
Abstract
Over the past two decades, we have witnessed an unprecedented explosion in available biological data. In the age of big data, large biological datasets have created an urgent need for the development of bioinformatics methods and innovative fast algorithms. Bioinformatics tools can enable data-driven hypothesis and interpretation of complex biological data that can advance biological and medicinal knowledge discovery. Advances in structural biology and computational modelling have led to the characterization of atomistic structures of many biomolecular components of cells. Proteins in particular are the most fundamental biomolecules and the key constituent elements of all living organisms, as they are necessary for cellular functions. Proteins play crucial roles in immunity, catalysis, metabolism and the majority of biological processes, and hence there is significant interest to understand how these macromolecules carry out their complex functions. The mechanical heterogeneity of protein structures and a delicate mix of rigidity and flexibility, which dictates their dynamic nature, is linked to their highly diverse biological functions. Mathematical rigidity theory and related algorithms have opened up many exciting opportunities to accurately analyse protein dynamics and probe various biological enigmas at a molecular level. Importantly, rigidity theoretical algorithms and methods run in almost linear time complexity, which makes it suitable for high-throughput and big-data style analysis. In this chapter, we discuss the importance of protein flexibility and dynamics and review concepts in mathematical rigidity theory for analysing stability and the dynamics of protein structures. We then review some recent breakthrough studies, where we designed rigidity theory methods to understand complex biological events, such as allosteric communication, large-scale analysis of immune system antibody proteins, the highly complex dynamics of intrinsically disordered proteins and the validation of Nuclear Magnetic Resonance (NMR) solved protein structures.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

1 Introduction
In the current post-genomics era, advances in experimental and computational techniques have revolutionized biological and biomedical research. High-throughput technologies have paved the way to novel research avenues where we can systematically analyse whole genomes of organisms and individual or collection of proteins, including their structures and interactions with other proteins, which in many cases allow researchers to successfully decipher their biological functions. Proteins are macromolecules that are fundamental to most cellular function [1]. They comprise the highest levels of molecular and cellular structure and organization, and because the majority of physiological and disease processes are manifested within proteins, structural and computational biology research is focused on understanding protein function.
Proteins and other biomolecules are nanomachines. Accurate representation of their three-dimensional structure is a critical first step to understanding how they perform their functions. Advances in molecular biology, instrumentation, and imaging technologies such as X-ray crystallography, nuclear magnetic resonance (NMR), and electron microscopy have led to a revolution in structural biology. These techniques allow us to see beautiful yet complex three-dimensional shapes of protein structures and how they interact with other proteins and ligands. Protein imaging techniques are continuously improving, and for many proteins, we can now characterize their structures at an individual-atom-level resolution. A rapidly growing and revolutionary cryogenic-electron microscopy (cryo-EM) technique has been attracting significant attention, as very recently it has broken various resolution barriers [2] and can now discern individual atoms of very large protein structures (see Fig. 14.1). Cryo-EM complements X-ray crystallography because it reveals atomistic structural details without the need for a crystalline specimen. Protein Data Bank (PDB), a repository of experimentally solved protein structures, together with computationally determined protein structures, make up a rich source of protein structural data. Recent advances in AI and deep learning have provided significant improvements in inferring protein structures from a sequence of amino acids [3]. Deepmind’s Alphafold method has demonstrated that deep learning structure predictions can come astonishingly close to experimentally determined structures, and in the near future, we expect this will result in huge growth of macromolecular structural data. The increasing richness of the available protein structural data and the rapidly growing proteomics and bioinformatics big-data repositories open up possibilities to systematically analyse complex biological questions and gain novel biological insights. To facilitate data-driven biological knowledge discovery, many bioinformatics and computational biology tools, software packages, and databases have been developed [4].

