luminal concentration of perorally administered aspirin at the colorectal epithelium really is. However, based on such data, specific retardations of aspirin with dissolution in the colorectal intestinal segments could be developed, allowing higher luminal concentrations of aspirin at the colorectal epithelium and thereby possibly enhancing the anticancer effects, while potentially reducing the known gastroduodenal toxicity of the drug.
The aspirin targets COX-1 and 2 are associated with the upstream PI3K signaling axis, but because these enzymes are also inhibited by other NSAIDs, it is difficult to explain why the clinical effect of aspirin is specific to the drug, particularly in the context of prevention/treatment of PIK3CA-mutated CRC. Yet, increased generation of 15-(R)-HETE and the formation of aspirin-triggered lipoxins and resolvins distinguishes the pharmacodynamics of aspirin from that of other NSAIDs and selective COX-2 inhibitors. Moreover, as PIK3CA mutations, especially of the helical domain, render the enzyme independent of upstream RTK activation signals, it seems likely that the target responsible for its inhibition must be sought downstream. However, if the effect is prostaglandin-related, it is more likely connected to inhibition of cyclooxygenase expression mediated by hyperactive PI3K than to classical COX-acetylation. Nevertheless, further studies are required to decipher the importance of both COX isoforms in the context of PIK3CA-mutated malignancies and the effect of aspirin thereon.
But is there only one right answer? As such, the expected multiple interactions of aspirin and its metabolite salicylate with various complex networks of signaling pathways involving PI3K imply that numerous interactions must be considered to unravel the mechanisms of aspirin-mediated inhibition of PIK3CA-mutated cancer. The putative major signaling pathways responsible for carcinogenesis of PIK3CA-mutated CRC and the interactions of aspirin with them are shown in Fig. 4. For example, inhibitory effects of aspirin on signaling networks associated with the PI3K/Akt/mTOR pathway with known oncogenic potential, such as the NF-κB, Wnt/β-catenin, and Ras/Raf/MEK/ERK pathways (the latter shown in Fig. 2) have to be mentioned. In addition, new players have entered the field, adding to the level of complexity. For instance, the influence of non-coding RNAs, metabolism, gut microbiome, and CSCs on the development of CRC, as well as the reciprocal interactions of these factors with aspirin and PIK3CA mutations in the context of CRC, has long been overlooked and show promising potential in providing answers and novel treatment targets. Last, PIK3CA-independent actions of aspirin may contribute synergistically, for instance by inhibiting inflammation. Taken together, the sensitivity of PIK3CA-mutated cancers to aspirin has gained much attention, especially in recent years. Nevertheless, the key mechanisms of this effect are still unknown. However, deciphering the underlying mechanisms is of great importance, in particular with regard to identifying patients who will benefit from treatment or chemoprevention with aspirin. Further interesting research on this topic can, therefore, be expected in the coming years.

Potential key targets of aspirin in PIK3CA-mutated CRC. Mutations of the PIK3CA helical and kinase domain lead to an overactivity of PI3K (visualized by multiple arrows). Helical domain mutations decouple the enzyme from upstream signals by RTK, indicated by the red “X”, although it has to be noted that Ras-GTP activation by external signals may still be required for full PI3K activation. Kinase domain mutations are, however, independent from Ras-GTP activation. PI3K activates Akt and PDK1. mTOR is both an activator of Akt and a downstream target and, therefore, closely linked to its actions. mTOR has been identified as a direct acetylation target of aspirin (ASA), and is presumably inhibited by this. Akt is the central player connecting most pathways, however, glutaminolysis may be influenced by PDK1 Akt-independently. Aspirin has been reported to inhibit PDK1-mediated glutaminolysis by an, as yet, unresolved mechanism. Further, salicylate and aspirin have been observed to suppress NF-κB via IκB, thereby reducing chemoresistance in cancer cells. NF-κB is also involved in COX-2 expression. COX-2 has been reported to act upstream of PI3K via PGE2/EP4 and to modify the activity of growth factor receptors (e.g. IGFR or EGFR), inducing its own expression via a feedback loop. It is acetylated by aspirin at S516 which disables the production of PGH2, but enables the formation of aspirin-triggered lipoxin (AT-L) and resolvins (AT-Rv) in a transcellular fashion. In addition, salicylate may suppress the transcription of COX-2 mRNA. In addition, aspirin has been reported to inhibit Wnt/β-catenin-mediated signaling, although the exact target of aspirin in this context is unclear. Wnt/β-catenin are also substantially involved in the pathophysiology of cancer stem cells, which are highly associated with therapy-resistance and metastasis. Stemness of these cells may also be inhibited by aspirin via p300 and NANOG. NF-κB may also be involved. Gut bacteria are involved in inflammatory processes and the maintenance of intestinal homeostasis. In this context, it has been observed that aspirin can influence the growth of intestinal bacteria, possibly leading to a shift in the intestinal microbiome towards a more favorable composition. The named pathways and their potential interconnections are shown. Arrows illustrate the connections between them, arrow thickness is related to their hypothetical importance in the carcinogenesis of the PI3K pathway. The microbiome, inflammation, as well as AT-L and AT-Rv formation are depicted, despite not being directly involved in PI3K signaling. However, as they are influenced by aspirin, they might enhance the anticarcinogenic effect seen in patients with PIK3CA-mutated CRC
Data availability
Data sharing is not applicable to this article because no new data were created or analyzed in this study.
Abbreviations
- 4E-BP1:
-
Eukaryotic translation initiation factor 4E-binding protein 1
- 5-FU:
-
5-Fluorouracil
- ABD:
-
Adaptor binding domain
- AMPK:
-
Adenosine monophosphate–activated protein kinase
- APC:
-
Adenomatous polyposis coli
- ASA:
-
Acetylsalicylic acid
- ASCT2:
-
Alanine, serine, cysteine transporter 2, Neutral amino acid transporter B(0) (= SLC1A5)
- ATF4:
-
Activating transcription factor 4
- AT-L:
-
Aspirin-triggered lipoxins
- ATP:
-
Adenosine triphosphate
- AT-Rv:
-
Aspirin-triggered resolvins
- BAD:
-
Bcl-2 associated death promoter
- BH domain:
-
Breakpoint-cluster region homology domain (= RhoGAP domain)
- BIM:
-
Bcl-2-like protein
- circRNA:
-
Circular RNA
- COX:
-
Cyclooxygenases
- CRC:
-
Colorectal cancer
- CSC:
-
Cancer stem cell
- DVL:
-
Dishevelled
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- eIF4E:
-
Eukaryotic translation initiation factor 4E
- EMT:
-
Epithelial-mesenchymal transition
- eNOS:
-
Endothelial nitric oxide synthase (= NOS3)
- ERK:
-
Extracellular signal-regulated kinase
- FOXO:
-
Forkhead box protein O
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GLUT1:
-
Glucose transporter 1
- GPT2:
-
Glutamate pyruvate transaminase 2
- GSK3:
-
Glycogen synthase kinase 3
- HETE:
-
Hydroxyeicosatetraenoic acid
- HIF-1α:
-
Hypoxia-inducible factor 1 α
- HK2:
-
Hexokinase 2
- I/n/cSH2:
-
Inter/N-terminal/C-terminal Src homology 2 domain
- IFN-γ:
-
Interferon-γ
- IGF:
-
Insulin-like growth factor
- IGFR:
-
Insulin-like growth factor 1 receptor
- IκB:
-
IκB protein
- IκBα:
-
NFKB inhibitor alpha
- IKK-β:
-
IκB kinase-beta
- ILβ / 6:
-
Interleukin 1β / 6
- iNOS:
-
Inducible nitric oxide synthase (= NOS2)
- ISC:
-
Intestinal stem cell
- KRAS:
-
Kirsten rat sarcoma virus
- lncRNA:
-
Long noncoding RNA
- LPS:
-
