
Abstract
Gap junctions consist of arrays of intercellular channels that enable adjacent cells to communicate both electrically and metabolically. Gap junctions have a wide diversity of physiological functions, playing critical roles in both excitable and non-excitable tissues. Gap junction channels are formed by integral membrane proteins called connexins. Inherited or acquired alterations in connexins are associated with numerous diseases, including heart failure, neuropathologies, deafness, skin disorders, cataracts and cancer. Gap junctions are highly dynamic structures and by modulating the turnover rate of connexins, cells can rapidly alter the number of gap junction channels at the plasma membrane in response to extracellular or intracellular cues. Increasing evidence suggests that ubiquitination has important roles in the regulation of endoplasmic reticulum-associated degradation of connexins as well as in the modulation of gap junction endocytosis and post-endocytic sorting of connexins to lysosomes. In recent years, researchers have also started to provide insights into the physiological roles of connexin ubiquitination in specific tissue types. This review provides an overview of the advances made in understanding the roles of connexin ubiquitination in the regulation of gap junction intercellular communication and discusses the emerging physiological and pathophysiological implications of these processes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gap junctions are plasma membrane domains containing arrays of intercellular channels that allow for the direct transfer of ions and small molecules (< ~ 1.2 kDa) between cells [1]. In vertebrates, gap junction channels are formed by a family of transmembrane proteins called connexins, which in humans constitutes 20 members [2]. Connexins are expressed in almost all cell types of the human body and have essential roles during development as well as in the adult organism. In providing a pathway for electrical communication, gap junctions are fundamental to the function of excitable cells, such as neurons, cardiomyocytes and smooth muscle cells [3]. Moreover, by enabling the intercellular exchange of small metabolites and second messengers, gap junctions have a plethora of essential roles in non-excitable tissues, including the regulation of cell proliferation and differentiation and the maintenance of tissue homeostasis [4, 5]. There is also increasing evidence that connexins have important cell physiological functions that go beyond their ability to form canonical gap junctions. For instance, recent studies have demonstrated that connexins are involved in cell–cell communication via tunneling nanotubes and extracellular vesicles [6]. Moreover, connexin-based channels at the plasma membrane that are not assembled into gap junctions, also known as hemichannels, have central roles in autocrine and paracrine signaling by providing a pathway for the exchange of ions and small molecules between the intracellular and extracellular milieus [7]. In addition, the intracellular domains of connexins can interact with other proteins, including components of the cytoskeleton, such as tubulin, and cell signaling pathways, such as cyclin E and β-catenin [8,9,10,11]. Through such protein–protein interactions, connexins can modulate cell growth, differentiation, migration and other cellular processes in channel-independent manners. The multifaceted roles of connexins in human physiology are reflected by the fact that inherited or acquired alterations in connexins are associated with numerous diseases, including heart failure, neuropathologies, deafness, skin disorders, cataracts and cancer [12,13,14]. Obtaining a better understanding of the molecular basis underlying the loss of connexin function in disease pathogenesis may have important therapeutic implications [15,16,17].
The connexin pool that constitutes gap junctions is continuously replaced due to the formation of new intercellular channels at their edges and the removal of old channels from their center by endocytosis and subsequent degradation in lysosomes [18,19,20,21,22]. This dynamic nature of gap junctions is reflected by the observation that the connexins have a high turnover rate in most tissue types, typically displaying half-lives of 1.5-5 h [23,24,25]. Substantial evidence suggests that cells can modulate the connexin degradation rate in response to various extracellular or intracellular cues to alter the number of functional gap junction channels at the plasma membrane [20, 26,27,28,29,30,31,32].
An increasing body of experimental work suggests that post-translational modification of connexins by ubiquitination has an important role in regulating the level of gap junction intercellular communication. Ubiquitination has been suggested to be involved in controlling the degradation of de novo synthesized connexins at the endoplasmic reticulum, in modulating the rate of gap junction endocytosis, and in sorting connexins to lysosomes via either the autophagosomal or the endolysosomal pathways. In recent years, researchers have also started to provide insights into the physiological roles of connexin ubiquitination in specific tissue types and how dysregulation of this process may contribute to loss of gap junctions during disease pathogenesis. This review provides an up-to-date overview of the current understanding of the role of connexin ubiquitination in the regulation of gap junctional intercellular communication. We also discuss the emerging physiological and pathophysiological implications of connexin ubiquitination, focusing on the heart, lens and central nervous system. The implications of connexin ubiquitination for cancer pathogenesis have previously been reviewed elsewhere [33] and this topic will, therefore, not be covered here.
Connexin biosynthesis and assembly into gap junctions
Connexin proteins are named according to their approximate molecular weight (in kDa) [1]. The best studied and among the most ubiquitously expressed connexin isoforms in human tissues is connexin 43 (Cx43) [1]. The connexins are four-pass transmembrane proteins displaying their N- and C-terminal tails in the cytosol (Fig. 1a) [34]. The transmembrane domains and the two extracellular loops are highly conserved between the connexin family members. In contrast, the intracellular loop and N- and C-terminal tails exhibit great variation in their length and amino acid sequence between connexins. These regions have important roles in modulating gap junction channel gating and intracellular trafficking of connexins. For instance, the C-terminal tail of Cx43 contains multiple sites for protein–protein interactions and post-translational modifications that play important roles in the regulation of gap junctions [10, 35].

Connexins, connexons and gap junction channels. a Connexins have four transmembrane domains, which are connected by two extracellular loops and one intracellular loop. The N- and C-termini are both located in the cytosol. b Connexins form hexamers called connexons. Connexins can combine with either the same or different connexin isoforms, forming homomeric or heteromeric connexons, respectively. c Connexons form gap junction channels by interacting with either identical homomeric or heteromeric connexons in adjacent cells, forming homotypic channels, or with different homomeric or heteromeric connexons, forming heterotypic channels
Connexins are co-translationally inserted into the endoplasmic reticulum [36, 37]. A subpool of newly synthesized connexins undergoes endoplasmic reticulum-associated degradation (ERAD), a process in which they are retrotranslocated into the cytosol and degraded by the proteasome (Fig. 2) [38,39,40]. Connexins that are spared from ERAD are transported from the endoplasmic reticulum via the Golgi apparatus and the trans-Golgi network to the plasma membrane [41, 42]. Along the secretory pathway from the endoplasmic reticulum to the plasma membrane, connexins oligomerize into hexameric structures called connexons [43,44,45]. Connexons can consist of either six similar connexin isoforms or a combination of different connexin isoforms, referred to as homomeric and heteromeric connexons, respectively (Fig. 1b) [46, 47].

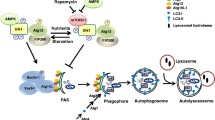
Intracellular trafficking and ubiquitination and SUMOylation of connexins. Connexins are co-translationally inserted into the endoplasmic reticulum and transported through the Golgi, through the trans-Golgi and to the plasma membrane. Along their trafficking to the plasma membrane, connexins oligomerize into connexons. A subpool of the newly synthesized connexins undergoes ERAD, in which connexins are retrotranslocated to the cytosol and degraded by proteasomes. In the plasma membrane, connexons can function as hemichannels or diffuse to the periphery of gap junctions, where they can dock with connexons in adjacent cells to form intercellular channels. During endocytosis of gap junctions, both membranes of the junctions are internalized into one of the cells, thereby forming a connexosome, also called an annular gap junction. Three different pathways for trafficking of connexins to lysosomes are illustrated in the figure: (1) direct fusion between connexosomes and lysosomes, (2) sequestration of the connexosome by a phagophore and subsequent fusion between an autophagosome containing the connexosome and a lysosome, and (3) transformation of the connexosome into a connexin-enriched, multivesicular endosome with a single limiting membrane, which is associated with the fusion between the connexosome and early endosomes. Connexins are then sorted from early endosomes via late endosomes to lysosomes. Connexons at the plasma membrane that are not assembled into gap junctions can also undergo endocytosis, but their intracellular trafficking is poorly characterized. They are possibly transported to early endosomes (indicated by a question mark). Following endocytosis, connexins may undergo recycling to the plasma membrane, possibly from the early endosomes (indicated by a question mark). Connexin ubiquitination has been suggested to be involved in ERAD of certain connexin isoforms, gap junction endocytosis, autophagy-mediated degradation of connexins and sorting of connexins from early endosomes to lysosomes. Eps15 has been suggested to bind to ubiquitinated Cx43 at the plasma membrane and to control gap junction endocytosis and autophagy-mediated degradation. Tsg101 is part of ESCRT and has been suggested to bind to ubiquitinated Cx43 at the limiting membrane of early endosomes and to regulate its sorting into the lumen of the endosome. p62 has been suggested to bind to ubiquitinated Cx43 after gap junction endocytosis and to be involved in the sequestration of connexosomes by autophagosomes. Cx43 has also been shown to be modified by SUMOylation, which has been suggested to stabilize Cx43 and consequently cause increased gap junction size
In the trans-Golgi network, connexons are packaged into vesicles that are transported along microtubules to the plasma membrane [18, 21, 22, 48]. Connexons can be delivered to regions of un-apposed plasma membrane where they diffuse laterally in the lipid bilayer until they reach the periphery of existing gap junctions [18, 21, 49, 50]. Alternatively, connexons can be sorted directly to, or in the immediate vicinity of, existing gap junctions [22]. The connexons then dock with connexons in the plasma membrane of neighboring cells to form gap junction intercellular channels. Connexons can form channels either with identical homomeric or heteromeric connexons in adjacent cells (homotypic channels) or with channels with different homomeric or heteromeric connexons (heterotypic channels) (Fig. 1c) [46, 47]. The connexin composition of the gap junction channels strongly affects the conductance and selectivity of the channels.
In addition to full-length Cx43, six N-terminally truncated Cx43 isoforms can form as a result of internal translation initiation [51,52,53,54]. Emerging evidence indicates that such truncated versions of Cx43 have important roles in controlling the intracellular trafficking of full-length Cx43 [41, 42]. For instance, a truncated 20-kDa Cx43 isoform, termed GJA1-20 k, has been shown to increase the delivery of Cx43 hemichannels to cardiac intercalated discs to increase the gap junction plaque size [55]. In addition, truncated Cx43 isoforms may have important functions in the nucleus. In accordance with this notion, GJA1-20 k was recently demonstrated to control neural crest cell migration in vivo by directly regulating N-cadherin transcription [56].