Cryo-EM snapshot structure of viral spike protein of SARS-CoV-2 (a key protein involved in COVID-19), which is a very large protein structure consisting of three chains (distinct colours), each consisting of nearly 1300 amino acids
Despite tremendous advances in bioinformatics, structural biology and imaging technologies which have generated hundreds of thousands of atomic snapshots of protein structures, many fundamental biological problems such as protein folding, allosteric regulation, receptor signalling, and enzyme catalysis, to name a few, still remain largely unresolved [5,6,7,8,9,10,11,12]. While the static high-quality representation of protein structures can offer clues to structure-function mechanisms, protein function is almost purely controlled by its dynamic character through a delicate mix of rigidity and flexibility. Research must move beyond static snapshot representations of proteins, as the mechanical heterogeneity of protein structures that dictates their dynamic nature is intimately linked to their highly diverse biological functions. Deep understanding of the connection between structures and internal protein flexibility, rigidity, and dynamics is absolutely critical, as it can lead to solutions to protein folding problem, elusive allosteric regulation and other dynamically driven biological secrets of protein regulation.
The primary desire of any protein researcher is to see proteins move in real time at the atomistic level while they carry out their biological functions. Yet, despite many advances in experimental techniques and molecular dynamics simulations, such a goal is still very far from being realized. Analysing and comprehending protein flexibility and dynamics has proven to be extremely difficult. One major challenge is that the main molecular simulation methods, such as classical molecular dynamics simulations, require a prohibitive amount of computational power and are not suitable to reach biologically relevant functional dynamics that occur on longer (millisecond-second) timescales. Furthermore, with rapid growth in the number of experimentally solved biomolecular structures and the increasing size of structural protein databases, including the expanding big-data size sets of computationally predicted protein structures, we are faced with a pressing need to develop fast algorithms and novel mathematical and computational techniques that simplify the classical force fields and can offer experimentally verified accurate predictions of protein flexibility and dynamics.
Techniques inspired from the field of mathematical-structural rigidity theory [13,14,15,16] have gained special attention as they are suitable for handling the many challenges with computational analysis of protein flexibility and its dynamics. Biological functions of protein structures are often related to their network (or graph) properties. Mathematical rigidity theory offers considerable promise in deciphering graph theoretical properties of protein networks to better understand protein function [13, 15,16,17,18]. In rigidity theory, proteins are modelled as geometric molecular frameworks consisting of atoms and various connecting intermolecular forces. Such frameworks are essentially multigraphs (networks), in which atoms are vertices and edges form various bonding and non-bonding constraints (see Sect. 14.3). The programme FIRST [15] and related methods [19] apply mathematical results that provide combinatorial characterization of rigidity and flexibility on a molecular multigraph, which can rapidly decompose a protein framework (i.e., multigraph) into flexible and rigid regions. Starting with a decomposition of a protein into rigid and flexible regions, fast Monte Carlo geometric simulation methods, such as FRODA and FRODAN [19,20,21,22], can sample the highly complex conformational space of proteins and simulate their functionally relevant motions. The main advantage of rigidity theory methods over classical molecular dynamics simulations is that their predictions of rigidity and flexibility are very fast, they are not affected by timescale issues (see Sect. 14.2), and they are suitable for high-throughput and big-data style analyses. Moreover, predictions based on rigidity theory have been widely shown to be consistent with experimental measures of protein flexibility and dynamics [11, 12, 15, 17,18,19, 22, 24,25,26].
In this chapter, we first discuss the importance of protein flexibility and dynamics for biological function (Sect. 14.2). We then provide a brief review of fundamental concepts in rigidity theory (Sect. 14.3) that enables us to perform fast predictions of flexibility and dynamics of protein structures. We next discuss how to represent biomolecules as a graph constraint network, the mathematical/algorithmic background for analysing protein networks, and the basic uses of rigidity theory software for analysing protein flexibility and its dynamics. We then review some major advances contributed by the author of this chapter, in which rigidity theory and algorithms were used to elucidate and provide new perspectives on very complex biological phenomena, such as long-range allosteric communication, enzyme catalysis, antibody dynamics, and NMR structural validation (Sect. 14.4). We conclude by reviewing some of these recent developments and some surprising breakthroughs that have led to rich protein function discoveries that were mainly driven by mathematical rigidity theory.
2 Protein Structural Flexibility and Dynamics
In this section, we briefly cover for non-biologists the background and the importance of predicting protein flexibility, which is arguably one of the most fundamental research topics in biochemistry, structural biology, and bioinformatics.
2.1 Protein Flexibility and Dynamics Is Central to Protein Function
Proteins are polypeptide chains composed of a linear sequence(s) of amino acids [1]. Through a complex protein folding process, forces are exerted on atoms which steer a polypeptide chain(s) into a defined three-dimensional biologically functional native-state structural ensemble. High-resolution X-ray crystallography and other techniques have revealed aesthetic structural complexity of protein structures and have revolutionized our understanding of their function, which have spearheaded the development of novel experimental and computational methods for examining protein function in atomistic detail. It is important to stress that solved protein structures are only snapshots or pictures of proteins at some low-energy state. This can often provide a misleading representation of proteins and potentially misinform about their function, which must include kinetic and thermodynamic descriptions [5] (see Fig. 14.2).
Proteins are composed of rigid and connecting flexible regions that can be highly dynamic, which facilitates sampling a wide variety of conformations spanning a complex multidimensional energy landscape. In this conformational biomolecular dance, proteins undergo dynamical fluctuations even under conditions that are preferentially biased towards a well-defined low-energy ’native’ state [5]. Such dynamically driven conformational states and fluctuations are critical to long-range allosteric regulations, ligand recognition, catalytic efficiency, antibody–antigen recognition and the majority of functional mechanisms. Understanding protein flexibility and rigidity and how it is modified by mutations and ligand binding is critical to understanding and modulating protein function [5, 7, 8, 11, 12]. Most globular proteins (excluding intrinsically disordered proteins) function through utilizing a delicate mix of rigidity and flexibility. Achieving appropriate balance between rigidity and flexibility is one of the most important keys for biological function. Protein rigidity is necessary, as it maintains overall structural fold, while flexibility and dynamics enable proteins to perform specific functions. Protein defects can lead to alterations in overall folding, or they can cause proteins to be overly flexible, interfering with protein function, or cause other extreme defects that can result in indestructible rigid protein. These scenarios are related to numerous medical conditions, including neurological disorders, Alzheimer’s disease, and Mad Cow disease [22, 27]. Hence, predicting and examining protein flexibility and dynamics is the most important, and probably the most complex, component of protein research. This is an active area of research in both experimental protein science and computational biology.

The structure of an enzyme (Protein Data Bank ID 2jz3) showing a protein snapshot representation and conformational ensemble depicting its dynamical characteristics b
Protein structures can have thousands of conformational degrees of freedom. It is therefore easy to imagine that their motions can be extremely complex, and determining flexible and rigid regions and how they move relative to one another can seem like a daunting task. Moreover, many proteins are oligomeric structures consisting of two or more interacting polypeptide chains, and in some cases the structures are very large, consisting of thousands of amino acids (see Fig. 14.1). Protein flexibility and rigidity are often regulated by interactions with small ligands, drugs, hormones, and cations (e.g., calcium and magnesium) and changes in temperature, pressure, and pH [11, 15, 17, 18, 24]. Internal motion and conformational change can be rapid and transient and result in a structural ensemble that can often be spectroscopically indistinguishable from the snapshot ground state determined by X-ray crystallography or other imaging techniques (see Fig. 14.2). Protein dynamics occur across a wide range of timescales, from very rapid short-amplitude motions caused by bond vibrations occurring on a femtosecond range, to side-chain motions on the picosecond to nanosecond timescale, all the way up to very slow larger-amplitude collective domain motions, which are often biologically most significant, occurring in the milliseconds to seconds range [5] (see Fig. 14.3). Dynamics on longer timescales (i.e., millisecond to second timescales) are functionally very important because many biological processes—including allostery, enzyme catalysis, receptor activations, and protein–protein interactions—occur on such timescales [5, 9, 11, 12, 24, 28]. Fluctuations between different low-energy states and the heights of their energy barriers can also be affected by mutations, ligand binding, and changes in temperature or pH. The timescale component of protein dynamics is one critical factor that complicates the computation examination of protein dynamics. Another important characteristic of protein dynamics is the amplitude and directionality of conformational fluctuations [5]. All these factors combine to contribute to the difficulty in obtaining knowledge about the flexibility and motion of proteins.

