
Abstract
With the globalization of seed trade and transgenic variety development, the application of molecular technologies for seed quality gained more significance in both the internal and international markets. Besides germination, genetic purity and seed health are the two most important seed quality parameters that determine the planting value of a seed lot. Compared to the conventional methods of testing, molecular marker technologies are more efficient for quality analysis as these are more accurate, robust, abundant, and faster. Among the various markers, simple sequence repeats (SSRs), due to their genome-wide presence, reproducibility, multi-allelic nature, and co-dominant inheritance, have emerged as the best markers, for establishing varietal distinctness, identity, and variety/hybrid seed purity testing. With the advent of the next-generation sequencing (NGS) technology, single nucleotide polymorphic (SNP) markers also became widely popular, and the closest to being an ideal marker besides SSRs, in seed genetic purity testing. With large-scale GM crop cultivation, testing for the adventitious presence and trait purity are two added components of seed quality testing. The methods of GM seed quality testing include DNA-based (conventional and real-time PCR), protein-based (lateral flow test and ELISA), and bioassay-based technologies. DNA-based methods including PCR/real-time PCR assays have been successfully employed to detect the adventitious presence of transgenic seeds in seed trade especially at international level, as well as in the national gene banks for germplasm conservation. ISTA plays a prominent role in international harmonization and providing universal guidelines on use of different methods to detect GM seeds. The BMT group of UPOV and the Working Group on DNA Methods of the Variety Committee of ISTA, work in tandem to standardize suitable molecular techniques for establishing variety identity and purity testing, respectively. In the area of seed health testing also, molecular detection assays such as, PCR (nested PCR, multiplex PCR, real-time PCR), loop-mediated isothermal amplification (LAMP), and DNA microarray with many advantages over the conventional assays have been proven highly useful. However, there is a need to validate the usefulness of molecular markers through stringent multi-laboratory tests for their reproducibility before recommending them in routine seed purity and health testing.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others
Keywords
- Variety identity
- Genetic purity testing
- ISTA
- UPOV
- PVP
- Hybrid purity
- DNA markers
- Trait purity
- ELISA
- GM detection
- PCR
- Transgenic crops
- Adventitious presence
- Seed-borne viruses
1 Introduction
With the globalization of the seed market, competition in the seed trade has increased manifold with more stringent quality standards. High seed quality can only be obtained by a thorough control of the entire seed production process, from planning to final delivery. Among the many parameters of seed quality; genetic purity and seed health are of high importance in determining the authenticity and planting value of a seed lot. While field-based grow-out method is still much in vogue for variety authentication, several DNA-based modern techniques have been developed to test the genetic purity and seed health. Genetic purity of a variety determines the extent of conformity of the submitted seed sample with the claimed variety or the extent of purity of a variety within its seed lot or purity of F1 hybrid in a given hybrid seed lot. Maintaining absolute (100%) varietal purity during production is difficult to achieve in spite of following the recommended steps, allowing some degree of varietal off-types, which occurs inadvertently due to outcrossing, incomplete roguing of the off-types, or physical admixture during harvest, storage, or seed handling (Bradford 2006). Hence, to ascertain the desired levels of purity, seed lots are subjected to post-control grow-out tests or laboratory-based tests for internal quality control by the seed companies/producer/supplier. The scope of seed purity testing got broadened with the introduction of transgenic varieties in the global market, making the tests for GM trait purity and the adventitious presence of GM seeds in non-GM seed lots an essential requirement. Seed health, the other major quality determinant, is a measure of freedom of seeds from incidence of fungi, bacteria, viruses, and (very serious error and need to be removed) pests such as nematodes and insects in the seed lot, many of which pose difficulty in detection due to lack of unambiguous, precise, and rapid methods. Molecular techniques offer high potential for testing variety identity, genetic purity of seed lots, trait purity, and adventitious presence of transgenes as well as ascertaining good seed health.
2 Variety Characterization/Identification and Genetic Purity Testing
With the proliferation of a large number of varieties and narrowing gene pool, characterization and identification of varieties and establishing distinctness among closely related genotypes have become crucial aspects in plant breeding, variety maintenance, seed production, seed trade and protecting the interests of farmers and consumers (Smith and Register 1998). Variety characterization based on genetically inherited markers (morphological, biochemical, or molecular) aids in the development of identification keys and in distinguishing each variety unambiguously. As the number of crop varieties in cultivation is rising every year, the need to identify these based on stable and robust genetic markers is becoming more challenging (Santhy and Meshram 2015). The scope of variety identification using molecular markers is unlimited and can be used to test the uniqueness of a variety using a small sample size. ISTA (International Rules for Seed Testing) (2021) termed ‘the combination of alleles determined for a specific set of DNA markers within a sample or variety’ as ‘allele profile’ which helps in identifying varieties. The characterization done at molecular level generating a marker profile is called DNA fingerprinting and can provide additional evidence for the uniqueness of a variety. While the number of bands generated by a polymorphic marker is used to differentiate varieties (Law et al. 1998), the number and genomic distribution of the markers determine the robustness of a DNA fingerprint. In general, for n number of varieties to be distinguished, the minimum number of polymorphic bands scored should be between n and 2n (Singh and Singh 2019). It is pertinent to mention that while one robust marker shall be enough for testing hybrid purity, more numbers or multiple markers with high discrimination are needed for varietal purity testing. The markers required are still higher for variety identification and, the number required may be the highest for variety registration as EDVs (Hwu 2013).
Varietal purity testing is a quality assurance tool for crop producers and suppliers to comply with seed regulations for both commercial marketing and international seed exchanges. Due to their stability, reliability, and abundance, molecular markers can be employed for seed purity-related issues such as (1) determining the genetic identity of a variety or parental lines and verifying if the variety offered for sale is the same or not; (2) testing purity of elite varieties/inbred lines, GM/non-GM seeds, and/or F1 hybrid seeds; (3) trait-specific testing of seed (GM seeds).
3 Molecular Markers for Varietal Identity and Genetic Purity
Molecular markers have several advantages in comparison with conventional markers such as, high polymorphic information content (PIC), insensitivity to environment, stability, developmental stage independence and abundance. These methods are best suited for unequivocal identification of varieties (Singh and Singh 2019) and to determine whether the allele profile of a sample is identical to that of an authentic reference variety (www.seedtest.org). Based on the loci studied, the marker can be multi-locus (RAPD, AFLP) or single locus (SSRs and SNPs), and based on dominance, they can be dominant (RAPD, AFLP) or co-dominant (SSRs, SNPs). Based on the method, they can be hybridization-based (RFLP) or amplification-based (RAPD, SSRs, AFLP). Based on the number of alleles, markers are biallelic (SNP) or multi-allelic (SSR). Markers can be those where prior genome information is required (SSR) or those where genome information is not required (RAPD, AFLP, SNP).
Among the various marker techniques available, a few markers which have been well studied for their potential in seed quality testing such as variety identification, distinctness testing, and hybridity determination are as follows: (1) Restriction Fragment Length Polymorphism (RFLP), (2) Randomly Amplified Polymorphic DNA (RAPD), (3) Amplified Fragment Length polymorphism (AFLP), (4) sequence Characterized Amplified Regions (SCAR), (5) Sequence Tagged Sites (STS), (6) Simple Sequence Repeats (SSRs)/microsatellites, (7) Inter-SSR markers (ISSR), (8) Sequence-Related Amplified Polymorphism (SRAP) and Single Nucleotide Polymorphism (SNP).