Endocytosis and degradation of gap junctions
During gap junction endocytosis, both membranes of the junction are internalized into one of the adjacent cells, thereby forming a double-membrane vesicle called an annular gap junction or connexosome [57, 58]. Either an entire gap junction or only a fragment of it can be internalized. The endocytosis of gap junctions involves clathrin [59,60,61,62]. Cx43 contains two functional tyrosine-based motifs (265YAYF268 and 286YKLV289) in its C-terminal tail that conform to the consensus YXXϕ (where Y is tyrosine, X is any amino acid and ϕ is an amino acid with a bulky hydrophobic side chain) [63, 64]. These motifs bind to the clathrin adaptor AP-2, which then recruits clathrin directly or indirectly via the clathrin adaptor Dab2 [63]. Other proteins shown to be involved in gap junction endocytosis include dynamin, 14-3-3 and the retrograde actin motor myosin-VI [62, 65,66,67].
Subsequent to gap junction endocytosis, connexins can follow different post-endocytic pathways before they are degraded in lysosomes (Fig. 2) [68,69,70]. Sometimes, connexosomes can fuse directly with lysosomes [30, 71,72,73]. In other situations, they can be sequestered by autophagosomes, which subsequently fuse with lysosomes [74,75,76,77,78]. Moreover, connexosomes have been suggested to be able to undergo a transformation into a connexin-enriched multivesicular endosome, which is associated with their fusion with early endosomes [79,80,81,82]. Some studies also indicate that following endocytosis, connexins can under certain conditions undergo recycling from endocytic compartments back to the plasma membrane [32, 75, 81, 83,84,85]. It has also been suggested that Cx43 can be transported directly from early secretory compartments to lysosomes without prior transport to the plasma membrane [30].
Post-translational modifications of connexins
Connexins are subjected to several different types of post-translational modifications, which contribute to the regulation of gap junctions by different mechanisms [86]. Among the post-translational modifications of connexins that have been identified are phosphorylation, ubiquitination, SUMOylation, acetylation, methylation, hydroxylation and nitrosylation [86].
Kinases known to catalyze the phosphorylation of Cx43 include mitogen-activated protein kinase (MAPK), protein kinase C (PKC), casein kinase 1, protein kinase A, c-Src and v-Src [87]. Phosphorylation regulates several stages of the connexin “life cycle”. For instance, phosphorylation of Cx43 on serine or tyrosine residues located in the C-terminal tail has been shown to regulate Cx43 trafficking from the Golgi apparatus to the plasma membrane, the oligomerization of Cx43 into connexons, the assembly of connexons into gap junctions, the gating of gap junction channels, and gap junction endocytosis and degradation [88]. As described below, increasing evidence suggests that gap junction endocytosis and connexin degradation are controlled by complex crosstalk between connexin phosphorylation and ubiquitination.
The ubiquitin system
Ubiquitin is a small globular protein that can be covalently attached to the ε-amino group of an internal lysine residue or free N-terminal α-amino group of the substrate, in a process known as ubiquitination [89]. A substrate can be subjected to different types of ubiquitin modifications, each of which can have different effects on the substrate. Proteins can be modified by a single ubiquitin, in a process known as monoubiquitination; by single ubiquitin moieties on several lysines, known as multiple monoubiquitination; or by a polyubiquitin chain, known as polyubiquitination. Ubiquitin has seven lysine residues that can all be involved in polyubiquitination, forming linear or branched ubiquitin chains. The distinct ubiquitin modifications are recognized by different effector proteins with specific ubiquitin-binding domains, thereby coupling the ubiquitination of a substrate to a downstream event, such as protein degradation, sorting to a specific subcellular location or assembly of a signaling complex [89]. For instance, modification of transmembrane proteins by polyubiquitin chains linked via Lys63 may act as a signal for their regulated endocytosis and subsequent targeting to lysosomes [89]. In contrast, modification of proteins by polyubiquitin chains linked via Lys48 may target proteins for degradation by the 26S proteasome [89].
Conjugation of ubiquitin to the target protein proceeds through a cascade mechanism that involves three enzymes: E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme and E3 ubiquitin ligase [90]. First, ubiquitin forms a high-energy thioester bond with a cysteine residue of an E1 ubiquitin-activating enzyme, a process requiring ATP. Ubiquitin is then transferred to an E2 ubiquitin-conjugating enzyme, forming a similar thioester linkage with a specific catalytic cysteine residue. E3 ubiquitin ligases recruit E2 ubiquitin-conjugating enzymes loaded with ubiquitin. The E3 ubiquitin ligase then recognizes a specific substrate and facilitates or directly transfers and conjugates ubiquitin to the target protein. E3 ubiquitin ligases can either conjugate ubiquitin to lysine residues in the target protein or conjugate ubiquitin to already attached ubiquitins, forming polyubiquitin chains [90].
The key regulatory determinants in protein degradation are the E3 ubiquitin ligases. While the human genome encodes two E1 activating enzymes and 37 E2 conjugating enzymes, it encodes over 600 E3 ubiquitin ligases [90]. E3 ubiquitin ligases are generally grouped into three subfamilies: the really interesting new gene (RING) type E3s, the U-box E3s and the homologous to E6-AP carboxyl terminus (HECT) domain-containing E3s [90].
Deubiquitinating enzymes are proteases that cleave ubiquitin off the substrate protein or within ubiquitin moieties in a polyubiquitin chain [91]. The human genome encodes approximately 100 deubiquitinating enzymes, the majority of which are cysteine proteases. The E3 ubiquitin ligases and deubiquitinating enzymes are key regulators of numerous intracellular processes and have been linked to the pathogenesis of a number of human diseases, including cancer and neurodegenerative disorders [92].
Connexin ubiquitination
The first experimental evidence to indicate that Cx43 is ubiquitinated was provided by Laing and Beyer [27]. Using E36 Chinese hamster ovary cells containing a temperature-sensitive defect in the ubiquitin-activating protein E1, the authors also demonstrated that efficient Cx43 degradation requires an intact ubiquitin conjugation system [27]. In accordance with the notion that ubiquitination is involved in the regulation of gap junctions, Rütz and Hülser demonstrated by electron microscopy that ubiquitin is present in gap junction plaques, as determined by immunogold labeling of freeze-fractured plasma membranes [93]. Subsequent studies carried out in our laboratory indicated that endocytosis of gap junctions and Cx43 degradation may be regulated by crosstalk between Cx43 phosphorylation and ubiquitination. Using as a model system IAR20 rat liver epithelial cells, which express Cx43 endogenously, we found Cx43 ubiquitination to be induced by epidermal growth factor (EGF) [28]. The EGF-induced ubiquitination of Cx43 was MAPK dependent and was associated with increased endocytosis and degradation of gap junctions. Subsequently, treatment of cells with the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA), a potent activator of PKC, was found to result in strongly increased Cx43 ubiquitination, which correlated with enhanced gap junction endocytosis and degradation of Cx43 in lysosomes [94]. As determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis, Cx43 in IAR20 cells appeared to be modified by one to four ubiquitin moieties [28, 94]. Analyses of the solubility of the ubiquitinated Cx43 pool in the detergent Triton X-100 indicated that Cx43 can be ubiquitinated while assembled into gap junctions and that Cx43 can remain ubiquitinated during gap junction endocytosis and along its sorting to early endosomes [81, 95].
Studies by Cuervo and colleagues have demonstrated that ubiquitination of Cx43 is induced in response to activation of macroautophagy by serum starvation [74]. Cx43 ubiquitination acts as a signal for gap junction endocytosis and autophagy-mediated degradation by recruiting the ubiquitin-binding protein Eps15 (epidermal growth factor receptor substrate 15) [74, 96] (Fig. 2). It has been suggested that Eps15 acts as an intermediate bridge molecule between ubiquitinated Cx43 and the autophagy machinery at the plasma membrane [74, 96]. Cx43 has also been shown to bind to the ubiquitin-binding autophagic adaptor p62 (also known as SQSTM1) [74, 76, 78]. The interaction between Cx43 and p62 increases in response to serum starvation or when a ubiquitin molecule is fused in-frame to the C-terminus of Cx43 to mimic Cx43 ubiquitination [74]. Cx43 co-localizes with p62 in intracellular vesicular compartments [74, 76, 78]. Moreover, depletion of p62 has been shown to result in accumulation of connexosomes [76]. These observations raise the possibility that ubiquitination of Cx43 may have a role in the sequestration of connexosomes by autophagosomes by recruiting p62 [74, 76, 78].
Cx43 ubiquitination has also been proposed to control its post-endocytic sorting from early endosomes to lysosomes [81]. Ubiquitinated forms of Cx43 have been suggested to be recognized by ubiquitin-binding proteins of the endosomal sorting complex required for transport (ESCRT), which is located at the limiting membrane of endosomes (Fig. 2). ESCRT is then thought to mediate the deubiquitination and subsequent sorting of Cx43 into the lumen of the endosomes [81]. In accordance with this notion, Cx43 has been shown to interact with the ubiquitin-binding protein Tsg101 (tumor susceptibility gene 101 protein), a member of ESCRT [97]. Depletion of Tsg101 results in increased Cx43 protein levels and enhanced levels of functional gap junctions at the plasma membrane, possibly due to increased recycling of Cx43 from early endosomes to the plasma membrane [81]. Moreover, under conditions in which Tsg101 is co-depleted with another ubiquitin-binding protein and member of ESCRT, Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate), Cx43 accumulates at the limiting membrane of early endosomes in a hyperubiquitinated form [81].
In addition to Cx43, several other connexin isoforms have been shown to be ubiquitinated (Table 1). As well as playing a role in modulating gap junction endocytosis and post-endocytic sorting of connexins to lysosomes, connexin ubiquitination has been shown to be involved in ERAD. However, the involvement of ubiquitination in ERAD appears to be connexin isoform specific. For instance, ERAD of Cx43 occurs independently of ubiquitination, whereas ERAD of Cx32 and Cx40 involves polyubiquitination [98,99,100]. Kelly et al. have shown that ERAD of connexins is inhibited by mild forms of cytosolic stress at a step before their dislocation into the cytosol [40]. To investigate the mechanisms underlying this inhibition of connexin degradation, a mutant version of Cx32 associated with Charcot–Marie–Tooth X-linked peripheral neuropathy was studied [99]. This mutant Cx32 is confined to the endoplasmic reticulum and thus not subject to lysosomal degradation, and it was found to be modified by polyubiquitination [99]. Moreover, its level of polyubiquitination was found to be reduced in response to cytosolic stress, which was associated with its reduced ERAD [99]. These observations suggest that the inhibition of ERAD of connexins induced by cytosolic stress may be due to reduced connexin polyubiquitination.