Lipopolysaccharide
- mCRC:
-
Metastatic colorectal cancer
- MACC1:
-
Metastasis‑associated colon cancer 1
- MEK:
-
Mitogen-activated protein kinase kinase
- miRNA:
-
Micro RNA
- mRNA:
-
Messenger RNA
- mTOR:
-
Mechanistic target of rapamycin kinase
- mTORC1 / 2:
-
Mechanistic target of rapamycin complex 1 / 2
- ncRNA:
-
Noncoding RNA
- NF-κB:
-
Nuclear factor κ B
- NO:
-
Nitric oxide
- NOS2:
-
Nitric oxide synthase 2 (= iNOS)
- NSAID:
-
Non-steroidal anti-inflammatory drug
- PDK1:
-
Phosphoinositide-dependent kinase 1
- PET:
-
Positron emission tomography
- PGE2 :
-
Prostaglandin E2
- PGF2 α :
-
Prostaglandin F2α
- PGH2 :
-
Prostaglandin H2
- PGHS:
-
Prostaglandin G/H synthase (= COX)
- PGI2 :
-
Prostacyclin
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIP2 :
-
Phosphatidylinositol 4,5-bisphosphate
- PIP3 :
-
Phosphatidylinositol 3,4,5-trisphosphate
- PKB:
-
Protein kinase B (= Akt)
- PP2A:
-
Protein phosphatase 2A
- PPP:
-
Pentose phosphate pathway
- PtdIns(3,4,5)P3 :
-
Phosphatidylinositol 3,4,5-trisphosphate (= PIP3)
- PtdIns(4,5)P2 :
-
Phosphatidylinositol 4,5-bisphosphate (= PIP2)
- PTEN:
-
Phosphatase and tensin homolog
- PTGIS:
-
Prostacyclin synthase
- PTGS:
-
Prostaglandin-endoperoxide synthase (= COX)
- Ras:
-
Rat sarcoma viral oncogene
- RBD:
-
Ras-binding domain
- RHEB:
-
Ras homolog enriched in brain
- RhoGAP:
-
Rho-GTPase-activating protein domain (= BH domain)
- RSK2:
-
Ribosomal S6 kinase 2
- RTK:
-
Receptor tyrosine kinase
- S6K:
-
Ribosomal protein S6 kinase
- SGK1:
-
Serum and glucocorticoid-regulated kinase 1
- SH3:
-
Src homology 3 domain
- SLC1A5:
-
Solute carrier family 1 member 5 (= ASCT2), Neutral amino acid transporter B(0)
- TBXAS1:
-
Thromboxane A synthase 1
- TNFα:
-
Tumor necrosis factor α
- TCA cycle:
-
Tricarboxylic acid cycle
- TP53:
-
Tumor protein 53
- TSC1/2:
-
Tuberous sclerosis 1/2
- TxA2 :
-
Thromboxane A2
- UTR:
-
Untranslated region
- VEGF-A:
-
Vascular endothelial growth factor A
References
Sung H, Ferlay J, Siegel RL et al (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249
Radtke R. Todesfälle aufgrund von Krebs in Deutschland bis 2019. https://de.statista.com/statistik/daten/studie/172573/umfrage/krebstote-in-deutschland/ (accessed 10/28/2021)
Ferlay J, Ervik M, Lam F, Colombet M, Mery L, Piñeros M et al. Global cancer observatory: cancer today. https://gco.iarc.fr/today (accessed 05/17/2022)
Barnes B, Bertz J, Buttmann-Schweiger N, Fiebig J, Jordan S, Kraywinkel K, Niemann H, Nowossadeck E, Poethko-Müller C, Prütz F, Rattay P, Schönfeld I, Starker A, Wienecke A, Wolf U (2016) Bericht zum Krebsgeschehen in Deutschland 2016. Robert Koch-Institut (RKI)
Vogelstein B, Fearon ER, Hamilton SR et al (1988) Genetic alterations during colorectal-tumor development. N Engl J Med 319:525–532
Smit WL, Spaan CN, Johannes de Boer R et al (2020) Driver mutations of the adenoma-carcinoma sequence govern the intestinal epithelial global translational capacity. Proc Natl Acad Sci 117:25560–25570
Rawla P, Sunkara T, Barsouk A (2019) Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Przeglad gastroenterologiczny 14:89–103
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7:606–619
Brown KK, Toker A (2015) The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000prime Rep 7:13
Barault L, Veyrie N, Jooste V et al (2008) Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer 122:2255–2259
Ogino S, Nosho K, Kirkner GJ et al (2009) PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol 27:1477–1484
Rosty C, Young JP, Walsh MD et al (2013) PIK3CA activating mutation in colorectal carcinoma: associations with molecular features and survival. PLoS One 8:e65479
Malinowsky K, Nitsche U, Janssen K-P et al (2014) Activation of the PI3K/AKT pathway correlates with prognosis in stage II colon cancer. Br J Cancer 110:2081–2089
Perrone F, Lampis A, Orsenigo M et al (2009) PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol 20:84–90
Jhawer M, Goel S, Wilson AJ et al (2008) PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Can Res 68:1953–1961
Wang Q, Shi Y-L, Zhou K et al (2018) PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis 9:739
Fariña Sarasqueta A, Zeestraten ECM, van Wezel T et al (2011) PIK3CA kinase domain mutation identifies a subgroup of stage III colon cancer patients with poor prognosis. Cell Oncol (Dordr) 34:523–531
Sartore-Bianchi A, Martini M, Molinari F et al (2009) PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Can Res 69:1851–1857
National Cancer Institute, Genomic Data Commons Data Portal, National Institute of Health. Distribution of most frequently mutated genes. https://portal.gdc.cancer.gov/exploration?canDistTable_size=60&filters=%7B%22op%22%3A%22and%22%2C%22content%22%3A%5B%7B%22op%22%3A%22in%22%2C%22content%22%3A%7B%22field%22%3A%22genes.is_cancer_gene_census%22%2C%22value%22%3A%5B%22true%22%5D%7D%7D%5D%7D&searchTableTab=genes (accessed 10/11/2021)
Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV (2005) The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene 24:7482–7492
Din FVN, Theodoratou E, Farrington SM et al (2010) Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut 59:1670–1679
Dovizio M, Bruno A, Tacconelli S, Patrignani P (2013) Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res 191:39–65
Elwood PC, Morgan G, Pickering JE et al (2016) Aspirin in the treatment of cancer: reductions in metastatic spread and in mortality: a systematic review and meta-analyses of published studies. PLoS One 11:e0152402
Friis S, Riis AH, Erichsen R, Baron JA, Sørensen HT (2015) Low-dose aspirin or nonsteroidal anti-inflammatory drug use and colorectal cancer risk: a population-based, Case-Control Study. Ann Internal Med 163:347–355
Baron JA, Cole BF, Sandler RS et al (2003) A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med 348:891–899
Bastiaannet E, Sampieri K, Dekkers OM et al (2012) Use of aspirin postdiagnosis improves survival for colon cancer patients. Br J Cancer 106:1564–1570
Li P, Wu H, Zhang H et al (2015) Aspirin use after diagnosis but not prediagnosis improves established colorectal cancer survival: a meta-analysis. Gut 64:1419–1425
Bibbins-Domingo K (2016) Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. preventive services task force recommendation statement. Ann Internal Med 164:836–845
Dehmer SP, Maciosek MV, Flottemesch TJ, LaFrance AB, Whitlock EP (2016) Aspirin for the primary prevention of cardiovascular disease and colorectal cancer: a decision analysis for the U.S. preventive services task force. Ann Internal Med 164:777–786
Liao X, Lochhead P, Nishihara R et al (2012) Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 367:1596–1606
Hedberg ML, Peyser ND, Bauman JE et al (2019) Use of nonsteroidal anti-inflammatory drugs predicts improved patient survival for PIK3CA-altered head and neck cancer. J Exp Med 216:419–427
Gu M, Nishihara R, Chen Y et al (2017) Aspirin exerts high anti-cancer activity in PIK3CA-mutant colon cancer cells. Oncotarget 8:87379–87389
Zumwalt TJ, Wodarz D, Komarova NL et al (2017) Aspirin-induced chemoprevention and response kinetics are enhanced by PIK3CA mutations in colorectal cancer cells. Cancer Prevent Res (Philadelphia, Pa.) 10:208–218
Domingo E, Church DN, Sieber O et al (2013) Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J Clin Oncol 31:4297–4305
Sheng H, Shao J, Townsend CM, Evers BM (2003) Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut 52:1472–1478
Hanahan D, Weinberg RA (2000) The Hallmarks of Cancer. Cell 100:57–70
Whitman M, Downes CP, Keeler M, Keller T, Cantley L (1988) Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332:644–6666
Otsu M, Hiles F, Gout I, Fry MJ, Ruiz-Larrea F, Panayotou G, Thompson A, Dhand R, Hsuan J, Toffy N, Smith AD, Morgan SJ, Courtneidge SA, Parker PJ, Waterfleld MD (1991) Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60 cs’c complexes, and PI3-kinase. Cell 65:91–104