Figure adapted from [5]
a A one-dimensional cross-sectional representation of a high-dimensional protein’s energy landscape. Proteins can be defined as multiple collections of low-energy conformational states (defined as minima in the energy surface), with many conformational ensemble substates interconverting between one another on very fast timescales. The time it takes a protein to transition from one low-energy state to another is dependent on the height of the energy barrier between the states. When the barrier is high, this can occur in a relatively long microsecond to second range. b Timescales of different dynamic processes in proteins and different experimental methods that can detect fluctuations on each timescale. Longer timescales are largely inaccessible to classical MD simulations. However, rigidity theory methods and simulations are not confined by this timescale issue.
Despite this complexity, functional motions will often involve large domain–domain motions (i.e., relative motions dominated by a few rigid bodies) and many degrees of freedom can be neglected or suppressed to study the functionally most important motions. Hence decomposition of a protein into rigid and flexible regions is a highly important aspect of deciphering protein dynamics.
2.2 Techniques for Analysing and Predicting Protein Flexibility and Dynamics
In terms of experimental techniques, NMR measurements such as order parameter measurements and chemical shifts are very useful in studying protein dynamics [24, 29]. Mass spectrometry, hydrogen–deuterium exchange, crystallographic B-values, etc. can also provide deep insights into the dynamical nature of protein structures [5, 11, 24, 25]. Fluorescence resonance energy transfer (FRET) [30] measures in particular have high practical value as they can characterize changes in distance for single molecules over time as well as possible corresponding conformational changes. However, the disadvantage of FRET is that only a single distance change is measured. Experimental measurements are useful as they can be used to infer specific information about dynamics across a specific range of timescales (see Fig. 14.3) and are specifically very helpful in supporting and validating computational predictions. The disadvantage of experimental tools is their high cost, susceptibility to uncertainty in measurements, and frequent inability to provide information about very dynamic regions of protein structures. Moreover, protein structures often have to be stabilized to extract structural and dynamical information. Experimental measurements can also take a long time to perform, as they require maintenance of very expensive equipment; yet, such measurements can rarely provide dynamical information about individual atoms.
Computationally, it should be theoretically possible to describe protein dynamics in their entirety. Molecular dynamics (MD) simulation has been the most widely used approach for simulating the motions of proteins and other biopolymers [28]. Molecular dynamics simulations of proteins have been a common tool in biochemistry and biophysics since the 1970s [31]. It has been successfully applied to protein folding problems, the impact of protein motions on enzyme catalysis, and the effects of mutations and ligand binding on protein motions [28]. Its uses have increased in recent years, pointing to the key importance of deciphering the relationships between complex motions and protein function. In molecular dynamics simulations, the trajectories of individual atoms in protein structures can be predicted by repeated numerical solutions of the Newtonian motion equation (i.e., \(F = ma\)), with forward integration in time, where F represents a force field (energy function). A force field models all potential forces and energies between the molecules and is supposed to be a simple parameterization of the energy surface of the protein. A number of different methods and force field models exist for parametrizing the potential energy surface. Assuming one can use an accurate description of a force field, a difficult and heavily debated concept, molecular dynamics simulation can be extremely useful in tracking the precise position of atoms over time. However, the major downside of molecular dynamics simulations is that they require prohibitively excessive computational power. Indeed, even despite today’s computational advances and special-purpose simulation machines [32], in the majority of cases molecular dynamics simulations are largely impractical for investigating biologically relevant protein motions on relatively long microsecond timescales. Stemming from the increase in protein structural data combined with the increasing size of solved structures, advances in emerging Cryo-EM technology and deep learning, it is clear that there is an urgent need to develop alternate efficient and accurate computational methods for molecular flexibility and dynamics simulations.
A large class of computational approaches that simplify classical force fields have been developed. Coarse-grained simulations, normal model analysis, principal component analysis, contact network analysis, and other related methods have become popular alternative approaches to classical MD simulations [33]. In coarse-grained and network approaches, physical units such as individual amino acids or a cluster of amino acids including rigid clusters can be treated as nodes (vertices), where edges indicate possible interactions or contacts. For more precise modelling, individual atoms should be treated as vertices and edges should model pairwise bonded and non-bonded contacts.
Arguably, one of the most powerful ways of analysing the flexibility and rigidity of protein structures, especially using an all atom representation, is based on mathematical rigidity theory [13,14,15,16, 19, 34]. Rigorous mathematical results in rigidity theory, whose details are explained below, can be used in combination with fast algorithms to rapidly decompose a protein constraint graph into rigid and flexible regions. Moreover, how rigidity is modified through protein–protein, protein–ligand, or other interactions can be quickly predicted. Such decompositions are very informative as they can be combined with other methods such as MD simulations, normal mode analysis, or Monte Carlo simulations [19, 22] to directly infer information about protein dynamics. This is discussed in more detail below. We now turn the discussion to mathematical formulations and the uses of rigidity theory for the analysis of protein structures.
3 Rigidity Theory
In this section, we present a basic introduction and results of rigidity theory that are essential for applications to protein structure and function analysis, with a focus on combinatorial rigidity theory concepts. For a thorough review of rigidity theory see [13, 19, 34].
3.1 Combinatorial Rigidity Theory and the Molecular Theorem
In general terms, flexibility is the ability of a material or framework to reversibly change the configuration of its joints, bodies, or building blocks. Rigidity, which is the opposite property of flexibility, describes a state in which no relative motions are allowed between the framework’s elements. In a rigid structure, only rigid body motions are possible (i.e., motions arising from congruences of space, rotations, translations, etc.). In biochemistry and biophysics, a notion related to rigidity is the concept of stability and robustness, where internal protein dynamics are not changed in response to small atomic fluctuations and the breaking of a few non-covalent interactions. Although to a non-expert, rigidity and stability may seem like related concepts, care should be taken to understand the potential differences and their implications.
Mathematical rigidity theory, sometimes called structural rigidity because of its close connections to structural and mechanical engineering, offers the most mathematically sound concepts and algorithms for analysis of rigidity and flexibility of frameworks [13, 14, 34]. Rigidity theory analyses the rigidity and flexibility of frameworks, as specified by geometric constraints such as fixed distances, directions, and volumes defined by a collection of points, lines, planes, or rigid bodies. Frameworks can be natural structures (molecules, crystals, proteins, etc.) or engineered structures (bridges, robots, etc.), and because rigidity is an essential property of most frameworks and materials, rigidity theory naturally has many applications in engineering, robotics, material science, and biology.
Rigidity theory has both geometric and combinatorial characteristics relying on techniques in linear algebra, discrete and algebraic geometry, graph theory, and combinatorics. Rigidity theory has a very long and rich history in mathematics, with early work appearing in the form of Euler’s (1766) conjectures on rigidity of polyhedra. Maxwell’s (1864) [14, 34] work on counting constraints in a framework for generic rigidity led to the birth of so-called ‘combinatorial rigidity’. Combinatorial characterization of rigidity theory, 140 years later, has turned out to be absolutely crucial for rapid flexibility analysis of materials such as glass networks and protein structures [14].
The classical and simplest frameworks studied in rigidity theory are the bar and joint frameworks (see Fig. 14.4), which are composed of universal (rotating) joints that are connected by bars that fix the distances between pairs of joints. A bar and joint framework is defined as a pair (G, p), where \(G = (V,E)\) is an undirected graph and \(p:V\rightarrow {\mathbb {R}}^d\), where vertices correspond to joints and edges correspond to bars that connect some pairs of joints; p represents a configuration of joints in \({\mathbb {R}}^d\). A framework (G, p) in \({\mathbb {R}}^d\) is rigid if the only edge-length-preserving continuous motions of the vertices are derived from isometries of \({\mathbb {R}}^d\). If \(d\ge 2\), it is NP-hard to determine if a bar and joint framework is rigid [34]. As determining the rigidity of frameworks is very difficult, a common approach is to linearize the problem by differentiating the length/bar constraints of the corresponding pair of connecting points/joints, which leads to a system of linear equations (one equation per edge) and a corresponding rigidity matrix. The solution to such a homogenous system can be captured by calculating the rank of the rigidity matrix, which indicates if a framework is infinitesimally rigid [34, 35]. However, in many applications and large frameworks such as proteins, this is not particularly practical owing to numerical errors and uncertainty in rank computations of the rigidity matrix.
A well-known fact within rigidity theory is that if the framework is generic (i.e., it does not have special singular geometry), then rigidity and infinitesimal rigidity coincide [34]. Generic frameworks are very important, as rigidity can be studied by pure graph and combinatorial techniques—a subfield of rigidity theory called combinatorial rigidity theory. A framework is generically rigid if it maintains rigidity even after minor changes to the position of its joints, and almost all frameworks are generic [13, 34, 36]. By assuming that a framework is in a generic position, one can neglect the geometric embedding of joints and actual distances of bars to focus on only the topology of the bar and joint framework and discuss the generic rigidity of (G, p) in terms of graph G.