An important question is which lysines in Cx43 and other connexin isoforms act as ubiquitination sites, and how ubiquitination of specific lysine residues affects gap junction levels. A study by Dunn et al. found that a mutant version of Cx43 with all lysines converted to arginines behaves similarly to Cx43-wt in the presence of proteasomal and lysosomal inhibitors [101]. The authors also demonstrated that proteasomal inhibition causes increased activation of Akt (protein kinase B), which is associated with enhanced Akt-mediated phosphorylation of Cx43 and, consequently, stabilization of Cx43 gap junctions [101]. The authors concluded that ubiquitination and proteasomal degradation of Akt, but not Cx43 ubiquitination, is involved in the regulation of Cx43 turnover. A subsequent study by Kells-Andrews, Margraf et al. indicated that two lysine residues located in the C-terminal tail of Cx43, Lys264 and Lys303 act as ubiquitin conjugation sites [102]. Mutation of these lysine residues to arginines resulted in the accumulation of Cx43 at the plasma membrane and increased its half-life. Under these conditions, Cx43 was found to be in a hyper-phosphorylated state, which could indicate that Cx43 ubiquitination is triggered by Cx43 phosphorylation under basal conditions. On the basis of these observations, the authors concluded that Cx43 ubiquitination is required for the constitutive gap junction internalization and degradation [102]. A recent study by Alaei et al. demonstrated that mutating five lysine residues in the C-terminal tail of Cx32 to arginines resulted in both reduced Cx32 acetylation and ubiquitination, and both these modifications were suggested to be involved in regulating Cx32 turnover [103]. Acetylation and ubiquitination were suggested to result in Cx32 stabilization and degradation, respectively [103]. The authors also demonstrated that mutating the five above-mentioned lysine residues to glutamine, which mimics acetylation, results in a stronger reduction in Cx32 ubiquitination as compared with when they are mutated to arginines [103]. These observations raise the possibility that Cx32 ubiquitination is negatively regulated by Cx32 acetylation, possibly through direct competition for the same lysine residues.
The available experimental evidence indicates that connexins can be subjected to different types of ubiquitin conjugation, including monoubiquitination (e.g., [104, 105]), multiple monoubiquitination (e.g., [94, 96]) and Lys63- or Lys48-linked polyubiquitination (e.g., [98,99,100, 102, 106,107,108,109]). It is likely that the type of ubiquitination that connexins are subjected to varies according to their subcellular localization. In future studies, it will be important to provide a more detailed understanding of the type of ubiquitin modifications that connexins are subjected to at different subcellular localizations and to better understand the functional consequences of the various types of modifications.
In addition to ubiquitination, Cx43 has been found to be modified by the ubiquitin-like protein SUMO (small ubiquitin-related modifier) [110]. In contrast to ubiquitination, SUMOylation of Cx43 has been proposed to stabilize Cx43 at the plasma membrane (Figs. 2 and 3; Table 2) [110]. This raises the possibility that gap junction endocytosis and degradation are controlled by an interplay between Cx43 ubiquitination and SUMOylation.

Overview of proteins involved in controlling gap junctions by connexin ubiquitination or SUMOylation. The figure depicts the proteins identified to date that participate in the regulation of gap junction intercellular communication through connexin ubiquitination or SUMOylation and the possible subcellular localizations where they may interact with connexins. For details, see Table 2 and the main text
Regulation of Cx43 gap junctions by NEDD4
The available data suggest that Cx43 is regulated by several E3 ubiquitin ligases (Fig. 3; Table 2). Among these, NEDD4 (neural precursor cell expressed developmentally down-regulated protein 4) is the best characterized. NEDD4 was originally discovered as a developmentally down-regulated gene in mouse brain [111]. NEDD4 is the founding member of the NEDD4 family of E3 ubiquitin ligases, which consists of nine members in humans [112]. The members of this protein family are characterized by a C-terminal catalytic HECT domain, a central protein–protein interaction region composed of 2–4 WW domains and an N-terminal membrane-targeting C2 domain. WW domains are protein modules that mediate protein–protein interactions by recognizing and binding proline-rich motifs and phosphorylated serine/threonine–proline sites in substrate proteins [113]. They are found in a variety of structural and signaling proteins and are involved in numerous cellular processes, including protein trafficking. WW domains consist of 38–40 amino acids and are characterized by two highly conserved tryptophan residues separated by 20–23 amino acids [113]. Based on their binding motif preference, WW domains are divided into five classes [113]. The WW domains of NEDD4 belong to class I, which bind to PY motifs (PPXY, where P is proline, X is any amino acid, and Y is tyrosine) in the substrate proteins. Cx43 contains a PY motif in its C-terminal tail (283PPGY286) [64].
In 2006, Leykauf et al. identified NEDD4 as the first E3 ubiquitin ligase to interact with Cx43. As determined by pull-down assays, rat NEDD4 (rNEDD4) was found to bind to the C-terminal tail of rat Cx43 (rCx43) and the interaction was shown to involve all three WW domains (WW1–3) of rNEDD4 [114]. Only WW2 was suggested to bind to the PY motif of Cx43, whereas the WW1 and WW3 domains were suggested to interact with other regions in the Cx43 C-terminal tail [114]. More recently, however, using various biophysical techniques, including nuclear magnetic resonance spectroscopy, Spagnol et al. provided evidence that all three WW domains of rNEDD4 bind to the PY motif of rCx43 [115]. WW2 was shown to have the highest binding affinity for the PY motif of rCx43, followed by WW3 and WW1. Mutation of the tyrosine in the PY motif (Y286A) led to a significant reduction in binding affinity for all WW domains, underscoring the importance of this residue in the interaction between the C-terminal tail of Cx43 and NEDD4.
Both the two aforementioned studies further indicated that phosphorylation of Cx43 is important for the interaction with NEDD4. Leykauf et al. found that treating cells with EGF, which causes MAPK-mediated phosphorylation of Cx43 at serine residues 255, 262, 279 and 282, increased the binding capacity of the WW3 domain of NEDD4 to Cx43 [114]. Interestingly, Ser279 and Ser282 are located just N-terminally to the PY motif, and phosphorylation of these residues (279pSPMpSPPGY286 where pS is phosphoserine) creates a potential class IV WW domain-binding motif [(pS/pT)P] adjacent to the PY motif [113]. Using peptides containing the PPXY motif, as well as the potential class IV motif, Spagnol et al. showed that phosphorylation of Ser282 significantly increased the affinity of all three WW domains of NEDD4 [115]. The binding affinity of WW2 was further increased when both Ser279 and Ser282 were phosphorylated, while phosphorylation of Ser279 alone had no influence. The contacts formed by phosphorylation of Ser279 and Ser282 were found to cause the region of the C-terminal tail of Cx43 encompassing the PY motif to form a horseshoe-shaped arrangement that fits into a groove formed by the NEDD4 WW2 domain. Taken together, these data raise the possibility that MAPK-mediated phosphorylation of the C-terminal tail of Cx43 increases the binding to NEDD4 in vivo, providing a possible mechanism for how the crosstalk between phosphorylation and ubiquitination of Cx43 is mediated.
The fact that the members of the NEDD4 family harbor multiple WW domains raises the possibility that tandem WW domain repeats may cooperate to enhance specificity and binding affinity [116]. For instance, the binding of the NEDD4 family member NEDD4-2 to its target SMAD2/3 first involves the phosphorylation-dependent binding of its WW3 domain to a site downstream of a PY motif, which positions the WW2 domain so that it can bind the PY motif, ultimately enhancing the total binding affinity [117]. The WW2 and WW3 domains of rNEDD4 have close proximity, raising the possibility that they could bind to the C-terminal tail of Cx43 in a cooperative manner. However, Spagnol et al. did not observe any cooperative binding of these two WW domains to a peptide containing the (pS/pT)P and PPXY motifs of Cx43, suggesting that they bind to Cx43 individually [115].
In accordance with the notion that NEDD4 binds to Cx43 via the PY motif, mutation of the first proline in this motif to a leucine (P283L) has been shown to result in reduced binding of NEDD4 to Cx43 as determined by co-immunoprecipitation [96], reduced Cx43 ubiquitination [96] and increased Cx43 protein levels [64]. Moreover, ectopic overexpression of NEDD4 in HeLa cells stably transfected with Cx43 or in C33A cervical carcinoma cells, which express Cx43 endogenously, has been shown to promote Cx43 ubiquitination and endocytosis, which is associated with a nearly complete loss of gap junctions [105]. The NEDD4-induced Cx43 endocytosis was found to be accompanied with loss of Cx43 protein levels due to its increased degradation via the endolysosomal pathway [105]. NEDD4 that contains an inactivating mutation in the catalytic cysteine of the HECT domain did not affect the Cx43 ubiquitination status, gap junction levels or the Cx43 protein level [105]. Thus, the ability of NEDD4 to promote ubiquitination, endocytosis and lysosomal degradation of Cx43 requires a functional HECT domain. NEDD4 is also involved in mediating the endolysosomal degradation of Cx43 in response to PKC activation [105]. In addition, NEDD4-induced ubiquitination of Cx43 has been suggested to act as a signal for gap junction endocytosis and autophagy-mediated degradation through recruitment of Eps15 [74, 96].
Other E3 ubiquitin ligases involved in regulating Cx43 gap junctions
SMURF2
SMURF1 and -2 (SMAD ubiquitination regulatory factor-1 and -2) are members of the NEDD4 family and were originally identified as important regulators of the transforming growth factor-β (TGF-β)/bone morphogenic protein signaling pathway by catalyzing the ubiquitination and degradation of SMAD proteins and the TGF-β receptor [118, 119]. Subsequent studies identified several other substrate proteins of SMURF1 and -2, and they were shown to play central roles in processes such as cell proliferation, differentiation, migration and senescence [120]. SMURF2 has been found to bind to Cx43, as determined by co-immunoprecipitation, and depletion of SMURF2 results in enhanced Cx43 levels and enlarged gap junctions, which is associated with increased gap junction intercellular communication [79]. SMURF2 also mediates the PKC-induced endocytosis and degradation of Cx43 [79]. In IAR20 cells, but not in HeLa cells, SMURF2 is also required for the remodeling of Cx43 gap junctions during mitosis [84]. However, it is unclear whether SMURF2 regulates gap junction endocytosis directly by catalyzing Cx43 ubiquitination or indirectly via another, yet unknown, protein [79].