Longenecker KL, Zhang B, Derewenda U et al (2000) Structure of the BH domain from graf and its implications for Rho GTPase recognition. J Biol Chem 275:38605–38610
Liu S, Knapp S, Ahmed AA (2014) The structural basis of PI3K cancer mutations: from mechanism to therapy. Can Res 74:641–646
Gabelli SB, Vogelstein B, Miller MS, Amzel LM (2014) Crystal Structure of p110α in complex with niSH2 of p85α
Luttrell LM, Della Rocca GJ, van Biesen T, Luttrell DK, Lefkowitz RJ (1997) Gbetagamma subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G protein-coupled receptor-mediated Ras activation. J Biol Chem 272:4637–4644
Jimenez C, Hernandez C, Pimentel B, Carrera AC (2002) The p85 regulatory subunit controls sequential activation of phosphoinositide 3-kinase by Tyr kinases and Ras. J Biol Chem 277:41556–41562
Castellano E, Downward J (2011) RAS interaction with PI3K: more than just another effector pathway. Genes Cancer 2:261–274
Zhang M, Jang H, Nussinov R (2019) The mechanism of PI3Kα activation at the atomic level. Chem Sci 10:3671–3680
Zhao L, Vogt PK (2008) Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci 105:2652–2657
Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM (1998) Regulation of the p85/p110 phosphatidylinositol 3’-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol 18:1379–1387
Zhang M, Jang H, Nussinov R (2019) The structural basis for Ras activation of PI3Kα lipid kinase. Phys Chem Chem Phys 21:12021–12028
Castel P, Ellis H, Bago R et al (2016) PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell 30:229–242
Gagliardi PA, Puliafito A, Primo L (2018) PDK1: at the crossroad of cancer signaling pathways. Semin Cancer Biol 48:27–35
Wendel H-G, de Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW (2004) Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 428:332–337
Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169:381–405
Hornsveld M, Dansen TB, Derksen PW, Burgering BMT (2018) Re-evaluating the role of FOXOs in cancer. Semin Cancer Biol 50:90–100
McCubrey JA, Steelman LS, Chappell WH et al (2012) Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 3:954–987
Jeong W-J, Ro EJ, Choi K-Y (2018) Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. NPJ Precis Oncol 2:5
Pandurangan AK (2013) Potential targets for prevention of colorectal cancer: a focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pacific J Cancer Prevent 14:2201–2205
Dong-xu W, Jia L, Su-juan Z (2015) MicroRNA-185 is a novel tumor suppressor by negatively modulating the Wnt/β-catenin pathway in human colorectal cancer. Indian J Cancer 52(Suppl 3):E182–E185
Duda P, Akula SM, Abrams SL et al (2020) Targeting GSK3 and associated signaling pathways involved in cancer. Cells 9(5):1110
Kashyap T, Pramanik KK, Nath N et al (2018) Crosstalk between Raf-MEK-ERK and PI3K-Akt-GSK3β signaling networks promotes chemoresistance, invasion/migration and stemness via expression of CD44 variants (v4 and v6) in oral cancer. Oral Oncol 86:234–243
Villegas SN, Gombos R, García-López L et al (2018) PI3K/Akt cooperates with oncogenic notch by inducing nitric oxide-dependent inflammation. Cell Rep 22:2541–2549
Lien EC, Lyssiotis CA, Cantley LC (2016) Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. Recent Results Cancer Res 207:39–72
Laissue P (2019) The forkhead-box family of transcription factors: key molecular players in colorectal cancer pathogenesis. Mol Cancer 18:5
VanderWeele DJ, Zhou R, Rudin CM (2004) Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther 3:1605–1613
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27:5497–5510
Alzahrani AS (2019) PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol 59:125–132
Asati V, Mahapatra DK, Bharti SK (2016) PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur J Med Chem 109:314–341
Hao Y, Samuels Y, Li Q et al (2016) Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat Commun 7:11971
Boku S, Watanabe M, Sukeno M et al (2020) Deactivation of glutaminolysis sensitizes PIK3CA-mutated colorectal cancer cells to aspirin-induced growth inhibition. Cancers 12(5):1097
Bai D, Ueno L, Vogt PK (2009) Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int J Cancer 125:2863–2870
Datta SR, Dudek H, Tao X et al (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241
Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C (2003) PI3K/Akt and apoptosis: size matters. Oncogene 22:8983–8998
Xie X, Hu H, Tong X et al (2018) The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168. Nat Cell Biol 20:320–331
Kandoth C, Mclellan MD, Vandin F et al (2013) Mutational landscape and significance across 12 major cancer types. Nature 502(7471):333–338
Lee JW, Soung YH, Kim SY et al (2005) PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 24:1477–1480
Samuels Y, Wang Z, Bardelli A et al (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science (New York, N.Y.) 304:554
Bachman KE, Argani P, Samuels Y et al (2004) The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther 3:772–775
Saal LH, Holm K, Maurer M et al (2005) PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res 65:2554–2559
Cizkova M, Susini A, Vacher S et al (2012) PIK3CA mutation impact on survival in breast cancer patients and in ERα, PR and ERBB2-based subgroups. Breast Cancer Res 14:R28
Campbell IG, Russell SE, Choong DYH et al (2004) Mutation of the PIK3CA gene in ovarian and breast cancer. Can Res 64:7678–7681
Chen L, Yang L, Yao L et al (2018) Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat Commun 9:1357
García-Escudero R, Segrelles C, Dueñas M et al (2018) Overexpression of PIK3CA in head and neck squamous cell carcinoma is associated with poor outcome and activation of the YAP pathway. Oral Oncol 79:55–63
Levine DA, Bogomolniy F, Yee CJ et al (2005) Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res 11:2875–2878
Ng PK-S, Li J, Jeong KJ et al (2018) Systematic functional annotation of somatic mutations in cancer. Cancer Cell 33:450–462 (e10)
Samuels Y, Diaz LA, Schmidt-Kittler O et al (2005) Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 7:561–573
Bader AG, Kang S, Vogt PK (2006) Bader 2006 PIK3CA mutations oncogenic in vivo// Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci USA 103:1475–1479
Gymnopoulos M, Elsliger M-A, Vogt PK (2007) Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci USA 104:5569–5574
Leontiadou H, Galdadas I, Athanasiou C, Cournia Z (2018) Insights into the mechanism of the PIK3CA E545K activating mutation using MD simulations. Sci Rep 8:15544
Mandelker D, Gabelli SB, Schmidt-Kittler O et al (2009) A frequent kinase domain mutation that changes the interaction between PI3Kα and the membrane. Proc Natl Acad Sci 106:16996–17001
Burke JE, Perisic O, Masson GR, Vadas O, Williams RL (2012) Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). Proc Natl Acad Sci 109:15259–15264
Gkeka P, Evangelidis T, Pavlaki M et al (2014) Investigating the structure and dynamics of the PIK3CA wild-type and H1047R oncogenic mutant. PLoS Comput Biol 10:e1003895
Papadatos-Pastos D, Rabbie R, Ross P, Sarker D (2015) The role of the PI3K pathway in colorectal cancer. Crit Rev Oncol Hematol 94:18–30
Alqahtani A, Ayesh HSK, Halawani H (2019) PIK3CA gene mutations in solid malignancies: association with clinicopathological parameters and prognosis. Cancers 12(1):93
Reggiani Bonetti L, Barresi V, Maiorana A, Manfredini S, Caprera C, Bettelli S (2018) Clinical impact and prognostic role of KRAS/BRAF/PIK3CA mutations in stage I colorectal cancer. Dis Markers 2018:2959801
Aoife Carr (2021) Targeting the phosphatidylinositol-3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways to enhance chemoradiotherapy responsiveness in colorectal cancer
Kim JS, Kim JE, Kim K et al (2017) The impact of cetuximab Plus AKT- or mTOR- inhibitor in a patient-derived colon cancer cell model with wild-type RAS and PIK3CA mutation. J Cancer 8:2713–2719
