
Abstract
Efficient exogenous DNA integration can be mediated by Cas9 through the non-homology end-joining pathway. However, such integrations are often imprecise and contain a variety of mutations at the junctions between the external DNA and the genomic loci. Here we describe a microhomology-dependent targeted integration method, designated MITI, for precise site-specific gene insertions. We found that the MITI strategy yielded higher knock-in accuracy than Cas9 HITI for the insertion of external DNA and tagging endogenous genes. Furthermore, in combination with negative selection and four different CrRNAs targeting donor vectors and genome-targeted sites with a CrRNA array, MITI facilitated precise ligation at all junctions. Therefore, our Cas12a-based MITI method increases the repertoire of precision genome engineering approaches and provides a useful tool for various gene editing applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Site-specific transgene integration is primarily implemented by homology-directed repair (HDR) and non-homology end joining (NHEJ) pathways [1]. The precise introduction of foreign DNA to endogenous target sites through the HDR pathway is time-consuming and cumbersome as it requires the cloning of the homology arms for every gene. Also, since this method can only exert its effect in the S/G2 cell phase, its efficiency is low, especially in non-dividing cells [2]. Accurate site-specific gene insertions could also be mediated via a method called Obligate Ligation-Gated Recombination (ObLiGaRe) through the efficient NHEJ pathway using zinc-finger nucleases (ZFNs) or Tale nucleases (TALENs) [3], but its wide application is limited by both high cost and complicated design. CRISPR/Cas9 also enables convenient and efficient knock-in of external DNA cassettes at target sites in zebrafish [4, 5], mammalian cells [6,7,8], and so on via NHEJ pathway; however, the junctions between the external DNA and the genome usually contain various mutations. Several efforts have aimed to optimize this system, such as controlling the expression of Cas9 protein via fusion of FKBP12-L106P destabilization domain to Cas9 [9] and cleaving both donor and target sites with the same sgRNA via homology-independent target integration (HITI) strategy [10].
Cas12a, as a newly identified RNA-guided nuclease of the Cas12 family, has several distinct advantages in comparison with Cas9, such as lower mismatch tolerance and greater specificity [11, 12]. Cas12a can process its CRISPR RNAs (CrRNAs) array into mature CrRNAs, and this facilitates concurrent targeting of multiple genes, which distinguishes it from Cas9 [13, 14]. Another difference between these two nucleases is that Cas9 and Cas12a generate blunt and sticky ends, respectively [15, 16]. Cas12a mainly cuts at the 18th base site of the non-complementary strand and the 23rd base site of the complementary strand downstream of the PAM sequence, generating a 5-nt overhang when the spacer length is 20 nt or longer. When the spacer length is shorter than 20 nt, Cas12a mainly cleaves after the 14 nt on the non-complementary strand and the 22 nt on the complementary strand. This feature makes Cas12a a useful tool similar to restriction enzymes for precise in vitro DNA assembly [17, 18]. The blunt-ended DSB generated by Cas9 is compatible with the Gibson assembly method, which relies on sequence homology for in vitro molecular cloning [19, 20]. Based on these in vitro comparison studies, we hypothesized that Cas12a may be used in mammalian cells for precise gene knock-in, with better accuracy than Cas9.
In this study, we devised a Cas12a-based method for precise gene integration. In this method, the recognition site is readily engineered into the donor vector by a simple PCR or T4 ligation. The orientation of Cas12a target sequences in the donor vector must be opposite to that of the genome target, but the 5 base pairs distal to the PAM are identical to the last 5 bp sequences of the genome target site, which enables the generation of seamless junctions between endogenous and exogenous DNA via complementary sticky sequences. Since our new method depends on the 5 bp microhomology, we denoted it as microhomology-dependent targeted integration (MITI). MITI could increase the accuracy of knock-in outcomes compared with the Cas9 HITI approach at several loci. Therefore, MITI is a useful tool for precise targeting.