TRIM21
TRIM21 (cytosolic Fc receptor tripartite motif 21) is a member of the tripartite motif-containing (TRIM) protein family of RING E3 ubiquitin ligases [121]. TRIM21 links Fc-mediated antibody recognition to the ubiquitin proteasome system and has also been linked to the initiation of autophagy [121]. In addition, TRIM21 has been shown to regulate cell proliferation and cell death, and among its known substrate proteins is the cyclin-dependent kinase inhibitor p27 [122, 123]. Chen and colleagues have demonstrated that Cx43 interacts with TRIM21, as determined by both liquid chromatography tandem mass spectrometry and co-immunoprecipitation [104]. Through confocal microscopy, TRIM21 was found to co-localize with Cx43 at gap junctions. Analysis of TRIM21-mediated Cx43 ubiquitination in vitro with cell lysates of C6 rat glioma cells that exogenously expressed Cx43 (C6-Cx43) or astrocytes showed that mono- and diubiquitinated forms of Cx43 accumulated approximately 30 min after the initiation of the procedure. After 3 h, most Cx43 was modified by polyubiquitination and/or multiple monoubiquitination. Analysis of TRIM21/Cx43 complexes by high-performance size exclusion chromatography showed that complexes that consisted of highly phosphorylated Cx43 also contained ubiquitinated Cx43. Moreover, inhibition of the trafficking of Cx43 from the endoplasmic reticulum to the Golgi apparatus with Brefeldin A resulted in the near complete loss of both phosphorylated and ubiquitinated forms of Cx43. Mathematical modeling demonstrated that EGF-induced phosphorylation of Cx43 was associated with increased Cx43 ubiquitination. Collectively, these observations support the notion that Cx43 degradation is controlled by crosstalk between Cx43 phosphorylation and ubiquitination.
In addition to the above-mentioned E3 ubiquitin ligases, another member of the NEDD4 family, WWP1, has been shown to regulate Cx43 gap junctions in cardiomyocytes [124] and the E3 ubiquitin ligases LNX1 and -2 (ligand of NUMB Protein-X1 and -2) were recently found to have important roles in controlling the level of Cx36 gap junctions in the central nervous system [125], as is further discussed below.
Regulation of Cx43 gap junctions by deubiquitinating enzymes
Cx43 ubiquitination is a reversible process, and two deubiquitinating enzymes, AMSH [associated molecule with the SH3 domain of STAM (signal transducing adaptor molecule)] and USP8 (ubiquitin carboxyl-terminal hydrolase 8) have been found to regulate Cx43 deubiquitination and lysosomal degradation (Fig. 3; Table 2) [107, 126].
AMSH
AMSH has been shown to bind to Cx43, as determined by co-immunoprecipitation, and to partly co-localize with Cx43 at the plasma membrane and in intracellular compartments [126]. Ectopic overexpression of a catalytically inactive version of AMSH results in strongly increased levels of Cx43 modified by K63-linked polyubiquitin chains, which is associated with an increased Cx43 degradation rate. Upon overexpression of the catalytically inactive version of AMSH, Cx43 is observed on the limiting membrane of vesicular structures of the endolysosomal system, whereas the level of Cx43 at the plasma membrane is decreased [126]. A similar loss of Cx43 at the plasma membrane and an increase in Cx43 degradation are observed in response to AMSH depletion [126]. Accordingly, on overexpression of wild-type AMSH, the level of Cx43 at the plasma membrane is increased. Collectively, these data indicate that AMSH-mediated deubiquitination of Cx43 counteracts its endocytosis and lysosomal degradation, causing increased gap junction levels.
USP8
USP8 was recently shown to bind to the C-terminal tail of Cx43, as determined by co-immunoprecipitation [107]. Ectopic overexpression of USP8 was found to promote the loss of both Cx43 monoubiquitination and polyubiquitin chains linked via Lys48 or Lys63, which was associated with increased Cx43 protein levels. In accordance with this finding, depletion of endogenous USP8 was associated with increased Cx43 ubiquitination and reduced Cx43 protein levels due to increased degradation via the autophagosomal pathway. This was correlated with loss of gap junctional intercellular communication [107].
Emerging physiological and pathophysiological implications of connexin ubiquitination
Given the diverse physiological roles of gap junctions across different tissue types, connexin ubiquitination is expected to have a wide variety of important physiological implications. Below, we highlight recent studies that have started to elucidate the role of connexin ubiquitination in the regulation of gap junctions in the heart, the lens and the central nervous system and the implications of these findings for disease pathogenesis.
Role of connexin ubiquitination in the regulation of gap junctions in the heart
The normal heart rhythm depends fundamentally on the coupling of cardiac myocytes by gap junctions [127]. The gap junctions are located at the intercalated discs, where they mediate the intercellular electrical coupling responsible for synchronous contraction of the heart [127]. The principal connexins expressed in cardiac myocytes are Cx43, Cx40 and Cx45, of which Cx43 is the most predominant [127]. Cx43 in the heart has a high turnover rate, and regulation of Cx43 degradation may be an important mechanism for modulating gap junctional communication in the heart under normal and pathophysiological conditions [128, 129]. Cx43 is highly remodeled in the diseased heart [127]. During ischemia, Cx43 undergoes rapid dephosphorylation, which is associated with electrical uncoupling and alteration in the distribution of Cx43 to the sides of the myocyte and to intracellular pools [130, 131].
Nielsen and colleagues have shown that Cx43 in neonatal ventricular rat cardiomyocytes is subjected to ubiquitination and binds to NEDD4 [132]. Moreover, stimulation of α-adrenergic Gαq-coupled receptors with noreprinephrine was found to cause increased Cx43 ubiquitination, which was suggested to be associated with increased internalization of gap junctions. Studies from Matesic’s group have identified a role for the E3 ubiquitin ligase WWP1, a member of the NEDD4 family, in regulating Cx43 degradation and gap junction size in cardiomyocytes. The authors demonstrated that global- or cardiomyocyte-specific overexpression of WWP1 in mice results in lethal ventricular arrhythmias around 8 weeks of age because of a reduction in Cx43 protein levels in the heart muscle [124]. Overexpression of WWP1 did not affect the Cx43 mRNA levels, suggesting that the WWP1-induced loss of Cx43 occurs at the post-translational level. In line with this notion, WWP1 was found to partly co-localize with Cx43 in small intracellular vesicular compartments of cardiomyocytes. Since WWP1 did not co-localize with Cx43 at the plasma membrane, it was suggested that WWP1 may not regulate gap junction internalization, but instead promote the degradation of Cx43 after internalization. Notably, overexpression of WWP1 in cardiomyocytes did not affect the protein levels of Cx40 and Cx45, suggesting that WWP1 specifically acts on Cx43. Furthermore, WWP1 overexpression had no effects on N-cadherin or desmoplakin protein levels, further corroborating the specific effect of WWP1 on Cx43. As determined by co-immunoprecipitation, Cx43 and WWP1 were found to bind to each other when exogenously expressed in human embryonic kidney 293T cells [124]. The binding was suggested to occur via the PY motif of Cx43. In line with the notion that WWP1 promotes Cx43 degradation, the Cx43 protein level in 293T cells was reduced in response to co-expression of WWP1, whereas a catalytically inactive version of WWP1 did not affect the Cx43 protein level. Moreover, the WWP1-induced loss of Cx43 protein levels was associated with a robust increase in Cx43 ubiquitination, whereas the inactive version of WWP1 did not affect the Cx43 ubiquitination status [124].
Studies from Girão’s group indicate that Cx43 ubiquitination is involved in the remodeling of gap junctions during acute cardiac ischemia. Using the atrial cardiomyocyte cell line HL-1 and organotypic heart cultures as model systems, they found ischemia to be associated with autophagy-mediated degradation of Cx43 [133]. The ischemia-induced degradation of Cx43 was shown to correlate with increased Cx43 ubiquitination and binding to Eps15 and p62, and depletion of p62 partly counteracted the degradation of Cx43 under these conditions. Increased ubiquitination and autophagy-mediated degradation of Cx43 in response to ischemia were also observed in organotypic heart cultures [133]. Moreover, ischemia was associated with enhanced Cx43 ubiquitination, as determined by the rat heart Langendorff perfusion model, and under these conditions, Cx43 was found to be modified by K63-linked ubiquitin chains [106]. Notably, counteracting Cx43 degradation during ischemia with chemical inhibitors of autophagy also prevents the loss of ischemia-associated gap junction intercellular communication [133]. This finding raises the possibility that targeting autophagy-mediated degradation of Cx43 could represent a potential therapeutic approach for preventing the loss of functional gap junctions during heart ischemia [133, 134]. The increase in Cx43 ubiquitination in response to ischemia is accompanied by recruitment of NEDD4 to intercalated discs, where it forms a complex with Cx43 [106]. However, depletion of NEDD4 does not counteract Cx43 ubiquitination or degradation induced by ischemia, suggesting that another E3 ubiquitin ligase may be involved in this process [133].
Shaw and colleagues have provided further insights into the crosstalk between Cx43 phosphorylation and ubiquitination and its implications for the loss of gap junctions during acute ischemia [67]. Exposing Langendorff-perfused mouse hearts to global ischemia was shown to result in phosphorylation of Ser368, in accordance with previous studies suggesting that phosphorylation of Cx43 at Ser368 is involved in gap junction remodeling during ischemia [135,136,137]. When a Cx43 construct in which Ser373 was mutated to alanine (Cx43-S373A) was transiently transfected into HaCaT human keratinocytes, it was found to have lost phosphorylation of Ser368, as well as Ser255, and to display more Cx43 at the plasma membrane than Cx43-wt did. Compared with Cx43-wt, Cx43-S373A also displayed strongly reduced ubiquitination, both under basal conditions and in response to TPA treatment. In accordance with these observations, TPA was found to induce loss of Cx43-wt at the plasma membrane because of increased internalization, whereas it did not affect the level of Cx43-S373A [67]. Cx43-S373A also displayed loss of binding to 14-3-3τ, whose normal function is to promote Cx43 internalization [67]. Collectively, these data indicate that phosphorylation of Cx43 at Ser373 promotes both Cx43 ubiquitination and binding to 14-3-3τ, and both events may contribute to reduced gap junction levels by promoting increased Cx43 internalization.
Gemel et al. have shown that some Cx40 mutants linked to atrial fibrillation have reduced stability as compared to Cx40-wt, when expressed in HeLa cells or HL-1 cardiomyocytes [98]. Among the mutants studied, Cx40-G38D showed the most dramatic differences from Cx40-wt and was, therefore, characterized in further detail. Both Cx40-wt and Cx40-G38D were demonstrated to be subjected to polyubiquitination and this modification was more prominent for the mutant. Moreover, proteasomal inhibition caused strongly increased levels of Cx40-G38D, which was associated with restoration of functional gap junctions. Collectively, these observations suggest that mutations in Cx40 may lead to atrial fibrillation by causing increased polyubiquitination and ERAD of the mutant protein [98]. The findings also raise the possibility that restoring the function of certain Cx40 mutants linked to atrial fibrillation by reducing their proteasomal degradation may have therapeutical implications [98].