Morii Y, Tsubaki M, Takeda T et al (2021) Perifosine enhances the potential antitumor effect of 5-fluorourasil and oxaliplatin in colon cancer cells harboring the PIK3CA mutation. Eur J Pharmacol 898:173957
Narayan P, Prowell TM, Gao JJ et al (2020) FDA approval summary: alpelisib plus fulvestrant for patients with HR-positive, HER2-negative, PIK3CA-mutated, advanced or metastatic breast cancer. Clin Cancer Res 27(7):1842–1849
André F, Ciruelos E, Rubovszky G et al (2019) Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med 380:1929–1940
Tabernero J, van Geel R, Guren TK et al (2016) Phase 2 results: encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF- mutant colorectal cancer (BRAFm CRC). J Clin Oncol 34:3544
Aslam R, Toomey S, Hennessy B (2021) P-287 Preclinical evaluation of alpelisib (PI3K inhibitor) and its synergistic effect in combination with ribociclib (CDK 4/6 inhibitor) in colorectal cancer. Ann Oncol 32:S194–S195
Lee S. Alpelisib and Capecitabine in patients with PIK3CA Mutant mCRC patients (ALCAP). https://clinicaltrials.gov/ct2/show/study/NCT04753203 (accessed 04/26/2022)
Tabernero J. MEN1611 with cetuximab in metastatic colorectal cancer (C-PRECISE-01). https://clinicaltrials.gov/ct2/show/NCT04495621
Desborough MJR, Keeling DM (2017) The aspirin story—from willow to wonder drug. Br J Haematol 177:674–683
Wood JN (2015) From plant extract to molecular panacea: a commentary on Stone (1763) 'An account of the success of the bark of the willow in the cure of the agues'. Philos Trans R Soc Lond B Biol Sci 370(1666):20140317
World Health Organization (1978) WHO Expert Committee on the Selection of Essential Drugs, 2nd edn. The selection of essential drugs, Vol 1. Geneva: WHO
Weiss H, Aledort L (1967) IMPAIRED PLATELET/CONNECTIVE-TISSUE REACTION IN MAN AFTER ASPIRIN INGESTION. The Lancet 290:495–497
Schrör K (2016) Acetylsalicylic acid. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Silagy CA, McNeil JJ, Donnan GA, Tonkin AM, Worsam B, Campion K (1993) Adverse effects of low-dose aspirin in a healthy elderly population. Clin Pharmacol Ther 54:84–89
Huang W-Y, Saver JL, Wu Y-L, Lin C-J, Lee M, Ovbiagele B (2019) Frequency of intracranial hemorrhage with low-dose aspirin in individuals without symptomatic cardiovascular disease: a systematic review and meta-analysis. JAMA Neurol 76:906–914
Cryer B, Mahaffey KW (2014) Gastrointestinal ulcers, role of aspirin, and clinical outcomes: pathobiology, diagnosis, and treatment. J Multidiscip Healthc 7:137–146
Kawamura N, Ito Y, Sasaki M et al (2013) Low-dose aspirin-associated upper gastric and duodenal ulcers in Japanese patients with no previous history of peptic ulcers. BMC Res Notes 6:455
Sung JJY, Lau JYW, Ching JYL et al (2010) Continuation of low-dose aspirin therapy in peptic ulcer bleeding: a randomized trial. Ann Intern Med 152:1–9
McNeil JJ, Nelson MR, Woods RL et al (2018) Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med 379:1519–1528
Rainsford KD, Schweitzer A, Brune K (1982) Distribution of the acetyl compared with the salicyl moiety of acetylsalicylic acid. Biochem Pharmacol 32:1301–1308
Needs CJ, Brooks PM (1985) Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet 10:164–177
Leopoldt D. Acetylsalicylsäure. https://www.gelbe-liste.de/wirkstoffe/Acetylsalicylsaeure_41 (accessed 10/22/2021)
Mineshita S (1983) Influence of gastrectomy on aspirin absorption. Br J Clin Pharmacol 16:756–757
Ali B, Kaur S (1983) Mammalian tissue acetylsalicylic acid esterase(s): identification, distribution and discrimination from other esterases. J Pharmacol Exp Ther 226:589–594
Kim D-H, Yang Y-S, Jakoby WB (1990) Aspirin hydrolyzing esterases from rat liver cytosol. Biochem Pharmacol 40:481–487
Rowland M, Riegelman S, Harris PA, Sholkoff SD (1972) Absorption kinetics of aspirin in man following oral administration of an aqueous solution. J Pharm Sci 61:379–385
Voelker M, Hammer M (2012) Dissolution and pharmacokinetics of a novel micronized aspirin formulation. Inflammopharmacology 20:225–231
Ghahramani P, Rowland-Yeo K, Yeo WW, Jackson PR, Ramsay LE (1998) Protein binding of aspirin and salicylate measured by in vivo ultrafiltration*. Clin Pharmacol Ther 63:285–295
Aarons L, Clifton P, Fleming G, Rowland M (1980) Aspirin binding and the effect of albumin on spontaneous and enzyme-catalysed hydrolysis. J Pharm Pharmacol 32:537–543
Alfonso LF, Srivenugopal KS, Bhat GJ (2009) Does aspirin acetylate multiple cellular proteins? (Review). Mol Med Rep 2:533–537
Pinckard RN, Hawkins D, Farr RS (1968) In vitro acetylation of plasma proteins, enzymes and DNA by aspirin. Nature 219:68–69
Hawkins D, Pinckard RN, Farr RS (1968) Acetylation of human serum albumin by acetylsalicylic acid. Science 160:780–781
Ai G, Dachineni R, Kumar DR, Marimuthu S, Alfonso LF, Bhat GJ (2016) Aspirin acetylates wild type and mutant p53 in colon cancer cells: identification of aspirin acetylated sites on recombinant p53. Tumor Biol 37:6007–6016
Nam M-H, Smith AJO, Pantcheva MB et al (2020) Aspirin inhibits TGFβ2-induced epithelial to mesenchymal transition of lens epithelial cells: selective acetylation of K56 and K122 in histone H3. Biochem J 477:75–97
Marimuthu S, Chivukula RSV, Alfonso LF, Moridani M, Hagen FK, Bhat GJ (2011) Aspirin acetylates multiple cellular proteins in HCT-116 colon cancer cells: identification of novel targets. Int J Oncol 39:1273–1283
Wang J, Zhang C-J, Zhang J et al (2015) Mapping sites of aspirin-induced acetylations in live cells by quantitative acid-cleavable activity-based protein profiling (QA-ABPP). Sci Rep 5:7896
Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231(25):232–235
Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D (2014) Cyclooxygenase pathways. Acta Biochim Pol 61:639–649
Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA (2009) Prostanoids in health and disease. J Lipid Res 50(Suppl):S423–S428
Ricciotti E, FitzGerald GA (2011) Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31:986–1000
Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D (2009) Cyclooxygenase in normal human tissues–is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J Cell Mol Med 13:3753–3763
Rowlinson SW, Kiefer JR, Prusakiewicz JJ et al (2003) A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J Biol Chem 278:45763–45769
Lucido MJ, Orlando BJ, Vecchio AJ, Malkowski MG (2016) Crystal structure of aspirin-acetylated human cyclooxygenase-2: insight into the formation of products with reversed stereochemistry. Biochemistry 55:1226–1238
Ghlichloo I, Gerriets V (2022) StatPearls: nonsteroidal anti-inflammatory drugs (NSAIDs). Treasure Island (FL)
Kerola M, Vuolteenaho K, Kosonen O, Kankaanranta H, Sarna S, Moilanen E (2009) Effects of nimesulide, acetylsalicylic acid, ibuprofen and nabumetone on cyclooxygenase-1- and cyclooxygenase-2-mediated prostanoid production in healthy volunteers ex vivo. Basic Clin Pharmacol Toxicol 104:17–21
Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR (1993) Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA 90:11693–11697
Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG, Patrignani P (2007) Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat 82:85–94
McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA (1999) Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci 96:272–277
Gong L, Thorn CF, Bertagnolli MM, Grosser T, Altman RB, Klein TE (2012) Celecoxib pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics 22:310–318
Eckenstaler R, Ripperger A, Hauke M et al (2022) A Thromboxane A 2 receptor-driven COX-2–dependent feedback loop that affects endothelial homeostasis and angiogenesis. Arterioscler Thromb Vasc Biol 42(4):444–461
Wallace JL (2008) Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn’t the stomach digest itself? Physiol Rev 88:1547–1565
Miller SB (2006) Prostaglandins in health and disease: an overview. Semin Arthritis Rheum 36:37–49