Materials and methods
Plasmid cloning
The related primer sequences of plasmid and CrRNA constructions, knock-in detection, and T7E1 assays used in this study are noted in Supplementary Tables S1–S4. To generate the SA-IRES-GFP donor vector, the pZGs plasmid [21] was digested with NcoI, removing the LacZ-CAG promoter and allowing self re-ligation. The SV40-Puro fragment was PCR amplified and inserted into the AflII/BglII-digested pZGs backbone through Gibson assembly. The CrRNA or sgRNA target sequence of Cas12a or Cas9, respectively, was inserted into the XhoI/SpeI-digested donor backbone before the SA sequence to construct the single-cut donor vector, including D1, D2, and D3. To construct the D7 vector containing the HSV-TK negative selection gene, the linker carrying the other AAVS1 site CrRNA target sequence and the SalI enzyme cleavage site was inserted into the AscI/BglII-digested D2 backbone. A SalI/SpeI-digested TK1-Amp-ColE1 fragment from the pWS-TK7 plasmid was then ligated into the SalI/SpeI-digested vector backbone. Alternatively, donor vectors can be constructed by a simple PCR using the primers containing the target sequence to amplify the backbone fragment, followed by ligation with the reporter gene via Gibson assembly. The donor vector 3 × Flag-F2A-tdTomato-loxP-PGK-Puro-loxP was created through Gibson assembly, by combining the following five DNA fragments: (i) PCR-amplified 3 × Flag from pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid (Addgene 42230), (ii) PCR-amplified F2A fragment from pMaster12 plasmid (Addgene 58527), (iii) PCR-amplified tdTomato fragment from pRSET-B tdTomato plasmid, (iv) the PCR-amplified loxP-PGK-Puro-loxP fragment, and (v) NdeI/AscI-digested pPB-hNRASG12V [22] piggyBac backbone. The corresponding CrRNA target sequence was inserted into the NdeI/FseI-digested 3 × Flag-F2A-tdTomato-loxP-PGK-Puro-loxP vector to obtain the D4, D5, and D6 donor vectors. To construct the D4.1 vector, the linker containing the CLTA Cas12a target sequence identical to the genome was ligated into the AscI/AflII-digested D4 vector. The vector for expressing the AsCas12a CrRNA was constructed via linking the direct repeat sequence of AsCas12a and the BbsI enzyme site sequence into the BbsI/BamHI digested pCRISPR-sg4 backbone [22]. To get the all-in-one vector co-expressing the hU6-CrRNA and AsCas12a, the hU6-CrRNA (BbsI) PCR fragment was ligated into the NruI digested pY010 plasmid (Addgene 69982) backbone through Gibson assembly. The CrRNA array was constructed as previously described [14].
Cell culture and transfection
HEK293T cells, HeLa cells, HepG2 cells and the primary pig fetal fibroblasts (PFFs) cells were all grown at 37 °C, in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) containing l-glutamine and pyridoxine hydrochloride supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1 mM sodium pyruvate, 1 × NEAA and 1% penicillin and streptomycin. Cells were dissociated using 0.05% trypsin into single cells after washing in phosphate buffer saline and transfected using 4D-Nucleofector. For each transfection, 106 cells were mixed with 100 μL pre-warmed nucleofection reagents. The cell suspensions were then mixed with plasmids and electroporated with a 4D-Nucleofector Device (Lonza). Transfected cells underwent puromycin selection for 5 days to ensure successful transfection and were then analyzed by flow cytometry (BD FACS Calibur).
T7E1 assay
To prepare the genomic DNA from the cells for the T7E1 assay, cells were treated with 500 μL ES cells lysis buffer (100 mM NaCl; 20 mM Tris, pH 7.6; 10 mM EDTA; 0.5% SDS) and 15 μL of proteinase K (from 10 mg/mL stock) at 37 °C for 2–4 h without shaking. The genomic DNA was extracted as previously described [23]. Prior to PCR, 5 μL of genomic DNA was diluted in 15 μL HotShot lysis solution (25 mM NaOH, 0.2 mM EDTA), boiled for 15 min, and then neutralized with 15 μL of 40 mM Tris–Cl. The genomic region flanking the target site for each gene was PCR amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs, M0491S) following the manufacturer’s instructions. The 200–500 ng purified PCR fragments were mixed with 1 μL 10 × PfuUltra II Reaction Buffer (Agilent) and sterile ultra-pure water to a final volume of 10 μL and were subjected to a re-annealing process to enable heteroduplex formation: 95 °C for 10 min, 95 °C to 85 °C ramping at 2 °C/s, 85 °C to 25 °C at 0.25 °C/s, and 25 °C hold for 1 min. After re-annealing, the products were treated with 10 U of T7E1 enzyme (New England Biolabs) in 15 μL of the reaction mixture at 37 °C for 1 h, and the digested fragments were directly resolved on polyacrylamide gels. The Indel percentage was calculated with the following formula: 100 × (1 − sqrt (1 − (b + c)/(a + b + c))), where “a” represents the gray value of the intact band, and “b” and “c” are the gray values of each cleavage band. The gray values were determined using Image J software.