Recently, Green and colleagues demonstrated that Cx43 ubiquitination is involved in modulating gap junction levels in cardiac cells in response to loss of desmoplakin, a key component of desmosomes [138]. Desmosomes are cadherin-based intercellular adhesive junctions that have important roles in maintaining the integrity of the myocardium by tethering the intermediate filament cytoskeleton to sites of cell–cell adhesion [139]. Desmoplakin physically links the intermediate filament to the desmosomal cadherin complex and also regulates various intracellular signaling pathways [139]. Mutations in desmoplakin can cause cardiac disease, including deadly arrhythmias [139]. In cardiac cells, desmosomes are structurally and functionally associated with both adherens junctions and gap junctions. Conditional knock-out of desmoplakin in mouse cardiac cells results in a significant reduction in Cx43 protein levels [140, 141]. Mechanistically, loss of desmoplakin causes activation of the ERK1/2-MAPK pathway, which then stimulates phosphorylation of Ser279 and Ser282 within the C-terminal tail of Cx43 [138]. In neonatal rat ventricular cardiomyocytes, this phosphorylation event was found to be associated with increased Cx43 ubiquitination and degradation in lysosomes [138].
Role of connexin ubiquitination in the regulation of gap junctions in the lens
The lens is an avascular and transparent organ, whose cells are coupled by an extensive network of gap junctions that facilitates the exchange of ions and metabolites throughout the organ [142, 143]. The mature lens is composed of fiber cells covered with a single layer of epithelial cells on the anterior hemisphere. The lens continues to grow throughout life as epithelial cells at the lens equator differentiate into fiber cells. As new fiber cells arise, older cells are pushed toward the center and become mature lens fibers. The differentiation of epithelial cells into fiber cells involves cell elongation and loss of nuclei and organelles [142, 143].
The lens expresses three connexin isoforms in mammals: Cx43, Cx46 and Cx50 [142, 143]. These connexins display differential spatial distributions, which are related to their specific functions in different regions of the lens. Cx43 is predominantly expressed in epithelial cells, Cx46 is mainly expressed in fiber cells and Cx50 is expressed in both epithelial and fiber cells. Studies on mice with targeted deletion of the genes encoding Cx46 or Cx50 have demonstrated that Cx46 is essential for lens transparency, and Cx50 is important for lens growth and transparency [144, 145]. Mutations of the genes encoding Cx46 and Cx50 are one of the common causes of hereditary cataracts in humans [142, 143].
Takemoto and colleagues have demonstrated that Cx43 and Cx46 are expressed and regulated in a reciprocal manner in lens epithelial cells in a process that involves Cx43 ubiquitination [146]. Ectopic overexpression of Cx46 in NN1003A rabbit lens epithelial cells was found to cause reduced Cx43 protein levels due to proteasome-dependent degradation, which was associated with an increase in Cx43 ubiquitination. The level of Cx50 was not altered under these conditions, indicating that the effect was specific for Cx43. The study further demonstrated that the C-terminal tail of Cx46 was essential and sufficient to induce Cx43 degradation. Although the exact mechanisms involved in the Cx46-induced degradation of Cx43 remain to be determined, it was suggested that it could be due to increased ERAD [146].
Studies by Liu et al. have identified a novel connection between the ubiquitin–proteasome system, gap junctions and lens clarity [147]. Degradation of proteins by the ubiquitin–proteasome system in the lens was found to be perturbed by overexpression of a ubiquitin with a mutation in the Lys6 residue (ubiquitin-K6W). Although this mutant form of ubiquitin did not affect the ability of ubiquitin to form polyubiquitin chains, the ubiquitin conjugates formed under these conditions were not recognized by the proteasome and were thus resistant to proteasomal degradation [148]. Therefore, ubiquitin-K6W acts as a specific upstream dominant negative inhibitor of the ubiquitin–proteasome system. Expression of ubiquitin-K6W in lenses in vivo was shown to result in accumulation of ubiquitin conjugates, stabilization of numerous regulatory proteins and cell cycle arrest [149, 150]. It also slowed the differentiation of epithelial cells into fibers and the removal of nuclei [151]. Lenses expressing ubiquitin-K6W were found to have a significant increase in Ca2+ concentration [147]. The elevated Ca2+ concentration resulted in hyperactivation of calpain, one of the dominant proteases in mammalian lenses, and as a consequence increased cleavage of calpain substrates, causing developmental defects and cataracts. Interestingly, lenses expressing ubiquitin-K6W were found to have significantly higher levels of Cx43 protein, whereas the levels of Cx46 and Cx50 were decreased. As the mRNA levels for Cx43 were comparable in lenses expressing ubiquitin-K6W and ubiquitin-wt, it was suggested that the increased level of Cx43 in lenses expressing ubiquitin-K6W was due to impaired degradation rather than increased synthesis of Cx43. In accordance with this notion, lenses expressing ubiquitin-K6W were found to have accumulated ubiquitinated Cx43, much of which would be expected to have incorporated ubiquitin-K6W and, therefore, be resistant to degradation. The study further suggested that the increase in Ca2+ concentration in the lenses expressing ubiquitin-K6W was partly due to the increased Cx43 protein levels. Corroborating the in vivo data, ectopic expression of ubiquitin-K6W in human lens epithelial cells resulted in increased levels of ubiquitinated Cx43, which was associated with increased protein levels. Together, these observations suggest that Cx43 ubiquitination may have important roles in modulating Ca2+ concentration and, as a consequence, calpain activity in the lens. The findings also raise the possibility that exploiting the ubiquitin–proteasome system and the Ca2+-calpain pathway may provide new approaches to preventing or curing human cataracts [147].
In addition to Cx43, Cx45.6, the chick ortholog of mammalian Cx50, has been found to be modified by polyubiquitin chains when overexpressed in neuro2A (N2A) mouse neuroblastoma cells [109]. In accordance with this finding, the degradation of Cx45.6 in lens primary cultures was shown to be mediated primarily through the proteasomal pathway. Minogue et al. have shown that a mutant version of Cx50 that has a frameshift after amino acid 255 and causes recessive congenital cataracts undergoes strongly enhanced ERAD as compared to Cx50-wt when stably expressed in HeLa cells [152]. Accordingly, the cellular levels of the mutant Cx50 are considerably lower than that of Cx50-wt, and it rarely forms gap junctions. In line with the notion that the mutant Cx50 is subjected to ERAD, it is able to form functional gap junction plaques in response to proteasomal inhibition, which is associated with an increase in gap junction intercellular communication [152]. Moreover, the chemical inhibitor eeyarestatin I, which inhibits ERAD by targeting p97-associated deubiquitination [153], causes a considerably stronger increase in the cellular level of the mutant Cx50 than that of Cx50-wt. Under these conditions, both the mutant Cx50 and Cx50-wt are detected at higher apparent molecular masses in immunoblots, likely due to polyubiquitination [152].
Role of connexin ubiquitination in the regulation of gap junctions in the central nervous system
Cx36 is, to date, the most abundantly detected connexin in neurons in mammals, and it forms the majority of electrical synapses in the mammalian central nervous system [154]. Cx36 is expressed in multiple types of neurons in the retina, brain and spinal cord and is critical in establishing synaptic circuitry during development and in providing for synchronous neuronal activity in the adult brain [154,155,156,157]. Cx36 null mice display functional deficits in various neural systems, including visual, motor and memory impairments. Electrical synapses are highly dynamic, and, similar to other connexins, Cx36 has a half-life of 1–3 h. Cx36 contains a PDZ [PSD95, DLGA, zonula occludens-1 (ZO-1)] domain interaction motif in its carboxy terminus through which it binds to the PDZ domain-containing proteins such as ZO1 [158].
LNX1 and -2 are RING E3 ubiquitin ligases that have multiple roles in the central nervous system. In addition to the RING domain, LNX1 and -2 contain four PDZ domains through which they interact with their substrate proteins [159, 160]. Recently, Nagy and colleagues identified LNX1 and -2 as important regulators of Cx36 gap junctions in neurons [125]. LNX1 and LNX2 were found to partly co-localize with neuronal gap junctions formed by Cx36 in rodent brain, as well as in N2A cells transfected with Cx36-enhanced cyan fluorescence protein (Cx36-eCFP). In accordance with this finding, as determined by co-immunoprecipitation, LNX1 was found to form a complex with Cx36 in mouse brain. Through pull-down assays, LNX1 and LNX2 were found to directly interact with Cx36 via its carboxy terminus. Co-transfection of N2A cells with Cx36-eCFP and LNX1 caused a strong loss of Cx36-eCFP-containing gap junctions, whereas co-transfection with Cx36-eCFP and an LNX1 mutant that lacked E3 ubiquitin ligase activity did not affect Cx36-eCFP gap junction levels [125]. A similar loss of gap junctions was observed in response to overexpression of LNX2, whereas a catalytically inactive form of LNX2 did not affect gap junction levels. Moreover, LNX2-wt, but not a catalytically inactive version, was found to increase Cx36-eCFP ubiquitination. The LNX2-induced loss of gap junctions was associated with reduced Cx36-eCFP protein levels due to increased lysosomal degradation. Collectively, these data add Cx36 to the list of substrate proteins of LNX1 and -2 in neurons and suggest that ubiquitination has an important role in controlling the plasticity of electrical synapses formed by Cx36-containing neuronal gap junctions.
Conclusions and future perspectives
An increasing body of experimental data points toward a central role of connexin ubiquitination in the regulation of gap junction intercellular communication. The emerging picture is that connexins can become ubiquitinated at different subcellular localizations, such as the endoplasmic reticulum, the plasma membrane and endosomes, and that at each of these locations, connexin ubiquitination may play distinct roles. In accordance with this notion, connexins have been demonstrated to be modified with different types of ubiquitination. Each of these types of ubiquitin modifications of connexins may, in principle, be recognized by different effector proteins containing ubiquitin-binding domains that link connexin ubiquitination to a specific downstream event, such as proteasomal degradation, endocytosis or lysosomal sorting of connexins. In recent years, researchers have also started to shed light on the physiological roles of connexin ubiquitination in specific tissue types and how dysregulation of these processes may contribute to loss of functional gap junctions during disease pathogenesis.
Among the key challenges ahead is to obtain a more comprehensive understanding of the molecular basis by which E3 ubiquitin ligases and deubiquitinating enzymes, as well as other components of the ubiquitin system, control gap junction intercellular communication. The continuous technological developments in the fields of mass spectrometry and proteomics are likely to set the stage for new advances in this research area. In the future, it will also be interesting to further investigate the cell- and tissue-type specific roles of E3 ubiquitin ligases and deubiquitinating enzymes in the regulation of gap junctions using relevant mouse knock-out and knock-in models. A possible scenario is that the components of the ubiquitin conjugation machinery involved in controlling connexin ubiquitination and gap junction levels vary considerably between different cell types as well as between connexin isoforms.