Campbell WB, Falck JR, Okita JR, Johnson AR, Callahan KS (1985) Synthesis of dihomoprostaglandins from adrenic acid (7,10,13,16-docosatetraenoic acid) by human endothelial cells. Biochimica et Biophysica Acta (BBA) 837:67–76
Klein T, Reutter F, Schweer H, Seyberth HW, Nüsing RM (1997) Generation of the isoprostane 8-epi-prostaglandin F2α in vitro and in vivo via the cyclooxygenases. J Pharmacol Exp Ther 282:1658–1665
Meade EA, Smith WL, DeWitt DL (1993) Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem 268:6610–6614
Kohli P, Levy BD (2009) Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol 158:960–971
Serhan CN, Chiang N (2008) Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol 153(Suppl 1):S200–S215
Gilroy DW (2005) The role of aspirin-triggered lipoxins in the mechanism of action of aspirin. Prostaglandins Leukot Essent Fatty Acids 73:203–210
Austin Pickens C, Yin Z, Sordillo LM, Fenton JI (2019) Arachidonic acid-derived hydroxyeicosatetraenoic acids are positively associated with colon polyps in adult males: a cross-sectional study. Sci Rep 9:12033
Chen GG, Xu H, Lee JFY et al (2003) 15-hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cells via a peroxisome proliferator-activated receptor gamma-dependent pathway. Int J Cancer 107:837–843
Cabral M, Martín-Venegas R, Moreno JJ (2013) Role of arachidonic acid metabolites on the control of non-differentiated intestinal epithelial cell growth. Int J Biochem Cell Biol 45:1620–1628
Clària J, Serhan CN (1995) Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA 92:9475–9479
Ma J, Zhang L, Zhang J et al (2013) 15-lipoxygenase-1/15-hydroxyeicosatetraenoic acid promotes hepatocellular cancer cells growth through protein kinase B and heat shock protein 90 complex activation. Int J Biochem Cell Biol 45:1031–1041
Rauzi F, Kirkby NS, Edin ML et al (2016) Aspirin inhibits the production of proangiogenic 15(S)-HETE by platelet cyclooxygenase-1. FASEB J 30:4256–4266
Zhang B, Cao H, Rao GN (2005) 15(S)-hydroxyeicosatetraenoic acid induces angiogenesis via activation of PI3K-Akt-mTOR-S6K1 signaling. Can Res 65:7283–7291
Serhan CN, Hong S, Gronert K et al (2002) Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med 196:1025–1037
Sulciner ML, Serhan CN, Gilligan MM et al (2018) Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med 215:115–140
Du Y, Yang J, Su T, Shen Z, Li J (2021) Lipid mediator lipoxin A4 and its analog BML-111 exert antitumor effects in melanoma. Ann Transl Med 9:802
Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510:92–101
Gilligan MM, Gartung A, Sulciner ML et al (2019) Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc Natl Acad Sci 116:6292–6297
Xu XM, Sansores-Garcia L, Chen XM, Matijevic-Aleksic N, Du M, Wu KK (1999) Suppression of inducible cyclooxygenase 2 gene transcription by aspirin and sodium salicylate. Proc Natl Acad Sci USA 96:5292–5297
Cieslik K, Zhu Y, Wu KK (2002) Salicylate suppresses macrophage nitric-oxide synthase-2 and cyclo-oxygenase-2 expression by inhibiting CCAAT/enhancer-binding protein-beta binding via a common signaling pathway. J Biol Chem 277:49304–49310
Chae H-J, Chae S-W, Reed JC, Kim H-R (2004) Salicylate regulates COX-2 expression through ERK and subsequent NF-κB activation in osteoblasts. Immunopharmacol Immunotoxicol 26:75–91
Wang Z, Brecher P (1999) Salicylate inhibition of extracellular signal-regulated kinases and inducible nitric oxide synthase. Hypertension (Dallas, Tex.: 1979) 34:1259–1264
Kiss K, Kiss J, Rudolf E, Cervinka M, Szeberényi J (2004) Sodium salicylate inhibits NF-κB and induces apoptosis in PC12 cells. J Biochem Biophys Methods 61:229–240
Agarwal A, Das K, Lerner N et al (2005) The AKT/IκB kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-κB and β-catenin. Oncogene 24:1021–1031
Patel M, Horgan PG, McMillan DC, Edwards J (2018) NF-κB pathways in the development and progression of colorectal cancer. Transl Res 197:43–56
Slattery ML, Mullany LE, Sakoda L et al (2018) The NF-κB signalling pathway in colorectal cancer: associations between dysregulated gene and miRNA expression. J Cancer Res Clin Oncol 144:269–283
Hawley SA, Fullerton MD, Ross FA et al (2012) The ancient drug salicylate directly activates AMP-activated protein kinase. Science (New York, N.Y.) 336:918–922
Larsson SC, Giovannucci E, Wolk A (2006) Long-term aspirin use and colorectal cancer risk: a cohort study in Sweden. Br J Cancer 95:1277–1279
Frouws MA, Bastiaannet E, Langley RE et al (2017) Effect of low-dose aspirin use on survival of patients with gastrointestinal malignancies; an observational study. Br J Cancer 116:405–413
Frouws MA, van Herk-Sukel MPP, Maas HA et al (2017) The mortality reducing effect of aspirin in colorectal cancer patients: interpreting the evidence. Cancer Treat Rev 55:120–127
Wilson JC, Murray LJ, Hughes CM, Black A, Anderson LA (2013) Non-steroidal anti-inflammatory drug and aspirin use and the risk of head and neck cancer. Br J Cancer 108:1178–1181
Roh J-L, Kim EH, Jang H, Shin D (2017) Aspirin plus sorafenib potentiates cisplatin cytotoxicity in resistant head and neck cancer cells through xCT inhibition. Free Radical Biol Med 104:1–9
Jayaprakash V, Rigual NR, Moysich KB et al (2006) Chemoprevention of head and neck cancer with aspirin: a case-control study. Arch Otolaryngol 132:1231–1236
Zhao M, Wang T, Hui Z (2020) Aspirin overcomes cisplatin resistance in lung cancer by inhibiting cancer cell stemness. Thoracic Cancer 11:3117–3125
Holmes MD, Chen WY, Li L, Hertzmark E, Spiegelman D, Hankinson SE (2010) Aspirin intake and survival after breast cancer. J Clin Oncol 28:1467–1472
Ahmadi N, Goldman R, Seillier-Moiseiwitsch F, Noone A-M, Kosti O, Davidson BJ (2010) Decreased risk of squamous cell carcinoma of the head and neck in users of nonsteroidal anti-inflammatory drugs. Int J Otolaryngol 2010:424161
Vane J, Botting R (2003) The mechanism of action of aspirin. Thromb Res 110:255–258
Greenhough A, Smartt HJM, Moore AE et al (2009) The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 30:377–386
Schmitt M, Greten FR (2021) The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol 21:653–667
Wang D, DuBois RN (2010) The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 29:781–788
Sano H, Kawahito Y, Wilder RL et al (1995) Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Can Res 55:3785–3789
Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo XJ, Ramonetti JT (1996) Cyclooxygenase-2 overexpression and tumor formation are blocked by Sulindac in a Murine Model of Familial Adenomatous Polyposist. Cancer Res 56(11):2556–2560
DuBois RN, Radhika A, Reddy BS, Entingh AJ (1996) Increased cyclooxygenase-2 levels in carcinogen-induced rat colonic tumors. Gastroenterology 110:1259–1262
Peng L, Zhou Y, Wang Y, Mou H, Zhao Q (2013) Prognostic significance of COX-2 immunohistochemical expression in colorectal cancer: a meta-analysis of the literature. PLoS One 8:e58891
Sheng H, Shao J, Kirkland SC et al (1997) Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Investig 99:2254–2259
Grösch S, Tegeder I, Niederberger E, Bräutigam L, Geisslinger G (2001) COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J 15:2742–2744
Veettil SK, Nathisuwan S, Ching SM et al (2019) Efficacy and safety of celecoxib on the incidence of recurrent colorectal adenomas: a systematic review and meta-analysis. Cancer Manage Res 11:561–571
Valverde A, Peñarando J, Cañas A et al (2017) The addition of celecoxib improves the antitumor effect of cetuximab in colorectal cancer: role of EGFR-RAS-FOXM1-β-catenin signaling axis. Oncotarget 8:21754–21769
Chan TA, Morin PJ, Vogelstein B, Kinzler KW (1998) Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc Natl Acad Sci USA 95:681–686
Uddin S, Ahmed M, Hussain A et al (2010) Cyclooxygenase-2 inhibition inhibits PI3K/AKT kinase activity in epithelial ovarian cancer. Int J Cancer 126:382–394