Junction DNA sequencing by TA cloning
For junction sequence analysis after targeted knock-in, the genomic DNA of pooled cells or picked clones was extracted as described above. The target sites were PCR amplified using the Herculase II Fusion Enzyme (Agilent Technologies, Catalog #600677). For sequence analysis of the PCR product from pooled cells, GoTag polymerase (Agilent Technologies) was used to tail the PCR products, which were then cloned into the pMD19-T vector backbone (TakaRa, Catalog #6013) and sent for sequencing. For direct sequence analysis of the 5′ and 3′ junctions of picked clones, the PCR products were directly purified and sent for sequencing.
Immunofluorescence staining
The HepG2 cells with stably integrated 3 × FLAG-2A-tdTomato reporter were washed with PBS and fixed in 4% paraformaldehyde for 30 min at room temperature. After washing three times in PBS every 5 min, the cells were blocked with blocking buffer (Beyotime Biotechnology, P0102) for 1 h at room temperature. The cells were then incubated overnight at 4 °C with the anti-FLAG-tag primary antibody (Beyotime Biotechnology, AF0036, 1:100). Next, the cells were incubated with the goat anti-rabbit IgG secondary antibody (Invitrogen, 1:1000) for 30 min in the dark after removing the primary antibody. Finally, DAPI (1:10,000) in PBS was added into the cells for 1 min. The immunostained cells were examined by fluorescence microscopy.
Results
Cas12a MITI strategy could facilitate more precise targeted integration
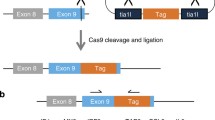
To test the effect of Cas12a sticky overhangs on the outcomes of targeted integrations, we designed two simple strategies: Cas12a HITI and Cas12a MITI, which are different in that the 5 bp PAM-distal sequences are oriented in opposite directions (Fig. 1a, b). The sticky end generated by Cas12a on the donor plasmid in MITI is complementary with that of the endogenous target site. For comparison, the accuracy of the Cas9 HITI strategy was also examined (Fig. 1c). We next tested these strategies for targeting the reporter gene cassette—SA-IRES-GFP-SV40-Puro—into an editable AAVS1 locus (Figs. 2a, S1). We first selected an overlapping target site at the AAVS1 locus that can be cleaved simultaneously by Cas9 and Cas12a. We transfected HeLa cells with the corresponding Cas12a or Cas9 targeting vectors and donor plasmids and performed flow cytometry analysis for GFP-positive cells after 5 days of puromycin selection. After Sanger sequencing the TA-cloned 5′- and 3′-junction PCR products of the Cas12a HITI, Cas12a MITI, and Cas9 HITI HeLa cells, we found that 70% of Cas12a MITI-mediated integrations were accurate at the 5′ junctions, whereas precision of Cas9 HITI integrations was only 16.67%, and Cas12a HITI had no precise integrations (Figs. 2b, S2A-D). At the 3′ junction, precision of Cas12a MITI-mediated integrations was less accurate (Fig. S2E), possibly due to re-digestion by Cas12a after integration. In brief, these results suggested Cas12a MITI facilitates efficient precise targeted integration. It should be noted that potential large deletions of GFP were not considered when analyzing the above results. However, this should not bias our conclusions since all three methods were analyzed the same way.