Bar and joint framework examples: a is flexible as it can deform its shape (note it is one edge too short in terms of Laman’s count, \(|E|< 2|V| - 3\)); b is minimally rigid in 2D (but flexible in 3D as one can rotate two triangles around the diagonal). c is redundantly rigid in 2D as it has a redundant (i.e., extra) edge and is minimally rigid in 3D
3.1.1 Counting for Rigidity and Flexibility
We now motivate the characterization of rigidity of generic frameworks using combinatorial arguments. For bar and joint frameworks in dimension d, each joint (point, vertex) has d conformational degrees; hence, N joints have a total of dN degrees of freedom. The number of trivial rigid body motions in dimension d or isometries is \(d(d+1)/2\). Therefore, in a generic rigid bar and joint framework, the number of bars \(\ge \) \(dN - d(d+1)/2\). This is known as Maxwell’s counting condition. In the plane (\(d = 2\)), Laman’s theorem [34] extends this result by proving that the \(2N - 3\) count is both necessary and sufficient for generic rigidity of two-dimensional bar and joint frameworks. More formally, a two-dimensional bar and joint framework is generically minimally rigidity if and only if |E| = \(2|N| - 3\) and, for all subsets of edges, \(|E'|\) \(\le \) \(2|N'| - 3\). In other words, this remarkable theorem says one can count the vertices and edges in a graph and their distributions over subgraphs to predict generic rigidity of two-dimensional bar and joint frameworks. A framework is minimally rigid if removal of any edge (bar) results in a flexible framework (see Fig. 14.4).

Maxwell’s counts in 3D do not guarantee rigidity. A bar and joint framework in 3D (known as the double banana graph) satisfies the \(3|N| - 6\) count condition but is flexible (two yellow rigid subgraphs can rotate about an imaginary hinge shown as a red dashed line)
Unfortunately, Maxwell’s counting results are not sufficient for minimally rigid bar and joint graphs in dimension 3 and higher. For example, a well-known counterexample is a graph of a double banana, which satisfies Maxwell’s \(3|N| - 6\) count but is flexible (see Fig. 14.5). Not only is there a lack of a Laman type of a theorem for generic bar and joint frameworks in dimension 3 and higher, there are no known polynomial time algorithms for testing rigidity for general three-dimensional graphs [34]. Extensive research has been conducted on this problem and, to date, only some partial results and approximation algorithms can be found [34, 35]. Fortunately, for different classes of frameworks, called body-bar and body-hinge frameworks, which includes molecular frameworks, there is a complete and rich combinatorial characterization of rigidity, which is discussed next.
3.1.2 Rigidity Model of Molecules and the Molecular Theorem
To build a computational method based on rigidity theory that can provide fast and accurate prediction of protein rigidity and flexibility, three requirements must be met: (i) a realistic physical model of a basic molecular framework; (ii) an accurate model of molecular interactions; and (iii) a fast algorithm for predicting rigidity/flexibility properties of the protein framework model.
Protein structures consist of atoms and various chemical interactions (forces) of different strengths. In rigidity theory, strong interactions between atoms are usually assumed to be fixed rigid constraints in terms of distances and angles. In such a rigidity model of a molecule, bonding interactions are assumed to fix distances between a pair of bonded atoms, and the angles between the bonds of an atom are fixed, allowing only dihedral angle rotations. High frequency motions such as bond vibrations are neglected. This is a sensible modelling assumption as single covalent bond lengths are essentially invariant. For example, the length of a covalent bond between two carbon atoms will vary less than a single percent from its equilibrium value of 1.53 angstroms [14]. Double bonds and peptide bonds lock dihedral angles, and non-covalent interactions such as hydrogen bonds and hydrophobic contacts also impose additional constraints.
A molecular framework in rigidity theory is a collection of atoms, which can be modelled as fully rigid bodies with six conformational degrees of freedom of a rigid body and bonds as rotatable hinges, which allow for rotational degrees of freedom between single-bonded atoms. Such frameworks in rigidity theory are a special case of body-hinge framework. Hinges (i.e., bonds) remove five degrees of freedom, and for algorithmic and theoretical reasons, it is useful to model hinges as a set of five rigid bars, where each bar (i.e., edge) generically removes a single degree of freedom between bonded atoms. This finally leads to a body-bar framework representation of a molecular body-hinge framework—that is, a collection of rigid bodies connected by linear bars. Special geometric criteria should be considered as bonds are not generic hinges (since bonds intersect at centre of atoms) and the five bars have to pass through the hinge axis to geometrically give the same model as a hinge, but such discussion is beyond the scope of this chapter (details can be found elsewhere; see [13]). Double bonds are modelled as a set of six bars between two atoms. Moreover, non-covalent interactions such as hydrogen bonds and hydrophobic interactions, which are important for overall protein structure folding and rigidity, can also be modelled as a set of one to five bars (where one bar indicates the bond is least restricting and five bars indicate it is most restricting) [25]. This overall model, consisting of rigid bodies for atoms and both covalent bonds and non-covalent interactions, defines the body-bar framework model of a protein structure (see Fig. 14.6).