Tury S, Becette V, Assayag F et al (2016) Combination of COX-2 expression and PIK3CA mutation as prognostic and predictive markers for celecoxib treatment in breast cancer. Oncotarget 7:85124–85141
Buchanan CM, Dickson JMJ, Lee W-J, Guthridge MA, Kendall JD, Shepherd PR (2013) Oncogenic mutations of p110α isoform of PI 3-kinase upregulate its protein kinase activity. PLoS One 8:e71337
Xu S, Zhou W, Ge J, Zhang Z (2018) Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMP-PKA/PI3K-Akt signaling pathway. Mol Med Rep 17:4702–4712
Di Popolo A, Memoli A, Apicella A et al (2000) IGF-II/IGF-I receptor pathway up-regulates COX-2 mRNA expression and PGE2 synthesis in Caco-2 human colon carcinoma cells. Oncogene 19:5517–5524
Hsu H-H, Lin Y-M, Shen C-Y et al (2017) Prostaglandin E2-induced COX-2 expressions via EP2 and EP4 signaling pathways in human LoVo colon cancer cells. Int J Mol Sci 18(6):1132
Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN (2006) Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci 103:1492–1497
Lee CS, Song IH, Lee A et al (2021) Enhancing the landscape of colorectal cancer using targeted deep sequencing. Sci Rep 11:8154
Yang J, Wang X, Gao Y et al (2020) Inhibition of PI3K-AKT signaling blocks PGE2-induced COX-2 expression in lung adenocarcinoma. Onco Targets Ther 13:8197–8208
Lee HK, Jeong S (2006) β-Catenin stabilizes cyclooxygenase-2 mRNA by interacting with AU-rich elements of 3’-UTR. Nucleic Acids Res 34:5705–5714
Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y (2013) The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev 65:1010–1052
Takahashi T, Ichikawa H, Morimoto Y, Tsuneyama K, Hijikata T (2019) Inhibition of EP2/EP4 prostanoid receptor-mediated signaling suppresses IGF-1-induced proliferation of pancreatic cancer BxPC-3 cells via upregulating γ-glutamyl cyclotransferase expression. Biochem Biophys Res Commun 516:388–396
Gray RT, Cantwell MM, Coleman HG et al (2017) Evaluation of PTGS2 expression, PIK3CA mutation, aspirin use and colon cancer survival in a population-based cohort study. Clin Transl Gastroenterol 8:e91
Tsuruo T, Fujita N (2008) Platelet aggregation in the formation of tumor metastasis. Proc Jpn Acad 84:189–198
Lucotti S, Muschel RJ (2020) Platelets and metastasis: new implications of an old interplay. Front Oncol 10:1350
Nie D, Lamberti M, Zacharek A et al (2000) Thromboxane A(2) regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem Biophys Res Commun 267:245–251
Gil-Bernabé AM, Lucotti S, Muschel RJ (2013) Coagulation and metastasis: what does the experimental literature tell us? Br J Haematol 162:433–441
Pradono P, Tazawa R, Maemondo M et al (2002) Gene transfer of thromboxane A(2) synthase and prostaglandin I(2) synthase antithetically altered tumor angiogenesis and tumor growth. Can Res 62:63–66
Lucotti S, Cerutti C, Soyer M et al (2019) Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J Clin Investig 129:1845–1862
Johnson KE, Ceglowski JR, Roweth HG et al (2019) Aspirin inhibits platelets from reprogramming breast tumor cells and promoting metastasis. Blood Adv 3:198–211
Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X (2003) Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem 278:30725–30731
Niho N, Kitamura T, Takahashi M et al (2006) Suppression of azoxymethane-induced colon cancer development in rats by a cyclooxygenase-1 selective inhibitor, mofezolac. Cancer Sci 97:1011–1014
Riehl TE, George RJ, Sturmoski MA et al (2006) Azoxymethane protects intestinal stem cells and reduces crypt epithelial mitosis through a COX-1-dependent mechanism. Am J Physiol 291:G1062–G1070
Wu WKK, Sung JJY, Wu YC et al (2009) Inhibition of cyclooxygenase-1 lowers proliferation and induces macroautophagy in colon cancer cells. Biochem Biophys Res Commun 382:79–84
Sakai H, Suzuki T, Takahashi Y et al (2006) Upregulation of thromboxane synthase in human colorectal carcinoma and the cancer cell proliferation by thromboxane A2. FEBS Lett 580:3368–3374
Peng X, Li J, Tan S et al (2017) COX-1/PGE2/EP4 alleviates mucosal injury by upregulating β-arr1-mediated Akt signaling in colitis. Sci Rep 7:1055
Li X-L, Zhou J, Chen Z-R, Chng W-J (2015) P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World J Gastroenterol 21:84–93
Yin MJ, Yamamoto Y, Gaynor RB (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(κ)B kinase-beta. Nature 396:77–80
Fu J, Xu Y, Yang Y, Liu Y, Ma L, Zhang Y (2019) Aspirin suppresses chemoresistance and enhances antitumor activity of 5-Fu in 5-Fu-resistant colorectal cancer by abolishing 5-Fu-induced NF-κB activation. Sci Rep 9:16937
McCarty MF, Block KI (2006) Preadministration of high-dose salicylates, suppressors of NF-κB activation, may increase the chemosensitivity of many cancers: an example of proapoptotic signal modulation therapy. Integr Cancer Ther 5:252–268
Fernandez HR, Lindén SK (2017) The aspirin metabolite salicylate inhibits lysine acetyltransferases and MUC1 induced epithelial to mesenchymal transition. Sci Rep 7:5626
Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810
Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 16:3797–3804
Bilic J, Huang Y-L, Davidson G et al (2007) Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316:1619–1622
Lecarpentier Y, Schussler O, Hébert J-L, Vallée A (2019) Multiple targets of the canonical WNT/β-Catenin signaling in cancers. Front Oncol 9:1248
Fevr T, Robine S, Louvard D, Huelsken J (2007) Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol 27:7551–7559
Tian X, Liu Z, Niu B et al (2011) E-cadherin/β-catenin complex and the epithelial barrier. J Biomed Biotechnol 2011:567305
Mittal V (2018) Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol 13:395–412
Leslie A, Carey FA, Pratt NR, Steele RJC (2002) The colorectal adenoma-carcinoma sequence. Br J Surg 89:845–860
Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P (1998) Downregulation of β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK3β. Curr Biol 8:573–581
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789
McManus EJ, Sakamoto K, Armit LJ et al (2005) Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J 24:1571–1583
Ding VW, Chen RH, McCormick F (2000) Differential regulation of glycogen synthase kinase 3β by insulin and Wnt signaling. J Biol Chem 275:32475–32481
Zeng H, Lu B, Zamponi R et al (2018) mTORC1 signaling suppresses Wnt/β-catenin signaling through DVL-dependent regulation of Wnt receptor FZD level. Proc Natl Acad Sci 115:E10362–E10369
Inoki K, Li Y, Zhu T, Wu J, Guan K-L (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657
Fang D, Hawke D, Zheng Y et al (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282:11221–11229
Ormanns S, Neumann J, Horst D, Kirchner T, Jung A (2014) WNT signaling and distant metastasis in colon cancer through transcriptional activity of nuclear β-Catenin depend on active PI3K signaling. Oncotarget 5:2999–3011
Prossomariti A, Piazzi G, Alquati C, Ricciardiello L (2020) Are Wnt/β-Catenin and PI3K/AKT/mTORC1 distinct pathways in colorectal cancer? Cell Mol Gastroenterol Hepatol 10:491–506
Du Q, Geller DA (2010) Cross-regulation between Wnt and NF-κB signaling pathways. Forum Immunopathol Dis Therapeutics 1:155–181
Bos CL, Kodach LL, van den Brink GR et al (2006) Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene 25:6447–6456
Kuo Y-C, Huang K-Y, Yang C-H, Yang Y-S, Lee W-Y, Chiang C-W (2008) Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem 283:1882–1892
Jin S, Wu X (2019) Aspirin inhibits colon cancer cell line migration through regulating epithelial-mesenchymal transition via Wnt signaling. Oncol Lett 17:4675–4682
Dunbar K, Valanciute A, Lima ACS et al (2021) Aspirin rescues Wnt-driven stem-like phenotype in human intestinal organoids and increases the Wnt Antagonist Dickkopf-1. Cell Mol Gastroenterol Hepatol 11:465–489
Rees WD, Tandun R, Yau E, Zachos NC, Steiner TS (2020) Regenerative intestinal stem cells induced by acute and chronic injury: the saving grace of the epithelium? Front Cell Development Biol 8:583919