Schematic of Cas12a MITI, Cas12a HITI, and Cas9 HITI gene knock-in strategies. a Schematic of the Cas12a MITI system. The Cas12a MITI-based method needs to introduce modified compatible CrRNA target sequences in the donor. b Schematic of the Cas12a HITI system. The Cas12a HITI-based method requires a CrRNA target site in the donor in reverse orientation to the genomic target sequence resembling the Cas9 HITI approach. c Schematic of the Cas9 HITI system. The Cas9 HITI strategy requires an identical sgRNA target site in the donor but in reverse orientation to the genome target sequences. Green “TTTN” is the PAM sequence for the Cas12a. The gray scissors represents the Cas12a nuclease, and the black scissors represents the Cas9 nuclease

Comparison between the Cas12a HITI, Cas12a MITI, and Cas9 HITI strategies-mediated reporter knock-in at the AAVS1 locus. a Schematic of the Cas9 HITI, Cas12a HITI and Cas12a MITI strategies at the AAVS1 locus. The dotted lines represent the cleavage site of Cas9 or Cas12a. The sequence of the cleavage site is shown in vermilion for Cas12a MITI and in orange for Cas12a HITI. The protospacer adjacent motif (PAM) sequence of Cas12a is shown in green, and the PAM sequences of Cas9 are in blue. b The precise integration efficiencies of the three knock-in strategies at the AAVS1 locus. The result was presented as mean ± SD, n = 3, *P < 0.05, **P < 0.01, ***P < 0.001, unpaired Student’s t test
MITI allows for more efficient seamless reporter gene tagging
To compare the accuracy of Cas12a MITI and Cas9 HITI, we next used a 2A-tdTomato reporter system to tag a constitutively expressed gene CLTA in 293T cells (Fig. 3a). By flow cytometry analysis, we found that Cas12a MITI had a significantly higher percentage of tdTomato positive cells (14.40 ± 2.40%) than Cas9 HITI (5.30 ± 1.10%) (P = 0.026, Fig. 3b) after 5 days of puromycin selection. This low frequency of precise integration may be due to the existence of an identical target sequence for both Cas12a and Cas9 in the CLTA pseudogene on chromosome 12. The 5′-end integration junction of unsorted cells was amplified for sequence analysis, which showed that Cas12a MITI had a higher in-frame and precise integration rate than Cas9 HITI (Figs. 3c, S3). We further tested Cas12a MITI for targeting the silenced gene GREB1L with 2A-tdTomato in porcine fibroblasts (Fig. S4A). After transfection and puromycin selection, we picked 24 cell clones for PCR and sequencing analysis (Fig. S4B). We found that 91.67% of clones had the targeted integration at the 5′ junction and 81.82% of the integrations had the expected junction sequence (Fig. S4C). The above results further demonstrated that Cas12a MITI can be strategically used for more precise reporter gene targeting.

Tagging the CLTA gene using the Cas12a MITI or Cas9 HITI strategy in HEK293T cells. a Schematic of CLTA gene targeting using the two approaches. Red “TGA” base pairs are the termination codon of the CLTA gene. b Relative precise knock-in efficiency of the two strategies in HEK293T cells at the CLTA locus tested via the percentage of tdTomato positive cells evaluated by flow cytometry analysis. c The precise integration efficiencies of the two knock-in strategies at the CLTA locus via TA cloning and Sanger sequencing. All the results were presented as mean ± SD, n = 3, *P < 0.05, **P < 0.01, ***P < 0.001, unpaired Student’s t test
Two cleavages in the donor help exclude unintended integration of the plasmid backbone
To avoid integration of unwanted donor plasmid backbone, which may result in pronounced transgene silencing [10], a second Cas12a recognition sequence with perfect homology to the genome target sequence was added to the CLTA donor plasmid behind the puromycin gene (Fig. 4a). After transfection into HepG2 cells and 5 days of puromycin selection, we picked 16 tdTomato-positive clones. PCR and sequencing results showed 10 out of 16 tdTomato-positive cells had the predicted 5′ junction sequence (Fig. 4b, c), and 8 out of 16 clones were successfully targeted without the integration of the plasmid backbone (Fig. 4b, d). Further verification of these successful tagging events was confirmed with immunostaining (Fig. S5). These results indicate that Cas12a MITI can readily avoid plasmid backbone integration.