3.1 Restricted Fragment Length Polymorphism (RFLP)
RFLP, the first DNA-based technique based on hybridization, produces polymorphisms inherited in a Mendelian fashion. They determine variation among varieties caused due to differences in the restriction sites resulting in the length difference of restricted fragments. The polymorphic bands are identified by Southern hybridization, thus identifying each cultivar and also determining the hybrid purity. The result obtained depends on the number of probes and the restriction enzymes used. RFLP markers have high reproducibility, have co-dominant inheritance, and are locus-specific. Disadvantages of the technique are, they are time-consuming, there is requirement of high quality and quantity of DNA, expensive radioactive probes, there is necessity to perform tedious Southern blotting method and prior sequence information is required for developing radiolabelled probes. The revolutionary invention of the polymorphic chain reactions (PCR) by Kary. B. Mullis in 1983 in making multiple copies of DNA segments led to a significant shift towards the use of markers based on amplification than those based on hybridization.
3.2 Randomly Amplified Polymorphic DNAs (RAPD)
The advent of RAPD markers provided a new tool for the molecular geneticist. RAPD uses low amounts of DNA with no need for high purity and does not require previous knowledge of the host genome. These have been employed extensively to discriminate crop varieties and, for the identification of parental lines and hybrids in, rice (Santhy et al. 2003), cotton (Ali et al. 2008), and vegetables (Kumar et al. 2008). In spite of its advantages, RAPD does not offer enough reliability since it uses short primers (of about ten nucleotides) and low annealing temperatures to amplify random regions in the genome, which makes the strategy non-reproducible across laboratories (Butler 2012). Due to its multiple bands and reproducibility issues across labs, RAPD markers were later replaced by other PCR-based markers such as CAPS, SCAR, AFLP, and SSR/microsatellite markers for plant variety identification and seed purity determination.
3.3 Cleaved Amplified Polymorphic Sequences
The CAPS markers are those in which DNA fragments after digestion with restriction enzymes are amplified with specific primers and separated on an agarose gel. CAPS markers closely linked to the gene of interest are helpful in crop breeding for marker-assisted selection and have been found useful in identifying the true female parents for authentic planting supply in few crops (Babu et al. 2017). The scoring of this type of marker is dependent on the variation in size of fragments following the digestion of the PCR product by a restriction enzyme.
3.4 Amplified Fragment Length Polymorphism (AFLP)
AFLP technology combines RFLP and PCR and is based on the selective amplification of a subset of genome restriction fragments using PCR. AFLP uses restriction enzymes recognizing frequent and rare restriction sites in the genome to generate fragments (that end with frequent or rare sticky ends or a combination of both), some of which are later specifically selected for PCR amplification. The selection is achieved with primers made of double-stranded adaptors linked to sequences complementing those generated by the restriction enzymes and, additionally carrying one to three nucleotides, to reduce the number of fragments for amplification. For higher specificity, this is usually completed in two steps, with a pre-amplification using only one nucleotide and a final amplification using two to three selective nucleotides. The use of AFLP can efficiently reveal multiple polymorphisms in a single reaction and is highly reproducible (Singh and Singh 2019). Their potential in fingerprinting and identification of inbred lines and hybrids has been demonstrated (Grzebelus et al. 2008). Due to the laborious procedure and requirement of high-quality DNA, it is not suited for routine purity testing.
3.5 Sequence Characterized Amplified Regions (SCAR)
To increase the reliability of a PCR-based marker, SCAR have been developed. These are obtained by eluting a selected fragment from RAPD or AFLP gel, cloned, and sequenced at its termini. A pair of primers (forward and reverse) specific for these termini is designed. This primer pair amplifies a single fragment under more restrictive annealing temperatures in PCR bringing higher reliability. Successful primer pairs give rise to SCAR markers. Direct application of these markers for hybrid purity testing in crop seeds has been demonstrated (Jang et al. 2004). Nevertheless, these markers are not adequate for the detection of seed admixtures or mislabelling.
3.6 Simple Sequence Repeats (SSRs)
SSRs or microsatellites are DNA stretches consisting of short, tandem repeats of short nucleotide sequences. SSR markers are also known as sequence-tagged microsatellite markers in which the above repeats are amplified using primers specific to sequences flanking these repeats. The amplification products are size separated by electrophoresis and visualized. The amplicons from different genotypes frequently show length polymorphisms due to allelic variation of the number of repeat motifs in the microsatellite.
SSRs/microsatellite markers became more valuable and reliable owing to their reproducibility, multi-allelic nature, co-dominant inheritance, genome-wide presence, robustness, higher polymorphism, and analytical simplicity (Abd El-Moghny et al. 2017).
SSR allelic profiles are conserved throughout the plant growth stage allowing unambiguous identification of crop varieties (Santhy et al. 2019; Ravishankar and Dinesh (2015). Being locus-specific and co-dominant, SSRs are the most suitable markers for seed hybridity testing as the heterozygosity of the hybrids can be easily determined by the presence of both the parental alleles (Tatiana et al. 2006; Selvakumar et al. 2010; Pallavi et al. 2011). The use of SSR markers for assessing seed purity has been reported in major crops like rice (Bora et al. 2016), maize (Daniel et al. 2012), and vegetable crops (Ravishankar et al. 2018). However, the target DNA region flanking each tandem repeat needs to be sequenced, primers have to be designed for the amplification of the repeat region, and marker screening needs to be done before they can be utilized. Occurrence of null alleles due to poor primer annealing and underestimation of heterozygosity is considered a major drawback associated with SSR markers.
3.7 Multiplex PCR
A greater throughput resolution of multiple markers can be achieved in a single PCR reaction making the technique much faster amplifying two or more loci simultaneously in the same reaction through multiplexing. In multiplex PCR, different segments from the same DNA get amplified simultaneously. We need to ensure that the length of amplified fragments does not overlap and different primer sets have the same melting temperature. Multiplexing of SSRs could discriminate parents of hybrids and prove the hybridity in brinjal and rice (Arun Kumar et al. 2014; Madhuchhanda et al. 2020).
3.8 Expressed Sequence Tags (ESTs)
The availability of enormous data for expressed sequence tags (ESTs) in the public domain made the marker-based studies shift from genomic SSRs to EST-SSRs. EST-SSRs amplify portions of expressed sequences in the genome which may be functionally associated with major component traits as compared to the non-EST-SSRs which may be randomly distributed across the genome. EST-SSRs based polymorphic markers have been identified in major crops (Parthiban et al. 2018) and have been proven useful for hybrid purity testing (Naresh et al. 2009).
3.9 Inter Simple Sequence Markers (ISSR)
Inter simple sequence markers (ISSR) are another set of PCR-based markers which have been widely studied for their utility in confirming F1 hybridity (Khajudparn et al. 2012). It is based on a single primer having a microsatellite sequence and detects variation in the size of the genomic region flanked by microsatellite sequences, generating multi-locus markers. In spite of being highly polymorphic, fast, and inexpensive, ISSR markers are associated with less reproducibility.