Guiu J, Hannezo E, Yui S et al (2019) Tracing the origin of adult intestinal stem cells. Nature 570:107–111
van der Heijden M, Vermeulen L (2019) Stem cells in homeostasis and cancer of the gut. Mol Cancer 18:66
Gupta R, Bhatt LK, Johnston TP, Prabhavalkar KS (2019) Colon cancer stem cells: potential target for the treatment of colorectal cancer. Cancer Biol Ther 20:1068–1082
Das PK, Islam F, Lam AK (2020) The roles of cancer stem cells and therapy resistance in colorectal carcinoma. Cells 9(6):1392
Chen J, Shao R, Li F et al (2015) PI3K/Akt/mTOR pathway dual inhibitor BEZ235 suppresses the stemness of colon cancer stem cells. Clin Exp Pharmacol Physiol 42:1317–1326
Wang J, Wang W, Cai H et al (2017) MACC1 facilitates chemoresistance and cancer stem cell-like properties of colon cancer cells through the PI3K/AKT signaling pathway. Mol Med Rep 16:8747–8754
Li H, Chen Y-X, Wen J-G, Zhou H-H (2017) Metastasis-associated in colon cancer 1: a promising biomarker for the metastasis and prognosis of colorectal cancer. Oncol Lett 14:3899–3908
Thakur B, Ray P (2017) Cisplatin triggers cancer stem cell enrichment in platinum-resistant cells through NF-κB-TNFα-PIK3CA loop. J Exp Clin Cancer Res 36:164
Zhou D, He Y, Li H, Huang W (2021) KLK6 mediates stemness and metabolism of gastric carcinoma cells via the PI3K/AKT/mTOR signaling pathway. Oncol Lett 22:824
Yoon C, Lu J, Yi BC et al (2021) PI3K/Akt pathway and Nanog maintain cancer stem cells in sarcomas. Oncogenesis 10:12
Zhang W, Sui Y, Ni J, Yang T (2016) Insights into the Nanog gene: a propeller for stemness in primitive stem cells. Int J Biol Sci 12:1372–1381
Kim JS, Kim BS, Kim J, Park C-S, Chung IY (2010) The phosphoinositide-3-kinase/Akt pathway mediates the transient increase in Nanog expression during differentiation of F9 cells. Arch Pharmacal Res 33:1117–1125
Storm MP, Bone HK, Beck CG et al (2007) Regulation of Nanog expression by phosphoinositide 3-kinase-dependent signaling in murine embryonic stem cells. J Biol Chem 282:6265–6273
Roudi R, Barodabi M, Madjd Z, Roviello G, Corona SP, Panahei M (2020) Expression patterns and clinical significance of the potential cancer stem cell markers OCT4 and NANOG in colorectal cancer patients. Mol Cell Oncol 7:1788366
Tamura S, Isobe T, Ariyama H et al (2018) E-cadherin regulates proliferation of colorectal cancer stem cells through NANOG. Oncol Rep 40:693–703
Chen Z, Li W, Qiu F et al (2018) Aspirin cooperates with p300 to activate the acetylation of H3K9 and promote FasL-mediated apoptosis of cancer stem-like cells in colorectal cancer. Theranostics 8:4447–4461
Shirakawa K, Wang L, Man N et al (2016) Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity. Elife 5:e11156
Yiannakopoulou E (2014) Targeting epigenetic mechanisms and microRNAs by aspirin and other non steroidal anti-inflammatory agents–implications for cancer treatment and chemoprevention. Cell Oncol 37:167–178
Huang W-C, Chen C-C (2005) Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol 25:6592–6602
Wang H, Liu B, Wang J et al (2017) Reduction of NANOG mediates the inhibitory effect of aspirin on tumor growth and stemness in colorectal cancer. Cell Physiol Biochem 44:1051–1063
Zou Z, Zheng W, Fan H et al (2021) Aspirin enhances the therapeutic efficacy of cisplatin in oesophageal squamous cell carcinoma by inhibition of putative cancer stem cells. Br J Cancer 125(6):826–838
Khoo BL, Grenci G, Lim JSY et al (2019) Low-dose anti-inflammatory combinatorial therapy reduced cancer stem cell formation in patient-derived preclinical models for tumour relapse prevention. Br J Cancer 120:407–423
Saha S, Mukherjee S, Khan P et al (2016) Aspirin suppresses the acquisition of chemoresistance in breast cancer by disrupting an NFκB-IL6 signaling axis responsible for the generation of cancer stem cells. Can Res 76:2000–2012
Pertea M (2012) The human transcriptome: an unfinished story. Genes 3:344–360
Palazzo AF, Lee ES (2015) Non-coding RNA: what is functional and what is junk? Front Genet 6:2
Anastasiadou E, Jacob LS, Slack FJ (2018) Non-coding RNA networks in cancer. Nat Rev Cancer 18:5–18
Chao JY, Chang H-C, Jiang J-K et al (2021) Using bioinformatics approaches to investigate driver genes and identify BCL7A as a prognostic gene in colorectal cancer. Comput Struct Biotechnol J 19:3922–3929
Slack FJ, Chinnaiyan AM (2019) The role of non-coding RNAs in oncology. Cell 179:1033–1055
Dragomir MP, Kopetz S, Ajani JA, Calin GA (2020) Non-coding RNAs in GI cancers: from cancer hallmarks to clinical utility. Gut 69:748–763
Grillone K, Riillo C, Scionti F et al (2020) Non-coding RNAs in cancer: platforms and strategies for investigating the genomic “dark matter.” J Exp Clin Cancer Res 39:117
O’Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol 9:402
Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15:509–524
Jia Z, An J, Liu Z, Zhang F (2022) Non-coding RNAs in colorectal cancer: their functions and mechanisms. Front Oncol 12:783079
Melé M, Mattioli K, Mallard W, Shechner DM, Gerhardinger C, Rinn JL (2017) Chromatin environment, transcriptional regulation, and splicing distinguish lincRNAs and mRNAs. Genome Res 27:27–37
Yao R-W, Wang Y, Chen L-L (2019) Cellular functions of long noncoding RNAs. Nat Cell Biol 21:542–551
Barrett SP, Salzman J (2016) Circular RNAs: analysis, expression and potential functions. Development (Cambridge, England) 143:1838–1847
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO (2013) Cell-type specific features of circular RNA expression. PLoS Genet 9:e1003777
Chen CY, Sarnow P (1995) Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268:415–417
Hansen TB, Jensen TI, Clausen BH et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495:384–388
Benetatos L, Voulgaris E, Vartholomatos G (2017) The crosstalk between long non-coding RNAs and PI3K in cancer. Med Oncol 34:39
Ellis BC, Graham LD, Molloy PL (2014) CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism. Biochem Biophys Acta 1843:372–386
Islam Khan MZ, Law HKW (2021) RAMS11 promotes CRC through mTOR-dependent inhibition of autophagy, suppression of apoptosis, and promotion of epithelial-mesenchymal transition. Cancer Cell Int 21:321
Pan S, Liu Y, Liu Q et al (2019) HOTAIR/miR-326/FUT6 axis facilitates colorectal cancer progression through regulating fucosylation of CD44 via PI3K/AKT/mTOR pathway. Biochimica et biophysica acta. Mol Cell Res 1866:750–760
Chen L, Qian X, Wang Z, Zhou X (2021) The HOTAIR lncRNA: A remarkable oncogenic promoter in human cancer metastasis. Oncol Lett 21:302
Shengnan J, Dafei X, Hua J, Sunfu F, Xiaowei W, Liang X (2020) Long non-coding RNA HOTAIR as a competitive endogenous RNA to sponge miR-206 to promote colorectal cancer progression by activating CCL2. J Cancer 11:4431–4441
Peng CL, Zhao XJ, Wei CC, Wu JW (2019) LncRNA HOTAIR promotes colon cancer development by down-regulating miRNA-34a. Eur Rev Med Pharmacol Sci 23(13):5752–5761
Li Y, Zeng C, Hu J et al (2018) Long non-coding RNA-SNHG7 acts as a target of miR-34a to increase GALNT7 level and regulate PI3K/Akt/mTOR pathway in colorectal cancer progression. J Hematol Oncol 11:89
Lyu J, Sun Y, Li X, Ma H (2021) MicroRNA-206 inhibits the proliferation, migration and invasion of colorectal cancer cells by regulating the c-Met/AKT/GSK-3β pathway. Oncol Lett 21:147
Chen X, Liu Y, Zhang Q et al (2021) Exosomal miR-590-3p derived from cancer-associated fibroblasts confers radioresistance in colorectal cancer. Mol Ther Nucleic Acids 24:113–126
El-Daly SM, Abba ML, Patil N, Allgayer H (2016) miRs-134 and -370 function as tumor suppressors in colorectal cancer by independently suppressing EGFR and PI3K signalling. Sci Rep 6:24720
Tu F-L, Guo X-Q, Wu H-X et al (2020) Circ-0001313/miRNA-510-5p/AKT2 axis promotes the development and progression of colon cancer. Am J Transl Res 12:281–291