Tagging the CLTA gene using the two-cut donor in HepG2 cells. a Schematics of the donor plasmid and targeting strategy for CRISPR/Cas12a-mediated insertion of the 3 × FLAG-2A-tdTomato reporter at the CLTA locus in HepG2 cells. The two-cut donor (D4.1) includes two Cas12a recognition sites, one is the MITI-modified CrRNA target sequence, and the other is identical to the genomic target. The genetically modified CLTA locus may result in two different situations. One is that only the reporter part is knocked into the target site, and the other one is that the entire vector containing the prokaryotic backbone is integrated into the target site. b PCR identification of tdTomato-positive HepG2 clones. c Identification of tdTomato-positive HepG2 picked clones bearing predicted integration of 3 × FLAG-tdTomato by PCR and Sanger sequencing at the 5′ junction. d The sequences of the 3′ junction site of the representative HepG2 clone integrated with the whole D4.1 vector
Improved accuracy of both junctions with two cleavage sites on MITI donors
To improve the sequence fidelity of both junctions between endogenous DNA and the exogenous gene, we selected two different neighboring genomic sites to target, and placed the corresponding MITI recognition sites around the foreign reporter gene in the donor vector. Meanwhile, we also introduced a herpes simplex virus thymidine kinase (HSV-tk) suicide cassette—a negative selection marker for avoiding random integration of the targeting vector [24, 25]—positioned outside of the reporter gene to exclude unintended genomic integration of donor plasmid (Fig. 5a). Two MITI cleavages on the donor plasmid eliminate the HSV-tk suicide gene along with cell sensitivity to Fialuridine (FIAU). After transfection and drug selection, we picked puromycin and FIAU dual resistant clones and analyzed both 5′ and 3′-end junction sequences by PCR sequencing. We found that in the puromycin and FIAU dual resistant clones, precise 3′ junction integration efficiency was improved (Fig. 5b, c). Our results suggest that two cleavage sites in combination with negative selection facilitate precise targeted integration of exogenous genes at both junctions. However, when using pairs of CrRNAs, complicated repair events may occur [26], and should be further examined in the future.

Two-cut MITI and negative selection strategy using Cas12a improved the accuracy of both junctions. a Schematics of the donor plasmid and targeting strategy for CRISPR/Cas12a-mediated insertion of the SA-IRES-GFP reporter at the AAVS1 locus. The donor plasmid carries two modified Cas12a target sites at both the 5′ and 3′ sides of SA-IRES-GFP (two-MITI donor, D7) which correspond to the genomic target sites, and a negative selection gene herpes simplex virus thymidine kinase (HSV-tk) in the prokaryotic backbone of the donor. b Detection of targeted integration of SA-IRES-GFP cassette from puromycin and FIAU dual resistant clones via PCR. Seven out of eleven clones had expected integration at the left and right junctions. c The sequencing results of the 5′- and 3′-integration junctions amplified from GFP-positive clones produced by the two-MITI donor
Discussion
In this study, we demonstrated that sticky ends generated by Cas12a could facilitate the precise integration of foreign DNA fragments into the target genomic locus. The MITI method relies on Cas12a-induced stable complementary sticky overhangs which can enhance the efficiency and precision of in situ reporter gene targeting, and consequently greater fidelity of targeted gene expression. Our side-by-side comparison experiment revealed that the accuracy of end joining with MITI tends to be higher than the Cas9 HITI strategy at the different loci tested. MITI could be an alternative tool for future gene therapy applications. However, it should be noted that the method we used has some limitations. The number of colonies analyzed in the current study is still small. The results could be further strengthened using NGS.
Using a dual MITI strategy, we demonstrated the feasibility of precisely replacing a segment of DNA with the desired external DNA sequence via simultaneous cleavage of two neighboring sites in the genome and two corresponding target sites in the donor vector flanked by a negative selection cassette. In contrast, the Cas9 HITI strategy is currently capable of inserting foreign DNA into only a single genomic locus. This dual MITI strategy may be useful in certain situations, such as adding the loxP sequences to both sides of the exon for constructing conditional knockout animals or substituting the whole target gene with the desired coding sequences.
When tagging endogenous genes with a reporter, Cas9-mediated homology-independent knock-in via HITI always deletes the last few codons of the gene, destroying its integrity, which might have an impact on the function of the gene. For example, in our study, when the reporter cassette was used to tag the human CLTA gene using a Cas9 HITI approach, six base pairs were inevitably deleted (Fig. S3). Our MITI approach avoided such genetic deletion by adding essential base pairs after the cleavage site in the donor making it more flexible than the Cas9 HITI method.