Dysregulation of protein ubiquitination is involved in numerous human diseases, including cancer, neurodegenerative diseases, and autoimmunity and inflammatory disorders [92]. Components of the ubiquitin system are, therefore, attractive drug targets [161]. So far, relatively few drugs targeting the ubiquitin system have been approved for clinical use [161]. However, new technical advances, combined with an increased knowledge about the ubiquitination process, are likely to spur progress in this area of drug development [92, 161]. As our understanding of the role of ubiquitination in the regulation of connexins increases, the development of new drugs that target the ubiquitin system could offer novel therapeutic opportunities for restoring functional gap junctions in human disease states.
References
Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC (2003) Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev 83:1359–1400
Beyer EC, Berthoud VM (1860) Gap junction gene and protein families: connexins, innexins, and pannexins. Biochim Biophys Acta 2018:5–8
Goodenough DA, Paul DL (2009) Gap junctions. Cold Spring Harb Perspect Biol 1:a002576
Aasen T (2015) Connexins: junctional and non-junctional modulators of proliferation. Cell Tissue Res 360:685–699
Vinken M (2015) Introduction: connexins, pannexins and their channels as gatekeepers of organ physiology. Cell Mol Life Sci 72:2775–2778
Ribeiro-Rodrigues TM, Martins-Marques T, Morel S, Kwak BR, Girao H (2017) Role of connexin 43 in different forms of intercellular communication—gap junctions, extracellular vesicles and tunnelling nanotubes. J Cell Sci 130:3619–3630
Saez JC, Leybaert L (2014) Hunting for connexin hemichannels. FEBS Lett 588:1205–1211
Kameritsch P, Pogoda K, Pohl U (1818) Channel-independent influence of connexin 43 on cell migration. Biochim Biophys Acta 2012:1993–2001
Laird DW (2010) The gap junction proteome and its relationship to disease. Trends Cell Biol 20:92–101
Leithe E, Mesnil M, Aasen T (1860) The connexin 43 C-terminus: a tail of many tales. Biochim Biophys Acta 2018:48–64
Sorgen PL, Trease AJ, Spagnol G, Delmar M, Nielsen MS (2018) Protein(-)protein interactions with connexin 43: regulation and function. Int J Mol Sci 10:E1428
Aasen T, Mesnil M, Naus CC, Lampe PD, Laird DW (2016) Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer 16:775–788
Delmar M, Laird DW, Naus CC, Nielsen MS, Verselis VK, White TW (2018) Connexins and disease. Cold Spring Harb Perspect Biol 4:a029348
Mesnil M, Aasen T, Boucher J, Chepied A, Cronier L, Defamie N, Kameritsch P, Laird DW, Lampe PD, Lathia JD, Leithe E, Mehta PP, Monvoisin A, Pogoda K, Sin WC, Tabernero A, Yamasaki H, Yeh ES, Dagli MLZ, Naus CC (1860) An update on minding the gap in cancer. Biochim Biophys Acta 2018:237–243
Aasen T, Leithe E, Graham SV, Kameritsch P, Mayan MD, Mesnil M, Pogoda K, Tabernero A (2019) Connexins in cancer: bridging the gap to the clinic. Oncogene 38:4429–4451
Laird DW, Lampe PD (2018) Therapeutic strategies targeting connexins. Nat Rev Drug Discov 17:905–921
Leybaert L, Lampe PD, Dhein S, Kwak BR, Ferdinandy P, Beyer EC, Laird DW, Naus CC, Green CR, Schulz R (2017) Connexins in cardiovascular and neurovascular health and disease: pharmacological implications. Pharmacol Rev 69:396–478
Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH (2002) Multicolor and electron microscopic imaging of connexin trafficking. Science 296:503–507
Jordan K, Solan JL, Dominguez M, Sia M, Hand A, Lampe PD, Laird DW (1999) Trafficking, assembly, and function of a connexin43-green fluorescent protein chimera in live mammalian cells. Mol Biol Cell 10:2033–2050
Laing JG, Tadros PN, Westphale EM, Beyer EC (1997) Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp Cell Res 236:482–492
Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM (2002) Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci USA 99:10446–10451
Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY (2007) Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128:547–560
Fallon RF, Goodenough DA (1981) Five-hour half-life of mouse liver gap-junction protein. J Cell Biol 90:521–526
Herve JC, Derangeon M, Bahbouhi B, Mesnil M, Sarrouilhe D (2007) The connexin turnover, an important modulating factor of the level of cell-to-cell junctional communication: comparison with other integral membrane proteins. J Membr Biol 217:21–33
Laird DW, Puranam KL, Revel JP (1991) Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J 273(Pt 1):67–72
Johnson KE, Mitra S, Katoch P, Kelsey LS, Johnson KR, Mehta PP (2013) Phosphorylation on Ser-279 and Ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol Biol Cell 24:715–733
Laing JG, Beyer EC (1995) The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J Biol Chem 270:26399–26403
Leithe E, Rivedal E (2004) Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci 117:1211–1220
Musil LS, Le AC, VanSlyke JK, Roberts LM (2000) Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem 275:25207–25215
Qin H, Shao Q, Igdoura SA, Alaoui-Jamali MA, Laird DW (2003) Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J Biol Chem 278:30005–30014
Rivedal E, Leithe E (2005) Connexin43 synthesis, phosphorylation, and degradation in regulation of transient inhibition of gap junction intercellular communication by the phorbol ester TPA in rat liver epithelial cells. Exp Cell Res 302:143–152
VanSlyke JK, Musil LS (2005) Cytosolic stress reduces degradation of connexin43 internalized from the cell surface and enhances gap junction formation and function. Mol Biol Cell 16:5247–5257
Leithe E (1865) Regulation of connexins by the ubiquitin system: implications for intercellular communication and cancer. Biochim Biophys Acta 2016:133–146
Sosinsky GE, Nicholson BJ (2005) Structural organization of gap junction channels. Biochim Biophys Acta 1711:99–125
Basheer W, Shaw R (1863) The “tail” of Connexin43: an unexpected journey from alternative translation to trafficking. Biochim Biophys Acta 2016:1848–1856
Ahmad S, Diez JA, George CH, Evans WH (1999) Synthesis and assembly of connexins in vitro into homomeric and heteromeric functional gap junction hemichannels. Biochem J 339(Pt 2):247–253
Zhang JT, Chen M, Foote CI, Nicholson BJ (1996) Membrane integration of in vitro-translated gap junctional proteins: co- and post-translational mechanisms. Mol Biol Cell 7:471–482
Mitra S, Annamalai L, Chakraborty S, Johnson K, Song XH, Batra SK, Mehta PP (2006) Androgen-regulated formation and degradation of gap junctions in androgen-responsive human prostate cancer cells. Mol Biol Cell 17:5400–5416
VanSlyke JK, Deschenes SM, Musil LS (2000) Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol Biol Cell 11:1933–1946
VanSlyke JK, Musil LS (2002) Dislocation and degradation from the ER are regulated by cytosolic stress. J Cell Biol 157:381–394
Aasen T, Johnstone S, Vidal-Brime L, Lynn KS, Koval M (2018) Connexins: synthesis, post-translational modifications, and trafficking in health and disease. Int J Mol Sci 19:E1296
Epifantseva I, Shaw RM (1860) Intracellular trafficking pathways of Cx43 gap junction channels. Biochim Biophys Acta Biomembr 2018:40–47
Das Sarma J, Wang F, Koval M (2002) Targeted gap junction protein constructs reveal connexin-specific differences in oligomerization. J Biol Chem 277:20911–20918
Martin PE, Blundell G, Ahmad S, Errington RJ, Evans WH (2001) Multiple pathways in the trafficking and assembly of connexin 26, 32 and 43 into gap junction intercellular communication channels. J Cell Sci 114:3845–3855
Musil LS, Goodenough DA (1993) Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 74:1065–1077
Cottrell GT, Burt JM (2005) Functional consequences of heterogeneous gap junction channel formation and its influence in health and disease. Biochim Biophys Acta 1711:126–141
Koval M (2006) Pathways and control of connexin oligomerization. Trends Cell Biol 16:159–166
Johnson RG, Meyer RA, Li XR, Preus DM, Tan L, Grunenwald H, Paulson AF, Laird DW, Sheridan JD (2002) Gap junctions assemble in the presence of cytoskeletal inhibitors, but enhanced assembly requires microtubules. Exp Cell Res 275:67–80
Simek J, Churko J, Shao Q, Laird DW (2009) Cx43 has distinct mobility within plasma-membrane domains, indicative of progressive formation of gap-junction plaques. J Cell Sci 122:554–562
Thomas T, Jordan K, Simek J, Shao Q, Jedeszko C, Walton P, Laird DW (2005) Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J Cell Sci 118:4451–4462
Joshi-Mukherjee R, Coombs W, Burrer C, de Mora IA, Delmar M, Taffet SM (2007) Evidence for the presence of a free C-terminal fragment of cx43 in cultured cells. Cell Commun Adhes 14:75–84
Salat-Canela C, Sese M, Peula C, Ramon y Cajal S, Aasen T (2014) Internal translation of the connexin 43 transcript. Cell Commun Signal 12:31
Smyth JW, Shaw RM (2013) Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep 5:611–618
Ul-Hussain M, Olk S, Schoenebeck B, Wasielewski B, Meier C, Prochnow N, May C, Galozzi S, Marcus K, Zoidl G, Dermietzel R (2014) Internal ribosomal entry site (IRES) activity generates endogenous carboxyl-terminal domains of Cx43 and is responsive to hypoxic conditions. J Biol Chem 289:20979–20990
Basheer WA, Xiao S, Epifantseva I, Fu Y, Kleber AG, Hong T, Shaw RM (2017) GJA1-20k arranges actin to guide Cx43 delivery to cardiac intercalated discs. Circ Res 121:1069–1080
Kotini M, Barriga EH, Leslie J, Gentzel M, Rauschenberger V, Schambony A, Mayor R (2018) Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat Commun 9:3846
Falk MM, Bell CL, Kells Andrews RM, Murray SA (2016) Molecular mechanisms regulating formation, trafficking and processing of annular gap junctions. BMC Cell Biol 17(Suppl 1):22