3.10 Sequence-Related Amplified Polymorphisms
SRAP markers are appropriate molecular markers for genotype identification since they are based on the amplification of coding regions in the genome utilizing two primers. The primers are 17 or 18 nucleotides long and contain a 13–14-base-long core sequence, followed by the sequences CCGG in the forward primer and AATT in the reverse primer, with the first 10 or 11 bases at the 5′ end being sequences of no specific structure (filler sequences). At the 3′ terminus, three selected nucleotides follow the core. The forward and reverse primers’ filler sequences must be distinct from one another. This marker reveals more polymorphic fragments than AFLP markers and is more compatible with genotype morphological variability (Ferriol et al. 2003). These markers were found highly efficient and reproducible for genetic purity testing of cabbage commercial hybrid seeds (Liu et al. 2007).
3.11 Single Nucleotide Polymorphisms (SNPs)
An SNP is a single nucleotide base difference between two DNA sequences or individuals. SNPs can be categorized according to nucleotide substitutions as either transitions (C/T or G/A) or transversions (C/G, A/T, C/A, or T/G) or insertions/deletions in the genome (Jiang 2013). These provide the ultimate/simplest form of molecular markers as a single nucleotide base is the smallest unit of inheritance. They may be present within coding and/or non-coding, intergenic regions of the genome at different frequencies. They are co-dominat markers, often linked to genes, can be easily automated, multiplexing possible making it cost-effective, enabling quick detection of high polymorphisms with lower error rate. The potential SNP markers in detecting varietal polymorphism and in reliable genetic purity testing of Indonesian rice varieties were proven by Utami et al. (2016). Given the availability of whole-genome sequence in many crops (rice, soybean, maize, etc.), SNPs are expected to be the standard marker of choice in the future.
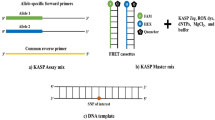
A high-density oligonucleotide SNP array has hundreds to thousands of probes arrayed on a small chip allowing many SNPs to be tested simultaneously. The probes are so designed to have the SNP sites in several locations matching with target DNA as well as not matching to the SNP allele present in the target DNA at several other locations. By comparing the differential amount of hybridization of the target DNA to each of these probes, it is possible to determine specific homozygous and heterozygous alleles. Microarray-based SNP genotyping is more time-consuming, and only hundreds of samples can be genotyped with thousands of SNPs. Compared to SSRs, the information obtained using SNPs are low, and hence, there is need to employ them in large numbers. Some of the high-throughput SNP genotyping methods include KASP (competitive allele-specific PCR) (Peng et al. 2021) and Fluidigm assays (Park et al. 2021).
Target SNP-seq combines the advantages of high-throughput sequencing and multiplex PCR amplification. It uses genome-wide perfect SNPs with conserved flanking sequences captured uniquely in PCR amplification (Zhang et al. 2020). This approach has 1000 times more coverage in a very short time and at a low cost, making it more competitive than the current SNP genotyping methods.
The SNP markers have been reliably employed for variety identification, distinctness testing, fingerprinting, genetic purity testing, assessing the parent-offspring relationship, and diversity analysis (Zhang et al. 2020; Kim et al. 2021; Josia et al. 2021).
An ideal DNA marker should be uniform, have a wide genome distribution, be co-dominant, have multiple alleles, have less DNA requirement, and be simple, easy to execute, less error-prone, reproducible, and amenable to high-throughput automation. SNP markers which fulfil most of these criteria are the closest to being ideal (Jiang 2013) (Fig. 1).

Types of molecular markers used in seed genetic purity testing
The International Seed Testing Association (ISTA) and the International Union for the Protection of New Varieties of Plants (UPOV) are working in a synchronized manner and sharing information on the use of DNA-based methods in variety testing, though with different objectives. While the BMT working group of UPOV aims the use of molecular markers in variety characterization for registration and protection, the DNA working group of ISTA aims for their use in routine seed testing/certification and protocol development. ISTA has identified a suitable set of SSRs for discriminating varieties in rice, maize, wheat, and soybean (www.upov.int).
4 Use of Molecular Markers for Plant Variety Protection
Plant variety registration and protection has attained critical importance all over the world and emphatically comes under the purview of seed regulation for quality control. Testing for DUS is an essential component of variety registration advocated by the International Union for the Protection of New Varieties of Plants (UPOV). The current plant variety protection system relies on the morphological description of plant varieties, which at times is not sufficient to discriminate a large number of varieties being developed in crops (Jamali et al. 2019). The potential of DNA markers in testing variety distinctness has been tested and proven (Santhy et al. 2000; Shengrui et al. 2020). The UPOV’s Working Group on Biochemical and Molecular Techniques (BMT), after thorough research to identify marker-trait associations which are robust, has proposed the use of molecular markers that are directly linked to conventional characteristics. It was proved that molecular markers developed for a subset of DUS traits (genomic DUS) can be robustly used as a tool for determining the distinctness, uniformity, and stability of crop varieties (Yang et al. 2021). BMT-UPOV also proposed to develop/calibrate threshold levels for molecular markers against the minimum distance in phenotype traits for their use in testing for essentially derived varieties. This provides a system combining phenotypic and molecular distances as a tool to improve the efficiency of distinctness evaluation (Jones et al. 2013). The BMT group of UPOV is putting efforts in harmony with the DNA working group of ISTA and the International Association of Plant Breeders (ASSISNEL) to study the implications of the use of molecular markers in testing the distinctness of varieties for granting protection.
Within UPOV’s system, breeders can freely use protected varieties in breeding programmes. However, breeders of protected varieties may seek to share the ownership of essentially derived varieties once it is proven that these, except for a few distinctive DUS trait(s), conform to parental varieties in essential characteristics. DNA markers can be a good replacement for morphological traits in defining boundaries between independent and essentially derived varieties. With the advent of new breeding technologies that allow minor modification in varieties with outcomes of specific merit or utility, detecting distinctness between varieties may become increasingly challenging (Yu and Chung 2021). Extensive studies have been undertaken in maize regarding the use of SSR and SNP markers for EDV identification (UPOV BMT 2007; Yang et al. 2021), and technical guidelines to help determine EDV status using SSRs and SNPs in maize have been suggested (Rousselle et al. 2015).
5 Transgenic/Genetically Modified Seeds
Biotechnological advances have offered tremendous scope for creating novel transgenic plants to combat biotic and abiotic stresses more efficiently and to improve the yield and quality. Plants derived by transfer of genes for specific traits from diverse organisms such as bacteria, viruses, animals, etc. using molecular or recombinant DNA/genetic engineering techniques are called GMOs (genetically modified organisms)/genetically engineered/transgenic plants (Phillips 2008). The potential of transgenics to effectively address many specific problems such as resistance to pests and diseases and tolerance to drought and herbicides made it gain wider acceptance all over the world. Currently, the global area under GM crops is 190.4 million hectares cultivated in 26 countries which include 21 developing and 5 industrial countries (ISAAA 2019). Major traits which have been utilized for developing transgenics include insect resistance, herbicide tolerance, and improved nutritional quality. GM crops that have been commercialized in the recent past include tomato, corn, soybean, cotton, canola, rice, potato, squash, melon, and papaya, of which herbicide-tolerant soybeans; insect- and herbicide-tolerant corn; bollworm-resistant, herbicide-tolerant cotton; and herbicide-tolerant canola are the major ones.
6 Methods of GM Seed Testing
There are four different levels of GM assessment in seed samples as mentioned below:
-
GM detection: Screening a seed sample for detecting the presence of transgenic seeds. Primers are used to detect a general genetic element such as the constitutive promoter or a selection marker, which is frequently found in all GMOs, thereby detecting their presence within a seed lot.