Nonetheless, further examination of potential limitations and optimization of the MITI method is required. For example, widespread nickase activity of Cas12a [27,28,29] has been reported and may cause random insertion of foreign genes. However, since the fidelity of Cas12a is higher than Cas9 nuclease [11, 12], MITI is likely to have fewer off-target integrations than Cas9 HITI. Furthermore, when using pairs of CrRNAs for dual Cas12a targeting at two different loci, complicated repair events may occur, similar to that seen for CRISPR/Cas9, including the formation of extrachromosomal circular DNA, DNA inversion, and translocation [26]. This should be further tested and verified in the future. Considering there are many kinds of Cas12 with different specificities, potencies and PAM dependence [17, 30,31,32,33], it is possible to extend editable sequence space and enable further optimization of our MITI method.
Abbreviations
- MITI:
-
Microhomology-dependent targeted integration
- HITI:
-
Homology-independent targeted integration
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- CrRNAs:
-
CRISPR RNAs
- DSB:
-
Double-strand break
- NHEJ:
-
Non-homology end joining
- HDR:
-
Homology-directed repair
- GOI:
-
Gene-of-interest
- ZFNs:
-
Zinc finger nucleases
- ObLiGaRe:
-
Obligate ligation-gated recombination
- TALENs:
-
Tale nucleases
- PFFs:
-
Pig fetal fibroblasts
- HSV-tk :
-
Herpes simplex virus thymidine kinase
References
Yamamoto Y, Gerbi SA (2018) Making ends meet: targeted integration of DNA fragments by genome editing. Chromosoma 127:405–420. https://doi.org/10.1007/s00412-018-0677-6
Heyer W-D, Ehmsen KT, Liu J (2010) Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44:113–139. https://doi.org/10.1146/annurev-genet-051710-150955
Maresca M, Lin VG, Guo N, Yang Y (2013) Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res 23:539–546. https://doi.org/10.1101/gr.145441.112
Auer TO, Duroure K, De Cian A et al (2014) Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res 24:142–153. https://doi.org/10.1101/gr.161638.113
Kimura Y, Hisano Y, Kawahara A, Higashijima SI (2014) Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci Rep 4:6545. https://doi.org/10.1038/srep06545
Bachu R, Bergareche I, Chasin LA (2015) CRISPR-Cas targeted plasmid integration into mammalian cells via non-homologous end joining. Biotechnol Bioeng 112:2154–2162. https://doi.org/10.1002/bit.25629
He X, Tan C, Wang F et al (2016) Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res 44:e85. https://doi.org/10.1093/nar/gkw064
Lackner DH, Carré A, Guzzardo PM et al (2015) A generic strategy for CRISPR-Cas9-mediated gene tagging. Nat Commun 6:10237. https://doi.org/10.1038/ncomms10237
Geisinger JM, Turan S, Hernandez S et al (2016) In vivo blunt-end cloning through CRISPR/Cas9-facilitated non-homologous end-joining. Nucleic Acids Res 44:e76. https://doi.org/10.1093/nar/gkv1542
Suzuki K, Tsunekawa Y, Hernandez-Benitez R et al (2016) In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540:144–149. https://doi.org/10.1038/nature20565
Kim D, Kim J, Hur JK et al (2016) Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 34:863–868. https://doi.org/10.1038/nbt.3609
Kleinstiver BP, Tsai SQ, Prew MS et al (2016) Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol 34:869–874. https://doi.org/10.1038/nbt.3620
Fonfara I, Richter H, BratoviÄ M et al (2016) The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532:517–521. https://doi.org/10.1038/nature17945
Zetsche B, Heidenreich M, Mohanraju P et al (2017) Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 35:31–34. https://doi.org/10.1038/nbt.3737
Jinek M, Chylinski K, Fonfara I et al (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. https://doi.org/10.1126/science.1225829
Zetsche B, Gootenberg JS, Abudayyeh OO et al (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771. https://doi.org/10.1016/j.cell.2015.09.038