Laird DW (2006) Life cycle of connexins in health and disease. Biochem J 394:527–543
Gumpert AM, Varco JS, Baker SM, Piehl M, Falk MM (2008) Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett 582:2887–2892
Larsen WJ, Tung HN, Murray SA, Swenson CA (1979) Evidence for the participation of actin microfilaments and bristle coats in the internalization of gap junction membrane. J Cell Biol 83:576–587
Nickel BM, DeFranco BH, Gay VL, Murray SA (2008) Clathrin and Cx43 gap junction plaque endoexocytosis. Biochem Biophys Res Commun 374:679–682
Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM (2007) Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell 18:337–347
Fong JT, Kells RM, Falk MM (2013) Two tyrosine-based sorting signals in the Cx43 C-terminus cooperate to mediate gap junction endocytosis. Mol Biol Cell 24:2834–2848
Thomas MA, Zosso N, Scerri I, Demaurex N, Chanson M, Staub O (2003) A tyrosine-based sorting signal is involved in connexin43 stability and gap junction turnover. J Cell Sci 116:2213–2222
Gilleron J, Carette D, Fiorini C, Dompierre J, Macia E, Denizot JP, Segretain D, Pointis G (2011) The large GTPase dynamin2: a new player in connexin 43 gap junction endocytosis, recycling and degradation. Int J Biochem Cell Biol 43:1208–1217
Nickel B, Boller M, Schneider K, Shakespeare T, Gay V, Murray SA (2013) Visualizing the effect of dynamin inhibition on annular gap vesicle formation and fission. J Cell Sci 126:2607–2616
Smyth JW, Zhang SS, Sanchez JM, Lamouille S, Vogan JM, Hesketh GG, Hong T, Tomaselli GF, Shaw RM (2014) A 14-3-3 mode-1 binding motif initiates gap junction internalization during acute cardiac ischemia. Traffic 15:684–699
Berthoud VM, Minogue PJ, Laing JG, Beyer EC (2004) Pathways for degradation of connexins and gap junctions. Cardiovasc Res 62:256–267
Falk MM, Kells RM, Berthoud VM (2014) Degradation of connexins and gap junctions. FEBS Lett 588:1221–1229
Leithe E, Sirnes S, Fykerud T, Kjenseth A, Rivedal E (1818) Endocytosis and post-endocytic sorting of connexins. Biochim Biophys Acta 2012:1870–1879
Murray SA, Larsen WJ, Trout J, Donta ST (1981) Gap junction assembly and endocytosis correlated with patterns of growth in a cultured adrenocortical tumor cell (SW-13). Cancer Res 41:4063–4074
Naus CC, Hearn S, Zhu D, Nicholson BJ, Shivers RR (1993) Ultrastructural analysis of gap junctions in C6 glioma cells transfected with connexin43 cDNA. Exp Cell Res 206:72–84
Vaughan DK, Lasater EM (1990) Renewal of electrotonic synapses in teleost retinal horizontal cells. J Comp Neurol 299:364–374
Bejarano E, Girao H, Yuste A, Patel B, Marques C, Spray DC, Pereira P, Cuervo AM (2012) Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell 23:2156–2169
Carette D, Gilleron J, Denizot JP, Grant K, Pointis G, Segretain D (2015) New cellular mechanisms of gap junction degradation and recycling. Biol Cell 107:218–231
Fong JT, Kells RM, Gumpert AM, Marzillier JY, Davidson MW, Falk MM (2012) Internalized gap junctions are degraded by autophagy. Autophagy 8:794–811
Hesketh GG, Shah MH, Halperin VL, Cooke CA, Akar FG, Yen TE, Kass DA, Machamer CE, Van Eyk JE, Tomaselli GF (2010) Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ Res 106:1153–1163
Lichtenstein A, Minogue PJ, Beyer EC, Berthoud VM (2011) Autophagy: a pathway that contributes to connexin degradation. J Cell Sci 124:910–920
Fykerud TA, Kjenseth A, Schink KO, Sirnes S, Bruun J, Omori Y, Brech A, Rivedal E, Leithe E (2012) Smad ubiquitination regulatory factor-2 controls gap junction intercellular communication by modulating endocytosis and degradation of connexin43. J Cell Sci 125:3966–3976
Leithe E, Brech A, Rivedal E (2006) Endocytic processing of connexin43 gap junctions: a morphological study. Biochem J 393:59–67
Leithe E, Kjenseth A, Sirnes S, Stenmark H, Brech A, Rivedal E (2009) Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J Cell Sci 122:3883–3893
Malerod L, Pedersen NM, Sem Wegner CE, Lobert VH, Leithe E, Brech A, Rivedal E, Liestol K, Stenmark H (2011) Cargo-dependent degradation of ESCRT-I as a feedback mechanism to modulate endosomal sorting. Traffic 12:1211–1226
Boassa D, Solan JL, Papas A, Thornton P, Lampe PD, Sosinsky GE (2010) Trafficking and recycling of the connexin43 gap junction protein during mitosis. Traffic 11:1471–1486
Fykerud TA, Knudsen LM, Totland MZ, Sorensen V, Dahal-Koirala S, Lothe RA, Brech A, Leithe E (2016) Mitotic cells form actin-based bridges with adjacent cells to provide intercellular communication during rounding. Cell Cycle 15:2943–2957
Vanderpuye OA, Bell CL, Murray SA (2016) Redistribution of connexin 43 during cell division. Cell Biol Int 40:387–396
Axelsen LN, Calloe K, Holstein-Rathlou NH, Nielsen MS (2013) Managing the complexity of communication: regulation of gap junctions by post-translational modification. Front Pharmacol 4:130
Pogoda K, Kameritsch P, Retamal MA, Vega JL (2016) Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: a revision. BMC Cell Biol 17(Suppl 1):11
Solan JL, Lampe PD (2009) Connexin43 phosphorylation: structural changes and biological effects. Biochem J 419:261–272
Yau R, Rape M (2016) The increasing complexity of the ubiquitin code. Nat Cell Biol 18:579–586
Buetow L, Huang DT (2016) Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol 17:626–642
Leznicki P, Kulathu Y (2017) Mechanisms of regulation and diversification of deubiquitylating enzyme function. J Cell Sci 130:1997–2006
Popovic D, Vucic D, Dikic I (2014) Ubiquitination in disease pathogenesis and treatment. Nat Med 20:1242–1253
Rutz ML, Hulser DF (2001) Supramolecular dynamics of gap junctions. Eur J Cell Biol 80:20–30
Leithe E, Rivedal E (2004) Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem 279:50089–50096
Sirnes S, Leithe E, Rivedal E (2008) The detergent resistance of Connexin43 is lost upon TPA or EGF treatment and is an early step in gap junction endocytosis. Biochem Biophys Res Commun 373:597–601
Girao H, Catarino S, Pereira P (2009) Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res 315:3587–3597
Auth T, Schluter S, Urschel S, Kussmann P, Sonntag S, Hoher T, Kreuzberg MM, Dobrowolski R, Willecke K (2009) The TSG101 protein binds to connexins and is involved in connexin degradation. Exp Cell Res 315:1053–1062
Gemel J, Simon AR, Patel D, Xu Q, Matiukas A, Veenstra RD, Beyer EC (2014) Degradation of a connexin40 mutant linked to atrial fibrillation is accelerated. J Mol Cell Cardiol 74:330–339
Kelly SM, Vanslyke JK, Musil LS (2007) Regulation of ubiquitin-proteasome system mediated degradation by cytosolic stress. Mol Biol Cell 18:4279–4291
Kopanic JL, Schlingmann B, Koval M, Lau AF, Sorgen PL, Su VF (2015) Degradation of gap junction connexins is regulated by the interaction with Cx43-interacting protein of 75 kDa (CIP75). Biochem J 466:571–585
Dunn CA, Su V, Lau AF, Lampe PD (2012) Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J Biol Chem 287:2600–2607
Kells-Andrews RM, Margraf RA, Fisher CG, Falk MM (2018) Connexin-43 K63-polyubiquitylation on lysines 264 and 303 regulates gap junction internalization. J Cell Sci 131:jcs204321
Alaei SR, Abrams CK, Bulinski JC, Hertzberg EL, Freidin MM (2018) Acetylation of C-terminal lysines modulates protein turnover and stability of Connexin-32. BMC Cell Biol 19:22
Chen VC, Kristensen AR, Foster LJ, Naus CC (2012) Association of connexin43 with E3 ubiquitin ligase TRIM21 reveals a mechanism for gap junction phosphodegron control. J Proteome Res 11:6134–6146
Totland MZ, Bergsland CH, Fykerud TA, Knudsen LM, Rasmussen NL, Eide PW, Yohannes Z, Sorensen V, Brech A, Lothe RA, Leithe E (2017) The E3 ubiquitin ligase NEDD4 induces endocytosis and lysosomal sorting of connexin 43 to promote loss of gap junctions. J Cell Sci 130:2867–2882
Martins-Marques T, Catarino S, Marques C, Matafome P, Ribeiro-Rodrigues T, Baptista R, Pereira P, Girao H (2015) Heart ischemia results in connexin43 ubiquitination localized at the intercalated discs. Biochimie 112:196–201
Sun J, Hu Q, Peng H, Peng C, Zhou L, Lu J, Huang C (2018) The ubiquitin-specific protease USP8 deubiquitinates and stabilizes Cx43. J Biol Chem 293:8275–8284
Xiao D, Chen S, Shao Q, Chen J, Bijian K, Laird DW, Alaoui-Jamali MA (2014) Dynamin 2 interacts with connexin 26 to regulate its degradation and function in gap junction formation. Int J Biochem Cell Biol 55:288–297
Yin X, Liu J, Jiang JX (2008) Lens fiber connexin turnover and caspase-3-mediated cleavage are regulated alternately by phosphorylation. Cell Commun Adhes 15:1–11
Kjenseth A, Fykerud TA, Sirnes S, Bruun J, Yohannes Z, Kolberg M, Omori Y, Rivedal E, Leithe E (2012) The gap junction channel protein connexin 43 is covalently modified and regulated by SUMOylation. J Biol Chem 287:15851–15861
Kumar S, Tomooka Y, Noda M (1992) Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun 185:1155–1161
Boase NA, Kumar S (2015) NEDD4: the founding member of a family of ubiquitin-protein ligases. Gene 557:113–122
Salah Z, Alian A, Aqeilan RI (2012) WW domain-containing proteins: retrospectives and the future. Front Biosci (Landmark Ed) 17:331–348
Leykauf K, Salek M, Bomke J, Frech M, Lehmann WD, Durst M, Alonso A (2006) Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci 119:3634–3642
Spagnol G, Kieken F, Kopanic JL, Li H, Zach S, Stauch KL, Grosely R, Sorgen PL (2016) Structural studies of the Nedd4 WW domains and their selectivity for the connexin43 (Cx43) carboxyl terminus. J Biol Chem 291:7637–7650
Dodson EJ, Fishbain-Yoskovitz V, Rotem-Bamberger S, Schueler-Furman O (2015) Versatile communication strategies among tandem WW domain repeats. Exp Biol Med (Maywood) 240:351–360