Wang J, Luo J, Liu G, Li X (2020) Circular RNA hsa_circ_0008285 inhibits colorectal cancer cell proliferation and migration via the miR-382-5p/PTEN axis. Biochem Biophys Res Commun 527:503–510
Chong X, Chen J, Zheng N et al (2022) PIK3CA mutations-mediated downregulation of circLHFPL2 inhibits colorectal cancer progression via upregulating PTEN. Mol Cancer 21:118
Lan F, Yue X, Han L et al (2012) Genome-wide identification of TCF7L2/TCF4 target miRNAs reveals a role for miR-21 in Wnt-driven epithelial cancer. Int J Oncol 40:519–526
Guo H, Liu J, Ben Q et al (2016) The aspirin-induced long non-coding RNA OLA1P2 blocks phosphorylated STAT3 homodimer formation. Genome Biol 17:24
Yokogami K, Wakisaka S, Avruch J, Reeves SA (2000) Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol 10:47–50
Chen M, Wu L, Zhan H, Liu T, He Y (2021) Aspirin-induced long non-coding RNA suppresses colon cancer growth. Transl Cancer Res 10:2055–2069
Forbes JD, Chen C-Y, Knox NC et al (2018) A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome 6:221
Marzano M, Fosso B, Piancone E, Defazio G, Pesole G, de Robertis M (2021) Stem cell impairment at the host-microbiota interface in colorectal cancer. Cancers 13:996
DeDecker L, Coppedge B, Avelar-Barragan J, Karnes W, Whiteson K (2021) Microbiome distinctions between the CRC carcinogenic pathways. Gut Microbes 13:1854641
Vacante M, Ciuni R, Basile F, Biondi A (2020) Gut microbiota and colorectal cancer development: a closer look to the adenoma-carcinoma sequence. Biomedicines 8:489
Fan X, Jin Y, Chen G, Ma X, Zhang L (2021) Gut microbiota dysbiosis drives the development of colorectal cancer. Digestion 102:508–515
Hussan H, Clinton SK, Roberts K, Bailey MT (2017) Fusobacterium’s link to colorectal neoplasia sequenced: a systematic review and future insights. World J Gastroenterol 23:8626–8650
Mehta RS, Nishihara R, Cao Y et al (2017) Association of dietary patterns with risk of colorectal cancer subtypes classified by fusobacterium nucleatum in Tumor tissue. JAMA Oncol 3:921–927
Toprak NU, Yagci A, Gulluoglu BM et al (2006) A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect 12:782–786
Kostic AD, Gevers D, Pedamallu CS et al (2012) Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22:292–298
Abdulamir AS, Hafidh RR, Abu BF (2011) The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J Exp Clin Cancer Res 30:11
de Almeida CV, Taddei A, Amedei A (2018) The controversial role of Enterococcus faecalis in colorectal cancer. Ther Adv Gastroenterol 11:1756284818783606
Bhatt AP, Redinbo MR, Bultman SJ (2017) The role of the microbiome in cancer development and therapy. Cancer J Clin 67:326–344
Zhao R, Coker OO, Wu J et al (2020) Aspirin reduces colorectal tumor development in mice and gut microbes reduce its bioavailability and chemopreventive effects. Gastroenterology 159:969-983.e4
Rogers MAM, Aronoff DM (2016) The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clini Microbiol Infect 22:178.e1-178.e9
Prizment AE, Staley C, Onyeaghala GC et al (2020) Randomised clinical study: oral aspirin 325 mg daily vs placebo alters gut microbial composition and bacterial taxa associated with colorectal cancer risk. Aliment Pharmacol Ther 52:976–987
Niccolai E, Russo E, Baldi S et al (2020) Significant and conflicting correlation of IL-9 with prevotella and bacteroides in human colorectal cancer. Front Immunol 11:573158
Yang J, McDowell A, Kim EK et al (2019) Development of a colorectal cancer diagnostic model and dietary risk assessment through gut microbiome analysis. Exp Mol Med 51:1–15
Wang F, Cai K, Xiao Q, He L, Xie L, Liu Z (2022) Akkermansia muciniphila administration exacerbated the development of colitis-associated colorectal cancer in mice. J Cancer 13:124–133
Cani PD, de Vos WM (2017) Next-Generation Beneficial Microbes: The Case of Akkermansia muciniphila. Front Microbiol 8:1765
Brennan CA, Nakatsu G, Gallini Comeau CA et al (2021) Aspirin Modulation of the Colorectal Cancer-Associated Microbe Fusobacterium nucleatum. MBio 12:e00547-21
Han H, Li Y, Qin W et al (2020) Fusobacterium nucleatum facilitates cetuximab resistance in colorectal cancer via the PI3K/AKT and JAK/STAT3 pathways
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23:27–47
Warburg O (1924) Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 12:1131–1137
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
DeBerardinis RJ, Chandel NS (2020) We need to talk about the Warburg effect. Nat Metab 2:127–129
Birkeland ES, Koch LM, Dechant R (2020) Another consequence of the warburg effect? Metabolic regulation of Na+/H+ exchangers may link aerobic glycolysis to cell growth. Front Oncol 10:1561
Brown RE, Short SP, Williams CS (2018) Colorectal cancer and metabolism. Curr Colorect Cancer Rep 14:226–241
Jiang J, Srivastava S, Zhang J (2019) Starve cancer cells of glutamine: break the spell or make a hungry monster? Cancers 11(6):804
Vikram R, Iyer RB (2008) PET/CT imaging in the diagnosis, staging, and follow-up of colorectal cancer. Cancer Imaging 8 Spec No A: S46–51
Cohen AS, Grudzinski J, Smith GT et al (2022) First-in-human PET imaging and estimated radiation dosimetry of l-5-11C-glutamine in patients with metastatic colorectal cancer. J Nuclear Med 63:36–43
Ling HH, Pan Y-P, Fan C-W et al (2019) Clinical significance of serum glutamine level in patients with colorectal cancer. Nutrients 11(4):898
Li H, Lu S, Chen Y et al (2019) AKT2 phosphorylation of hexokinase 2 at T473 promotes tumorigenesis and metastasis in colon cancer cells via NF-κB, HIF1α, MMP2, and MMP9 upregulation. Cell Signal 58:99–110
Zhuo B, Li Y, Li Z et al (2015) PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochem Biophys Res Commun 464:401–406
Jiang W, He T, Liu S et al (2018) The PIK3CA E542K and E545K mutations promote glycolysis and proliferation via induction of the β-catenin/SIRT3 signaling pathway in cervical cancer. J Hematol Oncol 11:139
Zhao Y, Feng X, Chen Y et al (2020) 5-Fluorouracil enhances the antitumor activity of the glutaminase inhibitor CB-839 against PIK3CA-mutant colorectal cancers. Can Res 80:4815–4827
Zhao Y, Zhao X, Chen V et al (2019) Colorectal cancers utilize glutamine as an anaplerotic substrate of the TCA cycle in vivo. Sci Rep 9:19180
Ilic N, Birsoy K, Aguirre AJ et al (2017) PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc Natl Acad Sci 114:E3434–E3443
Scalise M, Pochini L, Console L, Losso MA, Indiveri C (2018) The human SLC1A5 (ASCT2) amino acid transporter: from function to structure and role in cell biology. Front Cell Development Biol 6:96
Ai G, Dachineni R, Kumar DR, Alfonso LF, Marimuthu S, Bhat GJ (2016) Aspirin inhibits glucose-6-phosphate dehydrogenase activity in HCT 116 cells through acetylation: identification of aspirin-acetylated sites. Mol Med Rep 14:1726–1732
Cheng J, Huang Y, Zhang X et al (2020) TRIM21 and PHLDA3 negatively regulate the crosstalk between the PI3K/AKT pathway and PPP metabolism. Nat Commun 11:1880
Düvel K, Yecies JL, Menon S et al (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39:171–183
Porstmann T, Santos CR, Griffiths B et al (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 8:224–236
Hoxhaj G, Manning BD (2020) The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 20:74–88
Wang J-D, Chen W-Y, Li J-R et al (2020) Aspirin mitigated tumor growth in obese mice involving metabolic inhibition. Cells 9(3):569
Yan S, Hui Y, Li J, Xu X, Li Q, Wei H (2020) Glutamine relieves oxidative stress through PI3K/Akt signaling pathway in DSS-induced ulcerative colitis mice. Iran J Basic Med Sci 23:1124–1129
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hall, D.C.N., Benndorf, R.A. Aspirin sensitivity of PIK3CA-mutated Colorectal Cancer: potential mechanisms revisited. Cell. Mol. Life Sci. 79, 393 (2022). https://doi.org/10.1007/s00018-022-04430-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04430-y