a 3D body-hinge framework composed of seven rigid bodies connected by hinges (lines) can be modelled as a body-bar framework (with a corresponding body-bar multigraph shown). b A molecule consisting of two carbon atoms and a single bond can be viewed as a body-hinge structure where atoms are rigid bodies (one-valent hydrogen atoms are a part of a carbon atom rigid body, as their angles are fixed and can only spin around their axes) and a hinge is a rotatable bond, with corresponding body-bar multigraph. c A ring of seven carbon atoms (ignoring one-valent hydrogens) with a corresponding multigraph. (According to the molecular theorem a ring of seven atoms will have one internal degree of freedom. The total number of edges is \(7(5) = 35\), while we need \(6|7| - 6 = 36\)). d Protein structure can be modelled as a molecular body-bar multigraph with black, red, and green lines corresponding to covalent bonds, hydrogen bonds, and hydrophobic contacts, respectively
The topological structure of a body-bar (and body-hinge and molecular body-hinge) framework is a multigraph \(G = (V, E)\). Vertex set V corresponds to a set of bodies (i.e., atoms) and edge set E to a set of bars (i.e., bond constraints). In accordance with Laman’s theorem, an equivalent statement for body-bar frameworks was formulated by Tay [37]. Tay’s theorem confirms that the rigidity of generic body-bar frameworks in 3D (which works for all dimensions) can be checked using the \(6|V| - 6\) count in a body-bar multigraph. Tay’s theorem also extends to generic body-hinge structures [20]. It was proven by Katoh and Tanigawa [38] that the same counting condition stated in Tay’s theorem also characterizes the rigidity of generic molecular body-hinge frameworks. This result is known as the molecular theorem, which is here combined with Tay’s theorem into one statement.
Theorem 1
(Tay’s Theorem/Molecular Theorem) A generic three-dimensional body-bar framework (body-hinge/molecular framework where bonds (hinges) are replaced by five bars) on a multigraph \(G=(V,E)\) is minimally rigid if and only if |E| = \(6|V| - 6\), and for all subsets of edges, \(|E'|\) \(\le \) \(6|V'| - 6\).
In the stated original form, Tay’s theorem leads to an exponential algorithm, as it requires counting the number of edges in every subgraph. However, because these counts of G (same as Laman’s counts) define an independent set in a matroid [13, 35], this gives rise to greedy algorithms that can be used to efficiently track these counts. It is well known that all matroidal structures have greedy algorithms. A number of fast polynomial algorithms based on matroid unions, tree decompositions, and extension of bipartite matching algorithms, such as the pebble game algorithm, were subsequently developed for tracking these rigidity certifying counts (independence) in graph and subgraphs [16, 39].
3.1.3 Pebble Game Algorithm
The pebble game algorithm can very rapidly decompose a body-bar/molecular graph (i.e., protein structure) into rigid and flexible regions and quantify the overall number of degrees of freedom. The main step of the pebble game algorithm is to determine if a constraint (edge) is ’independent’ (i.e., removes degrees of freedom) or is ’redundant’ as its insertion has no effect on rigidity. The algorithm iteratively builds a maximal independent set of edges. We give a basic procedure of how the main steps of the pebble game algorithm are carried out for Tay’s theorem without full details or speedups, which can be found in previous publications [16, 39]. A similar procedure can be derived for Laman’s counts or other matroidal independence counting conditions. The implementation of the pebble game algorithm routine given here, which tracks counts in the molecular theorem, is important for the protein flexibility analysis that has been implemented in several software packages. such as FIRST (see below).

The pebble game algorithm described here tracks the independence of edges in graphs prescribed by the molecular theorem. The initialization of placing six free pebbles on each vertex (corresponds to six trivial rigid body motions) tracks the 6|V| part of the count. Pebbles are synonymous with degrees of freedom and removal of a pebble indicates the inserted constraint (edge) is independent. Redundant constraints do not remove degrees of freedom (pebbles) as their insertion (or deletion) from an already rigid region causes no change in rigidity. Every time an edge is pebbled, it grows the set of independent edges. Pebble game algorithms are building a maximal subsets that are independent; at every stage, the edges covered by pebbles will satisfy \(|E'|\) \(\le \) \(6|V'| - 6\) on all subsets. The requirement of at least seven free pebbles on the vertices before an edge is pebbled (i.e., declared independent) ensures the critical subtraction in \(6|V| - 6\) is respected on all subsets of edges. The algorithm is greedy. In other words, regardless of the order the edges are pebbled (i.e., are tested for independence), the algorithm will always give unique answers for total remaining free pebbles, the size of maximal independent I(G) and redundant \(\mathfrak {R}\)(G) set of edges. The pebble game algorithm is a very intuitive algorithm, which in the worst case runs in O(\(V^{2}\)) [39], and in practice, it runs in linear time [15] (Fig. 14.8).

A demonstration of a \(6|V| - 6\) pebble game algorithm on a 3D cyclohexane graph. Edges are pebbled one by one (when there is at least seven free pebbles on its end vertices (a, b). If we cannot locate seven free pebbles we can search for free pebbles along with the partially created directed graph, swapping pebbles back. The graph has six remaining free pebbles and all edges are pebbled, indicating it is minimally rigid

\(6|V| - 6\) Pebble game algorithm. a When we cannot pebble an edge, it indicates that edge is redundant and the corresponding failed search locates a redundantly rigid subgraph (b). Overall, the graph is flexible with one internal degree of freedom, as indicated by the remaining seven free pebbles. Rigid clusters are circled. Each one of the bonds can be moved with one internal rotational degree of freedom
There are many extensions one can extract from the pebble game [16]. For example, when we cannot locate the seventh free pebble, the failed search over the directed graph indicates a rigid cluster. By using this procedure, it is possible to find all the maximal rigid clusters and redundantly rigid clusters (Fig. 14.7). Prediction of a highly redundant rigid clusters provides useful importance to a biochemist as these regions will have additional robustness, and will not become unstable (flexible) due to one or few edges breaking. For example, when a hydrogen bond breaks in a significantly redundantly rigid region, it will not alter its rigidity. We can also extract the relative degree of freedom count for any subgraph in G. This is very useful in the prediction of flexibility of particular regions of interest in protein graphs, for example, in antibody protein flexibility studies and in allostery predictions, which is discussed in next section.
4 Protein Flexibility, Dynamics, and Function Analysis with Rigidity Theory
4.1 FIRST and Rigid Cluster Decomposition
The pebble game algorithm is the main component of the programme FIRST [15] and other related software for analysing protein rigidity and flexibility. Starting with a protein structure (experimentally or computationally determined structure) in Protein Data Bank File format, the programme FIRST begins by creating a molecular body-bar multigraph. The multigraph consists of all atoms (including hydrogen atoms) represented by vertices, with covalent bonds, hydrogen bonds, hydrophobic contacts, and electrostatic interactions represented by edges. Covalent bonds are modelled as five edges, with six edges for double bonds and peptide bonds (as they do not have bond rotation), while hydrogen bonds and hydrophobic interactions are modelled with between one and five edges [25]. Hydrophobic contacts are defined as a pair of carbon–carbon, carbon–sulfer, or sulfer–sulfer atoms in close contact. Each hydrogen bond is assigned an energy strength in kcal/mol using an energy potential based on hydrogen donor and acceptor geometries. Hydrogen bonds are very important to the overall protein shape and stability. A hydrogen bond cutoff energy value (which mimics temperature) is selected such that all bonds weaker than this cutoff are ignored in the graph. Once the final constraint multigraph is obtained (Fig. 14.6d), FIRST then uses the pebble game algorithm and molecular theorem to decompose the protein into rigid and flexible regions.