Li SY, Zhao GP, Wang J (2016) C-Brick: a new standard for assembly of biological parts using Cpf1. ACS Synth Biol 5:1383–1388. https://doi.org/10.1021/acssynbio.6b00114
Lei C, Li SY, Liu JK et al (2017) The CCTL (Cpf1-assisted Cutting and Taq DNA ligase-assisted Ligation) method for efficient editing of large DNA constructs in vitro. Nucleic Acids Res 45:e74. https://doi.org/10.1093/nar/gkx018
Jiang W, Zhao X, Gabrieli T et al (2015) Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat Commun 6:8101. https://doi.org/10.1038/ncomms9101
Wang JW, Wang A, Li K et al (2015) CRISPR/Cas9 nuclease cleavage combined with Gibson assembly for seamless cloning. Biotechniques 58:161–170. https://doi.org/10.2144/000114261
Wu S, Ying G, Wu Q, Capecchi MR (2007) Toward simpler and faster genome-wide mutagenesis in mice. Nat Genet 39:922–930. https://doi.org/10.1038/ng2060
Xu C, Qi X, Du X et al (2017) piggyBac mediates efficient in vivo CRISPR library screening for tumorigenesis in mice. Proc Natl Acad Sci USA 114:722–727. https://doi.org/10.1073/pnas.1615735114
Wu S, Ying G, Wu Q, Capecchi MR (2008) A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nat Protoc 3:1056–1076. https://doi.org/10.1038/nprot.2008.70
Mansour SL, Thomas KR, Capecchi MR (1988) Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 336:348–352. https://doi.org/10.1038/336348a0
Wu S, Wu Y, Zhang X, Capecchi MR (2014) Efficient germ-line transmission obtained with transgene-free induced pluripotent stem cells. Proc Natl Acad Sci USA 111:10678–10683. https://doi.org/10.1073/pnas.1409933111
Møller HD, Lin L, Xiang X et al (2018) CRISPR-C: circularization of genes and chromosome by CRISPR in human cells. Nucleic Acids Res 46:e131. https://doi.org/10.1093/nar/gky767
Fuchs RT, Curcuru J, Mabuchi M et al (2019) Cas12a trans-cleavage can be modulated in vitro and is active on ssDNA, dsDNA, and RNA. bioRxiv. https://doi.org/10.1101/600890
Murugan K, Seetharam AS, Severin AJ, Sashital DG (2019) Pervasive off-target and double-stranded DNA nicking by CRISPR-Cas12a. bioRxiv. https://doi.org/10.1101/657791
Fu BXH, Smith JD, Fuchs RT et al (2019) Target-dependent nickase activities of the CRISPR-Cas nucleases Cpf1 and Cas9. Nat Microbiol 4:888–897. https://doi.org/10.1038/s41564-019-0382-0
Sun H, Li F, Liu J et al (2018) A single multiplex crRNA array for FnCpf1-mediated human genome editing. Mol Ther 26:2070–2076. https://doi.org/10.1016/j.ymthe.2018.05.021
Tu M, Lin L, Cheng Y et al (2017) A ‘new lease of life’: FnCpf1 possesses DNA cleavage activity for genome editing in human cells. Nucleic Acids Res 45:11295–11304. https://doi.org/10.1093/nar/gkx783
Teng F, Cui T, Feng G et al (2018) Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov 4:63. https://doi.org/10.1038/s41421-018-0069-3
Strecker J, Jones S, Koopal B et al (2019) Engineering of CRISPR-Cas12b for human genome editing. Nat Commun 10:212. https://doi.org/10.1038/s41467-018-08224-4
Acknowledgements
We are grateful for continuous support from the Wu lab members. This work was supported by the Transgenic Research Grant 2016ZX08010001, the Transgenic Research Grant 2016ZX08009003, and Project for Extramural Scientists of State Key Laboratory of Agrobiotechnology (Grant no. 2016SKLAB1-8).
Author information
Authors and Affiliations
Contributions
SW, XD, PL, and CX conceived and designed the experiments. PL, LZ, and ZL performed the experiments. SW, XD, PL, and CX wrote and edited the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, P., Zhang, L., Li, Z. et al. Cas12a mediates efficient and precise endogenous gene tagging via MITI: microhomology-dependent targeted integrations. Cell. Mol. Life Sci. 77, 3875–3884 (2020). https://doi.org/10.1007/s00018-019-03396-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03396-8