-
Gene-specific detection: Identification of a specific GMO by testing for a specific transgene, using primers specific for the gene sequences. The presence of amplified fragments indicates that the seeds carry unique transgene being tested for.
-
Gene construct-specific detection: Identification of specific gene construct in a seed lot. This is done by using primers specific to sequences of promotor/terminator and part of the gene.
-
Event-specific detection: Is the detection of a gene event for which one should know about the flanking sequences of a targeted gene construct, i.e. host genome sequence which is close to the construct. The event is identified by using primers designed to detect the unique integration site of a specific GMO (Fig. 2).

Different levels of GM seed testing (Shrestha et al. 2010)
The two major purposes of GM seed testing include:
-
GM trait purity: Tests the extent of non-GM seeds in a GM seed lot.
-
Adventitious presence (AP) of GMOs: Detects the presence of GM seed in the non-GM seed lot. Mostly required for labelling in the global seed trade.
Hence, both qualitative and quantitative analytical methods are used for GM seed testing, which could be based on PCR, based on protein assays, or based on trait expression level (bioassays).
PCR-based methods | Protein-based methods | Bioassays |
|---|---|---|
Conventional PCR using DNA (endpoint qualitative PCR for the presence of gene) | Dipstick assays (lateral flow strip test): Tests for the presence of gene based on antigen-antibody reaction | Scoring based on trait expression in a specific condition provided artificially |
Real-time PCR using RNA/DNA: Follows progressive detection and is used to determine the quantity of DNA or gene copy number | ELISA test: Tests for the transgene presence and level of expression based on antigen-antibody reaction | Seed soak bioassays |
6.1 Lateral Flow Strip Test
The lateral flow strip test is based on immunoassay which uses a capillary paper immobilized with antibodies. The paper is inserted into a crude extract which allows the antigen to flow along the paper strip. The antigen in the extract binds with antibodies labelled with colloidal gold on the dipstick so as to form a distinct coloured band whenever antigen-antibody binds to each other (ISTA Rules 2021).
It is a qualitative rapid visual test and has been widely used for regulation of Bt cotton seed marketing in India (Kranthi 2013) (Fig. 3).

Dipstick method of testing Bt in seeds (Kranthi 2013)
6.2 ELISA (Enzyme-Linked Immunosorbent Assay) Test
ELISA is a sensitive immunoassay which uses solid-phase enzyme. In this immunoassay, the amount of unknown analyte in the sample is measured by adding the labelled antibodies. A capture antibody is coated on multi-well plate, each well loaded with crude seed extract. The labelled antibodies get attached with the analyte in the sample (antigen). The extra unbound labelled antibodies are washed away, and only antigen-bound labelled antibodies are present in the plate. When the plates are washed with a chromogenic substrate, there will be a reaction with the enzyme labelled antibodies wherever the antigen-antibody binding occurs and gives a fluorescence that is read by an ELISA reader. The intensity of the fluorescence is proportional to the quantity of the target protein (ISTA Rules 2021). In sandwich ELISA, the antigen (sample to be analysed) is sandwiched between two antibodies for more specific reactions (Kausar et al. 2017). It is very important to note that washing step after every reaction is very crucial after every reaction so as to wash away any extra materials. ICAR-CICR, Nagpur, Maharashtra, India, has developed simple ELISA test kits to enable farmers, seed testing officers, researchers, and seed companies to detect Bt seed quality (Kranthi 2013) (Fig. 4).

Schematic representation of the direct ELISA method (Kausar et al. 2017)
6.3 Bioassays
Bioassays are tests based on visual assessment of expression of a trait under specific growing conditions. For seed testing purpose, bioassays are used particularly for testing the herbicide tolerance trait in crops. Seeds are first exposed to the herbicide and then tested for germination and growth. If the seeds germinate and grow normally, they are scored as positive. The concentration of herbicide needs to be appropriate to obtain the intended difference in expression. Seed soak bioassays have been reported by researchers for identifying/detecting herbicide-tolerant seeds in soybean (Torres et al. 2003). Seed soak bioassays will only determine the GM trait and not the event.
6.4 Conventional PCR
Conventional PCR is employed for the general screening of GM seeds using primers that recognize common DNA which most GMOs harbour, e.g. Ca MV 35 S promoter and nos terminator. It can also be used for determining specific transgene/event purity. The procedure involves use of primers specific to the gene or event depending on the requirement. The test is positive if a band of appropriate size is observed on the gel. Multiplex PCR, a variant of conventional PCR, involving simultaneous amplification of multiple target sequences in a test sample is also employed for faster GM seed detection. A multiplex PCR assay was developed for the simultaneous amplification of transgene, (CaMV) 35S promoter, selectable marker gene, and nopaline synthase (nos) terminator along with β-fructosidase gene, an endogenous gene of Solanaceae family, for routine testing to detect GM tomato seeds in its germplasm collection (Randhawa et al. 2011). Multiplex PCR assays are also available to detect various GM events of maize (Degrieck et al. 2005) and cotton (Nadal et al. 2009).
6.5 Real-Time Polymerase Chain Reaction (RT-PCR/qPCR)
GMO quantification based on event-specific primers can be achieved through real-time PCR. It consists of amplification, simultaneous detection, and quantification of targeted DNA. In real-time PCR, DNA amplification activates fluorochromes attached to the primers, thus increasing the fluorescent signal with amplicons produced. This activation can be measured in real time giving an estimate of the DNA molecules being amplified in each cycle. In qualitative RT-PCR, positive score is given when the fluorescence detected is above the baseline. In quantitative RT-PCR, the target quantity is measured against a standard curve produced from certified reference material. There are several advantages real-time PCR has such as rapid cycling to reduce DNA amplification time, completion of PCR in a closed system to reduce the risk of cross-contamination, post-PCR electrophoresis not needed, and possibility of multiplexing by using probes containing different reporter dyes with distinct spectra (Fig. 5).

RT-PCR quantification of DNA using different spectral dyes with distinct spectra (Noli 2010)
7 ISTA and GM Seed Testing
The coexistence of GM and non-GM crops has raised a concern in many European countries, and the law requires that all GM food be traceable to its origin. Any product, food, or seed with GM content greater than 0.9% needs to be labelled. The GM quantification studies are done between 0% and 0.9% labelling thresholds, since the threshold levels vary within this range in different countries and regions around the world. When seeds are traded across borders between countries with different thresholds (http://norden.diva-portal.org), a reliable testing for determining these threshold levels has to be performed (Degrieck et al. 2005). As part of its role in harmonization of GM seed testing procedures, ISTA has been conducting proficiency test programmes since 2002 and workshops on GMO testing since 2001 and included a chapter on GM testing in the ISTA Seed Rules since 2014. ISTA developed methods which can be used for both testing adventitious presence (AP) of GM seeds and GM trait purity testing of seed lots (ISTA Rules 2021). ISTA international certificates, which can only be issued by ISTA-accredited laboratories, guarantee the identity of the seed lot with a single reference, the traceability of the analysis, the competence of the laboratory that did the analysis, the use of validated methods and standard units, and the use of standard reporting languages. Today, the ISTA Orange International Seed Lot Certificate (OIC) is widely used for international trade. Certified reference materials are used for the calibration or quality control of GMO quantification measurements typically carried out by quantitative real-time polymerase chain reaction (qPCR). Numerous sets of reference materials for different GM events in maize, soybean, potato, sugar beet, and cotton are offered for testing in the laboratories worldwide (www.ec.europa.eu). GM seed testing by ISTA is undertaken by (1) qualitative assay to assess trait purity in a GM seed sample by testing individual seeds in a sample, (2) semi-quantitative assay to assess adventitious presence of GM seeds in conventional seed lots by testing various seed bulks (sub-samples) within a seed lot, or (3) quantitative assay for quantitative assessment of adventitious presence (quantity of GM seed) in a non-GM seed lot done by analysing one single seed bulk from a seed lot (Enrico 2010). However, both AP GM seed testing and GM purity testing can be done on individual seeds or seed bulks.