Rigidity and flexibility analysis using FIRST and the pebble game algorithm on protein data from the Protein Data Bank (Protein Data Bank ID, 2jz3). The hydrogen bond dilution plot indicates how the protein breaks down as the hydrogen bond cutoff is increased (i.e., energy is increased), breaking hydrogen bonds one by one. Flexible regions are indicated by thin black lines and rigid regions are indicated by blocks, with separate colours indicating distinct rigid clusters. Flexible regions are coloured black on the protein structure. Initially, with inclusion of all potential hydrogen bonds, the protein is dominated by a few large rigid clusters (indicated by separate colours), and as hydrogen bonds are gradually broken with increasing energy, most of the protein becomes flexible (black) with a few remaining rigid clusters

Rigid cluster decomposition obtained with FIRST on a very large SARS-CoV-2 (in COVID-19) spike protein complex (Protein Data Bank ID 6vyb). At -1 kcal/mol energy cutoff, spike protein consists of more than 70 rigid clusters, each containing at least 20 atoms
Figures 14.9 and 14.10 show some examples of rigid cluster decompositions obtained with FIRST and the pebble game algorithm for two proteins. The rigid cluster decomposition on a very large Spike protein complex consisting of nearly 4000 residues was obtained in less than one second of running time (Fig. 14.10) We can monitor gradual changes in the rigid cluster decomposition as hydrogen bonds are removed one by one (i.e., by lowering the hydrogen bond energy threshold) in the order of increasing bond strength. The change in rigidity can be visualized using a hydrogen bond ’dilution plot’ (Fig. 14.9). Because the pebble game is a combinatorial integer algorithm (tracking molecular theorem counts) as opposed to a numeric algorithm, FIRST always gives a unique exact answer.
While tremendous computational power and resources are needed to simulate protein flexibility with MD simulations, FIRST can predict rigid clusters and flexible connections in less than one second on a typical PC/laptop. Because of its speed and efficiency, rigidity theory analysis using FIRST and other related programmes have been widely applied to analysing various aspects of protein function and flexibility analysis, such as viral capsids [40] (with enormous structures containing hundreds of copies of protein structures), protein engineering, and prediction and replication of experimental measures of dynamics such as hydrogen–deuterium exchange, allostery, and enzyme catalysis [11, 12, 15, 17,18,19, 23, 24, 26].
4.2 Large-Scale Rigidity and Flexibility Analysis
As an illustration of the efficiency and wider applicability of rigidity theory for large big-data high-throughput analyses of protein structures, we review a study where the author and colleagues carried out the largest study to date of flexibility predictions of antibody protein structures [41].

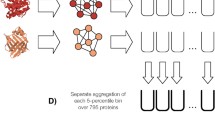
(figure on right adapted from [41])
Antibody is a large Y-shaped molecule. CDR H3 loop (shown in red) is located on the surface of each antibody arm, acting as a key region for antigen binding and recognition. In the study, authors applied extensions of the pebble game algorithm to analyse flexibility of the H3 loop using thousands of naïve and mature structures. There was no significant difference in flexibility between the naïve and mature H3 loops
Antibodies are proteins produced by B cells that play a main role in the adaptive immune system. They recognize a variety of pathogens and induce further immune response to protect the organism from external disturbance. Molecules that are bound by antibodies are called antigens. The focus of this study was to characterize flexibility of the key hyper-variable binding region on antibody called CDR H3 loop, which is the most important region in binding and recognition of various antigens. More specifically, we analysed whether the conformational flexibility of CDR H3 loop is changed as antibodies undergo affinity maturation. Antibodies can rapidly evolve to specific antigens, where affinity maturation drives this evolution through multiple cycles of mutation leading to enhanced antibody specificity and affinity. In this study, we utilized various extensions of the pebble game algorithm, initially developed in [16], which enables quantification of local flexibility of any subgraph, with focus on CDR H3 regions. By analysing thousands of mature and naïve antibody crystal structure and homology models, we found no clear statistically significant difference in the flexibility of CDR H3 loops (Fig. 14.11), which was also correlated with experimental measures of flexibility. Such large-scale analysis of the flexibility of protein structures could be carried out because of the speed of the underlying FIRST method and our various pebble game extensions.
4.3 Protein Allostery Analysis with Rigidity Theory
We now briefly discuss and review an important application of rigidity theory for analysis of allosteric signalling in protein structures. Allostery is one of the most powerful and fundamental mechanisms regulating protein function [8,9,10,11,12, 42,43,44]. Allostery refers to the regulation of protein function at a distance, where a perturbation of a protein structure at one part of protein structure (for example, due to a binding or mutational event) can affect conformations and dynamics at another distant site, resulting in regulation of protein function. Allostery is a common event in the cell, and most dynamic protein exhibit some form of allosteric control mechanism. Allostery has been referred to as ‘the second secret of life’, second only to the genetic code [8]. Monod and Jacob in 1960s [43] first introduced the allostery concept; however, most questions pertaining to allostery are still largely unresolved. Decoding the allosteric mechanism remains one of the key long-standing unsolved problems in the biological sciences.
One of the important areas in allostery research is describing the physical mechanism of distant coupled conformational changes. The utilization and extension of our earlier fundamental work in modelling allostery in frameworks and graphs [16] and a first rigidity-based mechanistic model of allosteric signalling has led to several important breakthroughs in understanding how allostery controls enzyme and receptor function [11, 12, 24, 44]. Our rigidity theory methods predict that if mechanical perturbation of rigidity at one site of the protein can transmit and propagate across a protein structure and, in turn, cause a change in the available conformational degrees of freedom and a change in the conformation and dynamics at a second distant site, resulting in allosteric transmission (Fig. 14.12a). Using various extensions of the pebble game algorithm, we can analyse how long-range conformational coupling occurs in protein structures, map out allosteric pathways (regions in protein that are important for allosteric signalling) and extract various other properties and features of long-range coupling.
A popular hypothesis is that dynamical effects play a central role in enzyme catalysis. Dynamical changes are often manifested in proteins through allosteric effects, where a substrate binding can cause changes in dynamics at remote parts of a protein. In a study published in Science [11] concerning bacterial homodimeric fluoroacetate dehalogenase enzyme, experimental NMR chemical shift data suggested that when a substrate binds to one monomer, the second empty monomer undergoes asymmetrically pronounced conformational changes through an increase in flexibility in dynamics, thereby entropically favouring the forward reaction. Our rigidity-based allostery theory was able to verify this and elucidate in great detail the key residues involved in the allosteric pathways responsible for changes in dynamics and how substrate binding enhances allosteric communication between two subunits (Fig. 14.12b). These findings also provided deep insights into the energetic nature of allosteric processes that drive catalysis.