Aragon E, Goerner N, Zaromytidou AI, Xi Q, Escobedo A, Massague J, Macias MJ (2011) A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev 25:1275–1288
Lin X, Liang M, Feng XH (2000) Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem 275:36818–36822
Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH (1999) A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400:687–693
Koganti P, Levy-Cohen G, Blank M (2018) smurfs in protein homeostasis, signaling, and cancer. Front Oncol 8:295
Rhodes DA, Isenberg DA (2017) TRIM21 and the function of antibodies inside cells. Trends Immunol 38:916–926
Espinosa A, Zhou W, Ek M, Hedlund M, Brauner S, Popovic K, Horvath L, Wallerskog T, Oukka M, Nyberg F, Kuchroo VK, Wahren-Herlenius M (2006) The Sjogren’s syndrome-associated autoantigen Ro52 is an E3 ligase that regulates proliferation and cell death. J Immunol 176:6277–6285
Sabile A, Meyer AM, Wirbelauer C, Hess D, Kogel U, Scheffner M, Krek W (2006) Regulation of p27 degradation and S-phase progression by Ro52 RING finger protein. Mol Cell Biol 26:5994–6004
Basheer WA, Harris BS, Mentrup HL, Abreha M, Thames EL, Lea JB, Swing DA, Copeland NG, Jenkins NA, Price RL, Matesic LE (2015) Cardiomyocyte-specific overexpression of the ubiquitin ligase Wwp1 contributes to reduction in connexin 43 and arrhythmogenesis. J Mol Cell Cardiol 88:1–13
Lynn BD, Li X, Hormuzdi SG, Griffiths EK, McGlade CJ, Nagy JI (2018) E3 ubiquitin ligases LNX1 and LNX2 localize at neuronal gap junctions formed by connexin36 in rodent brain and molecularly interact with connexin36. Eur J Neurosci 48:3062–3081
Ribeiro-Rodrigues TM, Catarino S, Marques C, Ferreira JV, Martins-Marques T, Pereira P, Girao H (2014) AMSH-mediated deubiquitination of Cx43 regulates internalization and degradation of gap junctions. FASEB J 28:4629–4641
Severs NJ, Bruce AF, Dupont E, Rothery S (2008) Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res 80:9–19
Beardslee MA, Laing JG, Beyer EC, Saffitz JE (1998) Rapid turnover of connexin43 in the adult rat heart. Circ Res 83:629–635
Saffitz JE, Laing JG, Yamada KA (2000) Connexin expression and turnover: implications for cardiac excitability. Circ Res 86:723–728
Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE (2000) Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 87:656–662
Matsushita T, Oyamada M, Fujimoto K, Yasuda Y, Masuda S, Wada Y, Oka T, Takamatsu T (1999) Remodeling of cell-cell and cell-extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ Res 85:1046–1055
Mollerup S, Hofgaard JP, Braunstein TH, Kjenseth A, Leithe E, Rivedal E, Holstein-Rathlou NH, Nielsen MS (2011) Norepinephrine inhibits intercellular coupling in rat cardiomyocytes by ubiquitination of connexin43 gap junctions. Cell Commun Adhes 18:57–65
Martins-Marques T, Catarino S, Zuzarte M, Marques C, Matafome P, Pereira P, Girao H (2015) Ischaemia-induced autophagy leads to degradation of gap junction protein connexin43 in cardiomyocytes. Biochem J 467:231–245
Martins-Marques T, Catarino S, Marques C, Pereira P, Girao H (2015) To beat or not to beat: degradation of Cx43 imposes the heart rhythm. Biochem Soc Trans 43:476–481
Miura T, Yano T, Naitoh K, Nishihara M, Miki T, Tanno M, Shimamoto K (2007) Delta-opioid receptor activation before ischemia reduces gap junction permeability in ischemic myocardium by PKC-epsilon-mediated phosphorylation of connexin 43. Am J Physiol Heart Circ Physiol 293:H1425–H1431
O’Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG (2011) A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res 108:704–715
Palatinus JA, Rhett JM, Gourdie RG (2011) Enhanced PKCepsilon mediated phosphorylation of connexin43 at serine 368 by a carboxyl-terminal mimetic peptide is dependent on injury. Channels (Austin) 5:236–240
Kam CY, Dubash AD, Magistrati E, Polo S, Satchell KJF, Sheikh F, Lampe PD, Green KJ (2018) Desmoplakin maintains gap junctions by inhibiting Ras/MAPK and lysosomal degradation of connexin-43. J Cell Biol 217:3219–3235
Broussard JA, Getsios S, Green KJ (2015) Desmosome regulation and signaling in disease. Cell Tissue Res 360:501–512
Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, Quarta G, Nobles M, Syrris P, Chaubey S, McKenna WJ, Tinker A, Lambiase PD (2012) Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin—a combined murine and human study. Eur Heart J 33:1942–1953
Lyon RC, Mezzano V, Wright AT, Pfeiffer E, Chuang J, Banares K, Castaneda A, Ouyang K, Cui L, Contu R, Gu Y, Evans SM, Omens JH, Peterson KL, McCulloch AD, Sheikh F (2014) Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum Mol Genet 23:1134–1150
Berthoud VM, Ngezahayo A (2017) Focus on lens connexins. BMC Cell Biol 18:6
Mathias RT, White TW, Gong X (2010) Lens gap junctions in growth, differentiation, and homeostasis. Physiol Rev 90:179–206
Gong X, Li E, Klier G, Huang Q, Wu Y, Lei H, Kumar NM, Horwitz J, Gilula NB (1997) Disruption of alpha3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell 91:833–843
White TW, Goodenough DA, Paul DL (1998) Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J Cell Biol 143:815–825
Banerjee D, Das S, Molina SA, Madgwick D, Katz MR, Jena S, Bossmann LK, Pal D, Takemoto DJ (2011) Investigation of the reciprocal relationship between the expression of two gap junction connexin proteins, connexin46 and connexin43. J Biol Chem 286:24519–24533
Liu K, Lyu L, Chin D, Gao J, Sun X, Shang F, Caceres A, Chang ML, Rowan S, Peng J, Mathias R, Kasahara H, Jiang S, Taylor A (2015) Altered ubiquitin causes perturbed calcium homeostasis, hyperactivation of calpain, dysregulated differentiation, and cataract. Proc Natl Acad Sci USA 112:1071–1076
Shang F, Deng G, Liu Q, Guo W, Haas AL, Crosas B, Finley D, Taylor A (2005) Lys6-modified ubiquitin inhibits ubiquitin-dependent protein degradation. J Biol Chem 280:20365–20374
Dudek EJ, Shang F, Valverde P, Liu Q, Hobbs M, Taylor A (2005) Selectivity of the ubiquitin pathway for oxidatively modified proteins: relevance to protein precipitation diseases. FASEB J 19:1707–1709
Liu Q, Shang F, Zhang X, Li W, Taylor A (2006) Expression of K6W-ubiquitin inhibits proliferation of human lens epithelial cells. Mol Vis 12:931–936
Caceres A, Shang F, Wawrousek E, Liu Q, Avidan O, Cvekl A, Yang Y, Haririnia A, Storaska A, Fushman D, Kuszak J, Dudek E, Smith D, Taylor A (2010) Perturbing the ubiquitin pathway reveals how mitosis is hijacked to denucleate and regulate cell proliferation and differentiation in vivo. PLoS One 5:e13331
Minogue PJ, Beyer EC, Berthoud VM (2013) A connexin50 mutant, CX50fs, that causes cataracts is unstable, but is rescued by a proteasomal inhibitor. J Biol Chem 288:20427–20434
Wang Q, Li L, Ye Y (2008) Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem 283:7445–7454
Nagy JI, Pereda AE, Rash JE (1860) Electrical synapses in mammalian CNS: past eras, present focus and future directions. Biochim Biophys Acta Biomembr 2018:102–123
Condorelli DF, Belluardo N, Trovato-Salinaro A, Mudo G (2000) Expression of Cx36 in mammalian neurons. Brain Res Brain Res Rev 32:72–85
Kamasawa N, Furman CS, Davidson KG, Sampson JA, Magnie AR, Gebhardt BR, Kamasawa M, Yasumura T, Zumbrunnen JR, Pickard GE, Nagy JI, Rash JE (2006) Abundance and ultrastructural diversity of neuronal gap junctions in the OFF and ON sublaminae of the inner plexiform layer of rat and mouse retina. Neuroscience 142:1093–1117
Miller AC, Pereda AE (2017) The electrical synapse: molecular complexities at the gap and beyond. Dev Neurobiol 77:562–574
Li X, Olson C, Lu S, Kamasawa N, Yasumura T, Rash JE, Nagy JI (2004) Neuronal connexin36 association with zonula occludens-1 protein (ZO-1) in mouse brain and interaction with the first PDZ domain of ZO-1. Eur J Neurosci 19:2132–2146
Nie J, McGill MA, Dermer M, Dho SE, Wolting CD, McGlade CJ (2002) LNX functions as a RING type E3 ubiquitin ligase that targets the cell fate determinant Numb for ubiquitin-dependent degradation. EMBO J 21:93–102
Rice DS, Northcutt GM, Kurschner C (2001) The Lnx family proteins function as molecular scaffolds for Numb family proteins. Mol Cell Neurosci 18:525–540
Huang X, Dixit VM (2016) Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res 26:484–498
Catarino S, Ramalho JS, Marques C, Pereira P, Girao H (2011) Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem J 437:255–267
Tsai CF, Cheng YK, Lu DY, Wang SL, Chang CN, Chang PC, Yeh WL (2018) Inhibition of estrogen receptor reduces connexin 43 expression in breast cancers. Toxicol Appl Pharmacol 338:182–190
Liao CK, Cheng HH, Wang SD, Yeih DF, Wang SM (2013) PKCvarepsilon mediates serine phosphorylation of connexin43 induced by lysophosphatidylcholine in neonatal rat cardiomyocytes. Toxicology 314:11–21
Zhang FF, Morioka N, Kitamura T, Hisaoka-Nakashima K, Nakata Y (2015) Proinflammatory cytokines downregulate connexin 43-gap junctions via the ubiquitin-proteasome system in rat spinal astrocytes. Biochem Biophys Res Commun 464:1202–1208
Liao CK, Jeng CJ, Wang HS, Wang SH, Wu JC (2013) Lipopolysaccharide induces degradation of connexin43 in rat astrocytes via the ubiquitin-proteasome proteolytic pathway. PLoS One 8:e79350
Acknowledgements
We apologize to authors whose work could not be cited due to space limitations. We acknowledge the financial support from the South-Eastern Norway Regional Health Authority (Grant Number 2016013) and the Kristian Gerhard Jebsen Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Totland, M.Z., Rasmussen, N.L., Knudsen, L.M. et al. Regulation of gap junction intercellular communication by connexin ubiquitination: physiological and pathophysiological implications. Cell. Mol. Life Sci. 77, 573–591 (2020). https://doi.org/10.1007/s00018-019-03285-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03285-0