The limit of detection (LOD) for quantitative tests and the limit of quantification (LOQ) for qualitative tests are critical criteria in deciding the sample size to be drawn (Broeders et al. 2014). LOD is defined as the smallest number of target seeds that has been demonstrated to be detected with a given level of confidence. The limit of quantification is the smallest amount of target analyte that has been demonstrated to be reliably measured with acceptable levels of accuracy and precision (ISTA Rules 2021). Obtaining a representative seed sample for testing GM seeds is important as in case of other quality parameters. Working sample size depends on the threshold requirements, method capability, and statistical confidence level and can be determined using appropriate statistical tools such as SeedCalc 8.0 (Remund et al. 2001). Testing methods developed using this tool can be used to estimate genetic purity of a seed lot, as well as a criterion to accept/reject a lot. The sample submitted to the laboratory must be at least the size of working sample and preferably larger than the working sample for accurate estimation.
An alternate PCR-based approach to test trait purity in bulk samples has been developed in which the absence of transgenic DNA is detected. For this, the insertion site of a transgene is characterized, and the corresponding sequence of the wild-type (wt) allele is used as primer binding site for amplification. This method could quantify the non-GM contamination as well as GM trait purity in RR soybean (Battistini and Noli 2009). Whatever the method is, ISTA suggests uniformity of result, and hence, a performance-based approach is followed for giving accreditation to any laboratory in GM seed testing. For detailed understanding of getting laboratory accreditation of ISTA in GM seed testing, readers may refer to ISTA Principles and Conditions for Laboratory Accreditation Under the Performance-Based Approach.
8 Molecular Markers in Germplasm Conservation and Maintenance
With the widespread cultivation of GM crops, conservation and exchange of germplasm have become a challenge in terms of determining the adventitious presence of GM seeds in conventional seeds. Conventional singleplex and multiplex PCR assays and real-time PCR targeting common screening elements or specific GM targets can be efficiently employed to check the unintended presence of GM seeds in any lot. DNA-based diagnostics including PCR/real-time PCR assays have been successfully employed to detect the adventitious presence of transgenic seeds in ex situ brinjal, okra, and cotton accessions conserved in National GeneBank (NGB) at the Indian Council of Agricultural Research-National Bureau of Plant Genetic Resources (ICAR-NBPGR), New Delhi (Kuwardadra et al. 2020; Randhawa et al. 2011). With technology advancement, the organization developed many cost- and time-efficient DNA-based GM detection technologies for the simultaneous detection of GM events in various crops (Randhawa et al. 2016). These include multiplex PCR, real-time PCR, visual LAMP, real-time LAMP, and multitarget TaqMan real-time PCR plate methods.
Minimizing the inclusion of duplicate accessions is important in germplasm conservation. It is estimated that there is an average of 50% duplication in different collections (Singh and Singh 2019). Molecular markers can be used for the unambiguous identification of duplicate accessions which can be safely removed from the holding for better maintenance. Genetic distance between the accessions can be determined from Nei’s dissimilarity matrix, and the ones which are having mean distance less than the threshold value can be considered as potential duplicates (Das et al. 2020).
9 Molecular Techniques for Seed Health Testing
Modern molecular methods hold great potential for improving pathogen detection in seeds, as it has many advantages over conventional assays in terms of specificity, sensitivity, rapidity, ease of implementation, and applicability.
9.1 Polymerase Chain Reaction (PCR)
The PCR technique had been successfully exploited for the detection of some of the seed-borne pathogens like Ascochyta lentis from lentil seeds (Hussain et al. 2000), Magnaporthe grisea (rice blast disease) (Chadha and Gopalakrishna 2006), Tilletia indica (karnal bunt disease) (Thirumalaisamy et al. 2011), and soybean yellow mottle mosaic virus (SYMMV) in French bean and mung bean seeds (Nagamani et al. 2020) (Fig. 6).

Confirmation of seed transmission of SYMMV in mung bean cv. Pusa Vishal. (a) Symptomatic mung bean plants showing less pod formation and stunted growth of the plants. (b) Infected leaf with mottling and puckering symptoms. (c–f) Seeds from infected plants showing brownish discolouration. (g) ISEM confirmation of SYMMV from infected mung bean seed. (h) PCR amplicons (1065 bp) obtained with coat protein (CP)-specific primers NS1 and NS2 (lanes 1–10 indicate RT-PCR from single whole infected seeds). (i) Detection of SYMMV through RT-PCR with CP primers in group of two (initial four lanes) and five seeds (last four lanes) where amplification was observed in whole seed and cotyledons. (j–l) Detection of SYMMV with CP primers in group of five seeds from various seed tissue parts (W, whole seed; Sc, seed coat; Co, cotyledons; E, embryo; H-RT, PCR from healthy seed; +ve, leaf tissue infected with SYMMV; M, GeneRuler 1 kb and 1 kb plus DNA ladders)
9.2 Bio-Polymerase Chain Reaction (bio-PCR)
Bio-PCR improves the efficiency and sensitivity of PCR by allowing target pathogen populations to increase in a pre-enrichment phase, before DNA extraction and PCR. This results in higher quantities of target DNA, which ultimately results in higher sensitivity. During the incubation and enrichment phase on artificial media, inhibitory compounds are adsorbed or diluted and do not interfere with DNA amplification. The seed samples are washed initially and crushed in suitable buffer to extract seed-borne bacteria. Aliquots are spread onto semi-selective media and incubated for 2–3 days. Colonies are then harvested for DNA extraction, and PCR is conducted with specific primers. In the case of seed-borne fungi, seeds are incubated under conditions of high relative humidity to increase target fungal mycelium mass before DNA extraction and PCR (Walcott 2003).
The advantages of Bio-PCR are that only viable colonies are detected, as the target organisms must grow on selective medium before it can be detected by PCR, and there is no need to identify the pathogen based on colony morphology as specific primers are used for amplification. Bio-PCR has been developed for the detection of bacterial fruit blotch of watermelon (Acidovorax avenae subsp. citrulli), halo blight (Pseudomonas syringae subsp. phaseolicola) of beans (Phaseolus vulgaris), bacterial ring rot (Clavibacter michiganensis subsp. sepidonicum) of potato (Solanum tuberosum), and black rot (Alternaria radicina) of carrot (Daucus carota) (Pryor and Gilbertson 2001; Schaad et al. 1999). The disadvantage of Bio-PCR includes the requirement of semi-selective medium for each pathogen for which specific knowledge about the nutritional requirements and chemical tolerances of the target organism is required. Also, the method requires 2–3 days for bacteria and 5–7 days for fungi to grow, thereby significantly increasing the time required for assay completion. Another drawback of Bio-PCR is that it is limited primarily to readily culturable bacteria and fungi and can’t be used for obligate parasites (e.g. viruses).