Rigidity theoretical model for allosteric communication. a Conformational changes in one region of the framework (or protein structures) can propagate and change conformations and rigidity at distant regions. b Rigidity theory allostery analysis showed that homodimeric fluoroacetate dehalogenase enzyme with substrate fluoroacetate molecule (shown as orange spheres) exhibits allosteric communication between the two subunits (shown in distinct colours), which is critical for enzyme catalysis [11, 24]. c In a study of human adenosine A2A receptor [12, 18]., a member of superfamily of receptors called G-protein-coupled receptors (GPCRs) a similar approach was used to discover that allosteric communication between receptors and different domains of G-protein is critical for full receptor activation
In a follow-up study [24], we showed that when there is a high concentration of substrate, the enzyme undergoes catalysis inhibition through the reduction in dynamics and dampening of interprotomer allosteric effects. Our computational rigidity predictions of allosteric networks and resulting changes in dynamics when additional substrates were bound to the enzyme were validated with NMR and functional experimental studies. These studies represented a major breakthrough in illustrating the role of dynamics and allostery in enzyme function.
Our rigidity-theoretical approaches have been extremely useful for studying allostery in other enzymes and proteins. Indeed, we were able to provide a major advancement and new level of insight regarding key allosteric processes in GPCR activation. GPCRs are situated in the plasma membrane, engage the G-protein and initiate cell signalling [45]. In several studies [12], we have shown how interactions between GPCR and its natural G-protein binding partner affect activation networks, as is critical for optimal GPCR activation (Fig. 14.12c), or how sodium, calcium, and magnesium can affect this activation process [18]. Our rigidity theory-based approaches offer a new perspective and opportunity to study the various facets of allosteric regulation of protein function, which will allow us to examine complicated signalling events in the cell.
4.4 Using Rigidity Theory to Simulate Protein Dynamics
So far, the discussion has focused on infinitesimal flexibility (which is equivalent to finite flexibility, assuming atom positions are in a generic configuration) and not on continuous motions. In other words, FIRST and the pebble game outputs do not simulate protein dynamics and indicate the amplitude of motions. One useful extension is to combine the rigid cluster decomposition with Monte Carlo-based geometric dynamics simulations [20, 21]. Rigid cluster decomposition can remove hundreds of degrees of freedom from the overall protein framework and serve as a natural coarse graining step to speed up protein dynamics simulations [19, 46]. For example, the all-atom geometric simulation method FRODA (Framework Rigidity Optimized Dynamic Algorithm) (which runs about 100,000 times faster than MD simulations) [20] uses rigid clusters as a preprocessing step to explore the conformational space of the protein motions. The rigid clusters, whose size and number depend on the selected energy threshold and the type of protein structure being analysed, can be kept fixed as rigid body geometrical components in the simulation motion (see Fig. 14.13). The atoms belonging to a rigid cluster can only move by utilizing trivial rigid body degrees of freedom. With this in mind, simulations can be focused on simulating the relevant degrees of freedom belonging to intermediate flexible regions. FRODA rapidly generates geometrically valid conformations that are consistent with bond lengths and angular constraints while maintaining all rigid clusters. In these protein motion simulations, we need to add the van der Waals collisions of atoms as constraints, where only allowed geometries (valid stereochemistry, bonding angles, Ramachandran plots etc.) accessible to protein motions are simulated. Figure 14.13b shows the output of FRODA for an antibody protein, which exemplifies large amplitude motions.

Geometric simulation as used in FRODA/FRODAN (a). A part of a 2D slice through the 3N-dimensional conformational space, where red indicates disallowed states and blue indicates allowed states [21]. A random move (green arrows) is accepted if it falls within a blue region (green dots) and rejected if it falls within a red region (yellow dots), followed by enforcement of the constraints (yellow arrows). The black path produces a valid geometric path within the allowed conformational space. Any rigid region (which can be potentially very large) identified with FIRST moves as a single rigid body within FRODA or very small rigid clusters or individual atoms within FRODAN. b FRODA was applied to a large antibody protein to explore the large-scale motions of arms (green and orange) of the Y-shaped antibody structure, where three distinct colours represent three separate large rigid bodies. c FRODAN dynamics simulation illustrating internal dynamics of a Spike protein [47]
We have applied and extended FRODA, using the related constrained geometric simulation programme FRODAN [21], which, like FRODA, provides very fast motion simulations but is better suited for proteins that are not dominated by large rigid clusters. In a FRODAN simulation, the rigid clusters are typically small, from single atoms up to small rigid cycles (e.g., proline rings and rigid loops). This makes FRODAN useful for simulations of protein motions that include substantial unfolding and refolding and analysing motions of intrinsically disordered proteins. Indeed, we have utilized a similar approach in combination with an experimental measure of dynamics, hydrogen–deuterium exchange, to characterize the highly complex motions and conformational ensemble of a large intrinsically disordered Tau protein [22]. Tau protein is a key protein in a number of pathologies and dementias such as Alzheimer’s disease, and its primary physiological role is to stabilize microtubules in neuronal axons at all stages of development. One of the main challenges in understanding the Tau structure–function relationship and finding successful therapeutics for Alzheimer’s disease is the poor understanding of the atomic structural ensemble and dynamics of the Tau protein. Moreover, Tau protein undergoes modifications to its shape and internal dynamics as mediated by a hyperphosphorylation defect. By performing FRODAN simulations and our various extensions, we were able to show an unprecedented first detailed view of the structural and dynamic characteristics of both the normal and the defective hyperphosphorylated forms of Tau [22]. This study provided a rich understanding of the structural basis of Tau pathology (see Fig. 14.14).

