
Abstract
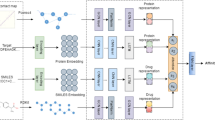
It is common knowledge that traditional new medication development is a costly, drawn-out procedure with greater safety uncertainty. Among them, drug target affinity prediction (DTA) is an important step in drug discovery and drug research. If we can significantly improve the accuracy of DTA prediction, it can help us potentially reduce the cost of new drug design and development significantly. Therefore, drug target affinity prediction is a very important topic. The precise and thorough characterization of medicines and proteins is the key to this subject. With the advancement of deep learning, it has become popular for academics to integrate deep learning into DTA prediction in an effort to increase accuracy. For example, DeepDTA, WideDTA, GraphDTA, etc., which are basically trained using information of drug molecules, information of protein molecules respectively, and does not make good use of their graph relationships as well as graph deep information, and with the increase in depth of the model over the years, what should have been excellent results are hardly excellent training results because of the difficulty of training. Inspired by Unet, this paper proposes a new method, UGraphDTA, which uses a new U-shaped architecture and Skip connection architecture to enable the model to understand deeper graph information. The novelty of this method is the use of skip connections in the convolutional network, which allows the model to utilize both the original molecular graph structure information and the information after convolution of the graph, enhancing the model's prediction ability for DTA tasks. The prediction performance of UGraphDTA is empirically proven to be better than other baseline models. This indicates that our proposed U-shaped convolutional architecture for drug target affinity prediction strategy that mines the deep information of drugs and proteins is effective.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


References
Ashburn, T.T., Thor, K.B.: Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discovery 3(8), 673–683 (2004)
Parenti, M.D., Rastelli, G.: Advances and applications of binding affinity prediction methods in drug discovery. Biotechnol. Adv. 30(1), 244–250 (2012)
Cheng, Z.Z., Zhou, S.G., Wang, Y., et al.: Effectively identifying compound-protein interactions by learning from positive and unlabeled examples. IEEE/ACM Trans. Comput. Biol. Bioinform. 15(6), 1832–1843 (2018)
Pahikkala, T., et al.: Toward more realistic drug–target interaction predictions. Brief. Bioinform. 16(2), 325–337 (2015)
He, T., Heidemeyer, M., Ban, F., Cherkasov, A., Ester, M.: SimBoost: a read-across approach for predicting drug–target binding affinities using gradient boosting machines. J. Cheminformatics. 9(1), (2017)
Öztürk, H., Özgür, A., Ozkirimli, E.: DeepDTA: deep drug–target binding affinity prediction. Bioinforamatics 34(17), i821–i829 (2018)
Öztürk, H., Ozkirimli, E., Özgür, A.: WideDTA: prediction of drug-target binding affinity. arXiv:1902.04166 [cs, q-bio, stat], (2019)
Nguyen, T., Le, H., Quinn, T. P., Nguyen, T., Le, T. D., Venkatesh, S.: GraphDTA: Predicting drug–target binding affinity with graph neural networks. Bioinformatics, (2020)
Kipf, T. N., Welling, M.: Semi-supervised classification with graph convolutional networks. arXiv:1609.02907 [cs, stat] (2017)
Ghosh, S., Ghosh, S: Exploring the ideal depth of neural network when predicting question deletion on community question answering. arXiv:1912.03585 [cs], (2019). Available https://arxiv.org/abs/1912.03585. Last Accessed 24 Aug 2022
Ronneberger, O., Fischer, P., Brox, T.: U-Net: Convolutional networks for biomedical image segmentation. arXiv.org, (2015)
Dang, L., Nie, Y., Long, C., Zhang, Q., Li, G.: MSR-GCN: Multi-scale residual graph convolution networks for human motion prediction. arXiv:2108.07152 [cs] (2021). Available https://arxiv.org/abs/2108.07152. Last Accessed 22 Aug 2022
He, K., Zhang, X., Ren, S., Sun, J.: Deep residual learning for image recognition. arXiv.org (2015)
Pahikkala, T., Airola, A., Pietil¨a, S., Shakyawar, S., Szwajda, A., Tang J., Aittokallio, T.: Briefings Bioinf. 16, 325–337 (2014). https://tdcommons.ai/multi_pred_tasks/dti/#davis. Last Accessed 22 Aug 2022
The, T., Heidemeyer, M., Ban, F., Cherkasov, A., Ester, M.: Cheminf. J. 9, 24 (2017). https://tdcommons.ai/multi_pred_tasks/dti/#kiba. Last Accessed 22 Aug 2022
Weininger, D.: Smiles, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 28 (1), 31–36 (1988)
Landrum, G.: RDKit: Open-source cheminformatics (2006)
DeepChem.: GitHub, (2022). https://github.com/deepchem/deepchem. Accessed 24 Aug 2022
Jiang, M., et al.: Drug–target affinity prediction using graph neural network and contact maps. RSC Adv. 10(35), 20701–20712 (2020)
PconsC4: GitHub, (2022). https://github.com/ElofssonLab/PconsC4. Accessed 24 Aug. 2022
Glorot, X., Bordes, A., Bengio, Y.: Deep sparse rectifier neural networks. Proceedings. mlr. press (2011)
Rosenblatt, F.X.: Principles of neurodynamics: perceptrons and the theory of brain mechanisms. Spartan Books, Washington DC (1961)
Srivastava, N., Hinton, G., Krizhevsky, A., Sutskever, I., Salakhutdinov, R.: Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 15(56), 1929–1958 (2014)
pytorch/pytorch.: GitHub (2021). https://github.com/pytorch/pytorch
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Chen, J., Dong, X., Yang, Z. (2023). Graph Convolutional Neural Networks for Drug Target Affinity Prediction in U-Shaped and Skip-Connection Architectures. In: Patnaik, S., Kountchev, R., Tai, Y., Kountcheva, R. (eds) 3D Imaging—Multidimensional Signal Processing and Deep Learning. Smart Innovation, Systems and Technologies, vol 349. Springer, Singapore. https://doi.org/10.1007/978-981-99-1230-8_24
Download citation
DOI: https://doi.org/10.1007/978-981-99-1230-8_24
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-1229-2
Online ISBN: 978-981-99-1230-8
eBook Packages: Intelligent Technologies and RoboticsIntelligent Technologies and Robotics (R0)