9.3 Multiplex Polymerase Chain Reaction (mPCR)
Multiplex PCR is a more reliable, fast, and cost-effective method for routine detection of seed-borne pathogens. Multiplex PCR (DNA targets) and multiplex RT-PCR (RNA targets) are useful for the simultaneous detection of multiple pathogens (broad-spectrum detection) in a single reaction containing more than one set of primers. Multiplex PCR was utilized for the simultaneous detection of three bacterial seed-borne diseases, viz. bacterial grain rot (Burkholderia glumae), bacterial leaf blight (Xanthomonas oryzae pv. oryzae), and bacterial brown stripe (Acidovorax avenae subsp. avenae), based on 16S and 23S rDNA and transposase A gene sequence in rice (Kang et al. 2016). Multiplex TaqMan real-time PCR assay was used for the detection of spinach seed-borne pathogens, viz. Peronospora farinosa f.sp. spinaciae, Stemphylium botryosum, and Verticillium dahliae, by using the primers based on internal transcribed spacer, intergenic spacer, and the elongation factor 1 alpha. Sensitivity of multiplex PCR is influenced by the number of target pathogens to be detected where a number of different primers are more important than the total amount of primer in the reaction mixture. Some of these limitations can be overcome with more precise specificity and sensitivity by improving the quality of nucleic acid extraction procedure and modification of PCR technology with the use of magnetic nanobeads and dual priming oligonucleotide primers or a nested reaction (Kwon et al. 2014).
9.4 Nested PCR
Nested PCR involves two pairs of amplification primers and two successive rounds of PCR. Initially, one primer pair is used in the first round of PCR with 15–30 cycles. The products of the first round of amplification are then subjected to a second round of amplification using the second set of primers which anneal to a sequence internal to the sequence amplified by the first primer set.
Nested PCR was successfully employed for the detection of some of the seed-transmitted pathogens, viz. tomato black ring virus (TBRV), Arabis mosaic virus (ArMV), cherry leafroll virus (CLRV), and grapevine fanleaf virus (GFLV), tobacco ring spot virus, and Ustilaginoidea virens (false smut disease of rice) (Danesh et al. 2014). The advantage of nested PCR is its increased sensitivity and specificity due to higher number of total cycles and annealing of the second primer set to sequences found only in the first round products. However, nested PCR assays are time-consuming and have an increased risk of cross-contamination which can create false-positive results.
9.5 Real-Time PCR
Inspite of many advantages real-time PCR has not been used much for the detection of seed-borne pathogens due to the requirement of expensive thermal cyclers that are equipped to detect fluorescence. Detection of seed-borne pathogens through real-time PCR was reported for Didymella bryoniae causing gummy stem blight of cucurbits (Ling et al. 2010), Clavibacter michiganensis subsp. michiganensis causing bacterial canker of tomato (Han et al. 2018), and soybean yellow mottle mosaic virus (Nagamani et al. 2020).
9.6 Loop-Mediated Isothermal Amplification (LAMP)
Loop-mediated isothermal amplification (LAMP), developed by Notomi et al. (2000), is a simple, cost-effective, and rapid method for the specific detection of genomic DNA that enables the synthesis of large amounts of DNA in a short period of time without the use of thermal cycler. LAMP technology uses a pair of four or six oligonucleotide primers and a thermophilic DNA polymerase for DNA amplification. LAMP products can be visualized by gel electrophoresis using magnesium pyrophosphatase which enhances the precipitation of amplified DNA or with a real-time turbidity reader or with the addition of an intercalating dye, such as SYBR Green I, which produces a colour change in the presence of target phytopathogen.
LAMP was used successfully for the detection of Fusarium graminearum (head blight disease) using the primers based on galactose oxidase gene (gaoA) (Niessen and Vogel 2010). Reverse transcription LAMP (RT-LAMP) assay was used for the detection of tomato brown rugose fruit virus in tomato and pepper seeds (Rizzo et al. 2021).
9.7 Microarray Technology
DNA chip or microarray technology has been applied to detect seed-borne pathogens. Microarray technology depends on the unique ability of nucleic acid molecules to hybridize specifically with molecules of complimentary sequences. In microarray technology, hundreds to thousands of oligonucleotides will be attached to specific locations on each chip. These oligonucleotides can be complementary to DNA sequences unique to certain pathogens and hence can be used to detect pathogens present in the seed sample. The DNA or RNA is extracted from the seed sample to be tested, amplified, and digested into smaller fragments that are labelled with fluorescent markers and hybridized with oligonucleotides fixed to the DNA chip. After hybridization, the chip is washed thoroughly, and fluorescence, which is directly proportional to the amount of nucleotide retained, is measured. If the pathogen of interest is present in the seed sample, then the oligonucleotide probe at the position on the chip that corresponds to that pathogen will display fluorescence.
DNA chip technology, which helps in the simultaneous detection of a wide range of pathogens within a short period of time (6 h), has great scope of application, and several such DNA chip assays for seed pathogen detection are already available.
10 Challenges in Using Molecular Techniques for Seed Quality Testing
Variety identification, seed genetic purity, and seed health testing are important parameters determining the seed quality. Being efficient, time-saving, less labour-intensive, reproducible, and amenable to high-throughput systems, the molecular marker systems would play an important role in cultivar identification and seed genetic purity testing.
Another important factor in genetic purity determination using molecular markers (e.g. SSR) is the number of core primers to be employed, which depends on the purpose of seed quality test. While one robust marker shall be enough for testing hybrid purity, multiple markers with high discrimination and repeatability are needed for varietal purity testing (ISTA 2021). Jamali et al. (2019) reviewed the potential deployment of DNA markers in plant variety protection and registration and summarized their efficacy, particularly in case of establishing the status of essentially derived varieties (EDVs). However, the number of markers required for establishing the EDV status may be even more. There is a need to standardize and identify minimum number of SNP/SSR markers, which have high reproducibility and can differentiate a good number of cultivars/genotypes precisely. These selected SSR/SNP markers can be analysed using high-throughput platforms, and these platforms also need to be flexible to accommodate variable number of samples.
Thorough mapping of crop genome by molecular markers that can adequately discriminate among elite adapted germplasm is required before these can be used in variety discrimination. Selection of markers has to be done using a large set of genotypes because a set of markers highly discriminating one set of genotypes with high PIC values need not necessarily be effective in discriminating another set of inbred lines. ISTA suggests that the identified markers are ought to produce sharp bands without null alleles and give similar allele patterns across repeats. Only common marker sets prescribed by ISTA have to be used in routine seed testing of a specific crop.
Tight linkages between the molecular marker loci and the loci expressing a morphological trait will facilitate its efficient use in purity testing/variety discrimination/protection. For example, molecular markers tightly linked to male sterile genes would facilitate an efficient and rapid transfer of ms genes into different genetic backgrounds through marker-assisted backcrossing, hybrid seed production, and genetic purity testing of hybrid seeds (Naresh et al. 2018).
Information about the homozygosity of polymorphic molecular marker loci in the parental inbred line is of importance, since segregation of these may create false interpretations of the purity of hybrid seed lots. The residual heterozygosity of useful marker loci needs to be kept at a minimum by adopting good maintenance breeding practices (Santhy et al. 2019). Hence, it is pertinent that there should be only minimum polymorphic loci available for purity checking so that it is easier to maintain the parental lines, and F1 profiles are also not varying during purity testing. Maintaining the genetic constitution of the reference material is of paramount importance when using markers in routine seed testing programmes. It is also important to ensure that the results obtained by the laboratories quantifying GMOs are comparable and results are based on the testing of a representative seed sample with an adequate sample size.
In seed health testing, it will be critical to rigorously evaluate molecular detection assays for their precision and accuracy through high-throughput testing of naturally infested seeds before adopting these for routine seed testing. To ensure that these detection assays work, they must be validated through stringent multi-laboratory tests which would evaluate their reproducibility, repeatability, and reliability.
References
Abd El-Moghny AM, Santosh HB et al (2017) Microsatellite marker-based genetic diversity analysis among cotton (Gossypium hirsutum) accessions differing for their response to drought stress. J Plant Biochem Biotechnol 26(3):366–370
Ali MA, Seyal MT et al (2008) Hybrid authentication in upland cotton through RAPD analysis. Aust J Crop Sci 2(3):141–149
Arun Kumar MB, Dadlani M et al (2014) Identification and validation of informative SSR markers suitable for ensuring the genetic purity of brinjal (Solanum melongena L.) hybrid seeds. Sci Hortic 171:95–100
Babu BK, Mathur RK et al (2017) Development, identification and validation of CAPS marker for SHELL trait which governs dura, pisifera and tenera fruit forms in oil palm (Elaeis guineensis Jacq.). PLoS One 12(2):e0171933
Battistini E, Noli E (2009) Real-time quantification of wild-type contaminants in glyphosate tolerant soybean. BMC Biotechnol 9:16
Bora A, Choudhury PR et al (2016) Assessment of genetic purity in rice (Oryza sativa L.) hybrids using microsatellite markers. Biotech 6:50
Bradford KJ (2006) Methods to maintain genetic purity of seed stocks. Agricultural biotechnology. In: California Series Publication 8189
Broeders S, Huber I et al (2014) Guidelines for validation of qualitative real-time PCR methods. Trends Food SciTechnol 37(2):115–126
Butler JM (2012) Random amplified polymorphic DNA. In: Advanced topics in forensic DNA typing methodology. Elsevier Academic Press, San Diego, CA, pp 473–495
Chadha S, Gopalakrishna T (2006) Detection of Magnaporthe grisea in infested rice seeds using polymerase chain reaction. J Appl Microbiol 100:1147–1153
Danesh YR, Tajbakhsh M et al (2014) Using nested PCR for detection of seed borne fungi Ustilaginoidea virens in rice fields of Iran. In: Turkey 5th seed congress. pp 670–672
Daniel IO et al (2012) Application of SSR markers for genetic purity analysis of parental inbred lines and some commercial hybrid maize (Zea mays L.). Aust J Exp Agric 2(4):597–606
Das A, Rajesh KS et al (2020) Identification of duplicates in ginger germplasm collection from Odisha using morphological and molecular characterization. Proc Natl Acad Sci India Sect B Biol Sci 90(5)
Degrieck I, de Andrada Silva E et al (2005) Quantitative GMO detection in maize (Zea mays L.) seed lots by means of a three-dimensional PCR based screening strategy. Seed Sci Technol 33(1):131–143
Ferriol M, Picó B, Nuez F (2003) Genetic diversity of a germplasm collection of Cucurbita pepo using SRAP and AFLP markers. Theor Appl Genet 107:271–282
Grzebelus D et al (2008) The use of AFLP markers for the identification of carrot breeding lines and F1 hybrids. Plant Breed 120(6):526–528
Han S, Jiang N et al (2018) Detection of Clavibacter michiganensis subsp. Michiganensis in viable but nonculturable state from tomato seed using improved qPCR. PLoS One 13(5):e0196525
Hussain S, Tsukiboshi T, Uematsu T (2000) Quick detection of Ascochyta lentis from lentil seeds using polymerase chain reaction (PCR) based techniques. Pak J Bot 32:45–56
Hwu K-K (2013) Overview of DNA technologies: current uses and applications. In: 30th ISTA Seed Congress held at Antalya, Turkey
ISAAA (2019) Global status of commercialized biotech/GM crops in 2019: biotech crops drive socio-economic development and sustainable environment in the new frontier (https:///resources/publications/pocket/16/)
ISTA (2021) International rules for seed testing (2021) Full Issue I–19-8 (300). https://doi.org/10.15258/istarules
Jamali SH, Cockram J, Lee T (2019) Insights on deployment of DNA markers in plant variety protection and registration. TheorAppl Genet 132(7):1911–1929
Jang I, Moon JH, Yoon JB et al (2004) Application of RAPD and SCAR markers for purity testing of F1 hybrid seed in chili pepper (Capsicum annuum). Mol Cells 18(3):295–299
Jiang GL (2013) Molecular markers and marker assisted breeding in plants. In: Andersen SB (ed) Plant breeding from laboratories to fields. Intech Open. https://www.intechopen.com/books/3060, London. https://doi.org/10.5772/3362
Jones H, Norris C et al (2013) Evaluation of the use of high-density SNP genotyping to implement UPOV model 2 for DUS testing in barley. Theor Appl Genet 126:901–911
Josia C, Mashingaidze K et al (2021) SNP-based assessment of genetic purity and diversity in maize hybrid breeding. PLoS One 16(8):e0249505
Kang IJ, Kang MH et al (2016) Simultaneous detection of three bacterial seed-borne diseases in rice using multiplex polymerase chain reaction. Plant Pathol J 32(6):575–579
Kausar M, Haleema S, Muhammad HB (2017) Protein based detection methods for genetically modified crops. https://doi.org/10.5772/Intechopen.75520
Khajudparn P, Prajongjai T et al (2012) Application of ISSR markers for verification of F1 hybrids in mungbean (Vigna radiata). Genet Mol Res 11(3):3329–3338
Kim M, Jung JK et al (2021) Genome-wide SNP discovery and core marker sets for DNA barcoding and variety identification in commercial tomato cultivars. Sci Hortic 276:109734
Kranthi KR (2013) Testing seed quality of Bt cotton. CAI Weekly Publication No. 29
Kumar V, Sharma S et al (2008) Genetic diversity in Indian common bean (Phaseolus vulgaris L.) using random amplified polymorphic DNA markers. Physiol Mol Biol Plants 14(4):383–387
Kuwardadra SI, Bhat KC et al (2020) Monitoring adventitious presence of transgenes in brinjal collections from the regions in India bordering Bangladesh: a case report. Genet Resour Crop Evol 67:1181–1192
Kwon JY, Hong JS et al (2014) Simultaneous multiplex PCR detection of seven cucurbit infecting viruses. J Virol Methods 206:133–139
Law JR, Donini P, RMD K et al (1998) DNA profiling and plant variety registration, III: the statistical assessment of distinctness in wheat using amplified fragment length polymorphisms. Euphytica 102:335–342
Ling KS, Wechter WP et al (2010) An improved real-time PCR system for broad-spectrum detection of Didymella bryoniae, the causal agent of gummy stem blight of cucurbits. Seed Sci Technol 38(3):692–703
Liu L et al (2007) Evaluation of genetic purity of F1 hybrid seeds in cabbage with RAPD, ISSR, SRAP, and SSR Markers. Hort Sci 42(3):724–727
Madhuchhanda P, Ngangkham U et al (2020) A multiplex PCR system for testing the genetic purity of hybrid rice (Oryza sativa L.). Indian J Genet Plant Breed 80(2):213–217
Nadal A, Esteve T, Pla M (2009) Multiplex polymerase chain reaction-capillary gel electrophoresis: a promising tool for GMO screening- assay for simultaneous detection of five genetically modified cotton events and species. J AOAC Int 92(3):765–772
Nagamani S, Tripathi A et al (2020) Seed transmission of a distinct soybean yellow mottle mosaic virus strain identified from India in natural and experimental hosts. Virus Res 280:197903
Naresh V, Yamini KN et al (2009) EST-SSR marker-based assay for the genetic purity assessment of safflower hybrids. Euphytica 170(3):347–353
Naresh P, Lin S et al (2018) Molecular markers associated to two non-allelic genic male sterility genes in peppers (Capsicum annuum L.). Front Plant Sci 9:1343
Niessen L, Vogel RF (2010) Detection of Fusarium graminearum DNA using a loop-mediated isothermal amplification (LAMP) assay. Int J Food Microbiol 140(2–3):183–191
Enrico Noli (2010) New Tools for measuring genetic quality in seed. In: 29th ISTA Seed Congress Cologne 2010. https://www.yumpu.com/en/document/view/8667848/aspects-of-purity-international-seed-testing-association
Notomi T, Okayama H, Masubuchi H et al (2000) Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28(12):e63
Pallavi HM, Gowda R, Shadakshari YG (2011) Identification of SSR markers for hybridity and seed genetic purity testing in sunflower (Helianthus Annuus L.). Helia 34(54):59–66
Park G, Choi Y et al (2021) Genetic diversity assessment and cultivar identification of cucumber (Cucumis sativus L.) using the Fluidigm single nucleotide polymorphism assay. Plants (Basel) 10(2):395
Parthiban S, Govindaraj P, Senthilkumar S (2018) Comparison of relative efficiency of genomic SSR and EST-SSR markers in estimating genetic diversity in sugarcane. 3 Biotech 8(3):144
Peng WANG, Zhejuan TIAN et al (2021) Establishment and application of a tomato KASP genotyping system based on five disease resistance genes. Yuan Yi Xue Bao 48(11):2211
Phillips T (2008) Genetically modified organisms (GMOs): transgenic crops and recombinant DNA technology. Nat Educ 1(1):213
Pryor BM, Gilbertson RL (2001) A PCR-based assay for detection of Alternaria radicina on carrot seed. Plant Dis 85:18–23
Randhawa G, Chhabra R, Singh M (2011) PCR based detection of genetically modified tomato with AVP1D gene employing seed sampling strategy. Seed Sci Technol 39(1):112–124
Randhawa G, Singh M, Sood P (2016) DNA-based methods for detection of genetically modified events in food and supply chain. Curr Sci 110(6):1000
Ravishankar KV, Dinesh MR (2015) Development and characterization of microsatellite markers in mango (Mangiferaindica) using next-generation sequencing technology and their transferability across species. Mol Breed 35:93
Ravishankar KV, Muthaiah G et al (2018) Identification of novel microsatellite markers in okra (Abelmoschus esculentus (L.) Moench) through next-generation sequencing and their utilization in analysis of genetic relatedness studies and cross-species transferability. J Genet 97(1):39–47
Remund K, Dixon D et al (2001) Statistical considerations in seed purity testing for transgenic traits. Seed Sci Res 11(2):101–120
Rizzo D, Lio DD et al (2021) Rapid and sensitive detection of tomato brown rugose fruit virus in tomato and pepper seeds by reverse transcription loop-mediated isothermal amplification assays (real time and visual) and comparison with RT-PCR end-point and RT-qPCR methods. Front Microbiol 12:640932
Rousselle Y, Elizabeth J et al (2015) Study on essential derivation in maize: III. Selection and evaluation of a panel of single nucleotide polymorphism loci for use in European and North American germplasm. Crop Sci 55:1170–1180
Santhy V, Meshram M (2015) Widening the character base for distinctness in cotton: emerging perspective. Curr Sci 109(11)
Santhy V, Mohapatra T et al (2000) DNA markers for testing distinctness of rice varieties. Plant Var Seeds 13:141–148
Santhy V, Dadlani M et al (2003) Identification of the parental lines and hybrids of rice (Oryza sativa L.) using RAPD markers. Seed Sci Technol 31(1):187–192
Santhy V, Meshram M et al (2019) Molecular diversity analysis and DNA fingerprinting of cotton varieties of India. Ind J Genet Plant Breed 79(4):719–725
Schaad NW, Berthier-Schaad Y et al (1999) Detection of Clavibacter michiganensis subsp. sepedonicus in potato tubers by BIO-PCR and an automated real-time fluorescence detection system. Plant Dis 83:1095–1100
Selvakumar P, Ravikeshavan R et al (2010) Genetic purity analysis of cotton (Gossypium spp.) hybrids using SSR markers. Seed Sci Technol 38:358–366
Shengrui Z, Li B et al (2020) Molecular-assisted distinctness and uniformity testing using SLAF-sequencing approach in soybean. Genes (Basel) 11(2):175
Shrestha HK, Hwu K-K, Chang M-C (2010) Trends Food Sci Technol 21(9):442–454
Singh BD, Singh AK (2019) Fingerprinting and gene cloning. In: Singh BD, Singh AK (eds) Marker assisted plant breeding principles and practices. Springer, New Delhi
Smith JSC, Register JC (1998) Genetic purity and testing technologies for seed quality: a company perspective. Seed Sci Res 8(2):285–294
Tatiana SA, Guchetl Z et al (2006) Development of marker system for identification and certification of sunflower lines and hybrids on the basis of SSR-analysis. Helia 29(45):63–72
Thirumalaisamy PP, Singh DV et al (2011) Development of species-specific primers for detection of Karnal bunt pathogen of wheat. Indian Phytopathol 64:164
Torres AC, Nascimento et al (2003) Bioassay for detection of transgenic soybean seeds tolerant to glyphosate. Pessqui Agropecu Bras 38(9):1053–1057
UPOV (2007) Working group on biochemical and molecular techniques, and DNA-profiling in particular
Utami DW, Rosdianti I et al (2016) Utilization of 384 Snp genotyping technology for seed purity testing of new Indonesian rice varieties, Inpari Blas and Inpari Hdb. SABRAO J Breed Genet 48(4):416–424
Walcott R (2003) Detection of seed borne pathogens. Hort Technol 13(1):40–47
Yang CJ, Russell J, Ramsay L et al (2021) Overcoming barriers to the registration of new plant varieties under the DUS system. Commun Biol 4:302
Yu JK, Chung YS (2021) Plant variety protection: current practices and insights. Genes (Basel) 12(8):1127. https://doi.org/10.3390/genes12081127
Zhang J, Yang J et al (2020) A new SNP genotyping technology target SNP-seq and its application in genetic analysis of cucumber varieties. Sci Rep 10(1):1–11
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this chapter

Cite this chapter
V., S., Sandra, N., Ravishankar, K.V., Chidambara, B. (2023). Molecular Techniques for Testing Genetic Purity and Seed Health. In: Dadlani, M., Yadava, D.K. (eds) Seed Science and Technology. Springer, Singapore. https://doi.org/10.1007/978-981-19-5888-5_15
Download citation
DOI: https://doi.org/10.1007/978-981-19-5888-5_15
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-5887-8
Online ISBN: 978-981-19-5888-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)