(The figure in b is adapted from [22])
a Tau protein is a large intrinsically disordered protein. Because of its high flexibility and disordered structure, it is able to take a wide variety of shapes, which makes it difficult to study with conventional MD simulations. b By performing large rigidity theory geometric simulations using FRODAN and its extensions, we were able to characterize the representative structures for the native and defective (i.e., hyperphosphorylated) forms of Tau, which was shown to be in agreement with HDX experimental data
FRODA, FRODAN and our various extensions can be applied to probe the dynamics of very large structures such as Spike proteins [47] or disordered proteins, which provides a significant advantage over traditional MD simulations. Probing motions of intrinsically disordered proteins with MD simulation is extremely challenging, if not essentially impossible, owing to their highly dynamic character. The rigidity theory-inspired methodologies FRODA/N discussed here can be run in either targeted and non-targeted modes, and we have recently combined these techniques with search algorithms in reinforcement learning (under review). The targeted mode employs biasing force during transitioning, while the non-targeted mode explores unbiased random fluctuations, which enables the exploration of a broad conformation space. Additionally, the targeted mode is useful for determining the conformational transition pathways between distinct conformations (i.e., opening and closing motions such as hinge-bending motions, GPCR activation, etc.).
5 Protein Structure Validation with Rigidity Theory
We now discuss another application of rigidity theory to structural biology. In a very recent study, we made an important breakthrough in the area of protein structure validation [48, 49].
Experimentally solved protein structures are only useful if they are known to be accurate and realistically represent the protein structures in their native environment. The vast majority of protein structures in the Protein Data Bank [50] have been solved by X-ray crystallography or NMR experiments. Both X-ray crystal structures and NMR structures are only model representations of experimental data, which are prone to uncertainties and errors. It is widely accepted that experimentally solved protein structures must be validated with (i) geometric tests and (ii) how well structures match input experimental data (restraints) [51]. Geometric criteria are easy to check for both X-ray and NMR structures, and measurements like R factor and Rfree values can be used to check how well X-ray structures match input X-ray diffraction data [48]. Unfortunately, no such validation criteria exist for NMR structures [51], and unlike crystal structures, validating the quality of NMR structures has been extremely difficult. In fact, since the first protein was determined by NMR in 1985 until now, there has been no effective method for NMR protein structural validation, which has largely limited the applications and use of NMR structures among protein researchers [51,52,53,54,55]. This has created a problem not only for users of structural information, but also for scientists who use NMR to computationally solve structures and want to know how accurate their solved structure is.
While structures solved by NMR represent less than 10% of all structures in PDB, they are extremely important, as not all proteins can be crystalized and NMR structures also include a high proportion of proteins with under-represented folds (shapes). NMR structures are determined in solution (a protein’s natural environment), whereas X-ray structures are determined in a crystalline environment, which arguably makes NMR structures more representative of in vivo structures. Hence, there has been a pressing need to find an acceptable validation measure for NMR structures.

(Figure adapted from [48]). c ANSURR output for an example NMR structure (Protein Data Bank ID, 2kpp) that has high accuracy for most models in the ensemble
a The ANSURR method evaluates the accuracy of nuclear magnetic resonance (NMR) protein structure (which are given as an ensemble of models) by comparing two measures of protein flexibility (orange predicted from structure, using mathematical rigidity theory using extensions of the method FIRST, and blue derived from the random coil index [RCI] using experimental NMR chemical shift data). b Analysis of ANSURR using four models from NMR (Protein Data Bank ID, 1e17). ANSURR provides two metrics for accuracy: a correlation score between FIRST (rigidity) and RCI and a root mean square difference (RMSD) score. The structures in the top right portion of the plot (high correlation and high RMSD scores) are high-quality NMR structures, and structures in the bottom left of the plot are considered poor structures
We have developed the method ANSURR (Accuracy of NMR Structures Using Random Coil Index and Rigidity) [48], which addresses this critical long-standing gap for NMR protein structure validation. ANSURR assesses the quality of NMR structures by comparing two measures of local protein rigidity, one derived from the original NMR input data and the other derived from rigidity theory prediction of protein flexibility using structural data. The measure of rigidity using input data is based on the Random Coil Index (RCI), which uses experimental NMR chemical shifts (a readily available data type for each NMR structure) to quantify the extent of disordered structure for each amino acid in solution. The second measure is based on FIRST and our rigidity theory extensions, which involves calculating the dilution plot (see Fig. 14.9) and extracting a flexibility score for each residue. ANSURR then compares these two measures of local rigidity and provides a residue-by-residue test of how well the rigidity of the structure (obtained from rigidity theory) compares to the experimentally determined (true, RCI chemical shift) rigidity. ANSURR provides two metrics for accuracy measurement. One is a correlation score between FIRST (rigidity) and RCI, which assesses the accuracy of protein folding (secondary structures), and the second is an RMSD score, which measures how well the overall rigidity and flexibility between FIRST and RCI match (Fig. 14.15).
Unlike crystal structures, NMR structures are always represented as an ensemble of (typically around 20) possible structural models. Because it is unclear which models are useful or accurate, this has created substantial and unnecessary confusion for users of NMR structures. A nice feature of ANSURR is its ability to estimate the accuracy of each model.
The performance of ANSURR was tested using several approaches [48]; first, ANSURR was applied to structures refined in an explicit solvent (which was found to be much better than unrefined structures), and then ANSURR was applied to a large set of good and bad structures (using decoy generations). ANSURR was also compared against previously proposed measures of accuracy (mostly restraint-based tests and geometric checks). Several of these indicators, such as restraint violations and restraints per residue, were shown to be poor measures of accuracy. On the other hand, a Ramachandran analysis (a standard check to determine if a protein backbone has a correct geometry) was found to be a useful geometric check of accuracy. A typical comparison of how well a structure compares to another structure is the backbone root mean square deviation, which can show if protein structures resemble each other when superimposed. However, this measure may miss many of the important structural differences found in amino acid side-chain orientations, which are responsible for forming critical hydrogen bonding interactions that have a direct impact on protein stability and functional aspects such as protein dynamics and enzyme catalysis. As rigidity measures are sensitive to side chains, ANSURR can also be used to assess the quality of side-chain atomic positions, which makes it a powerful tool for the assessment and refinement of protein structures.
Recent work [49] applied ANSURR to more than 7000 NMR structures in the PDB, showing that NMR structures span a wide range of accuracy. Most NMR structures have accurate secondary structures, but are too floppy, particularly in their loops. Our studies also indicate that both crystal structures and NMR structures have equally accurate secondary structural elements (helices, sheets), but crystal structures are typically too rigid in disordered regions, whereas NMR structures are too flexible overall.
Development of ANSURR is a major advancement in the long-standing problem of protein structure validation, as it provides the first workable measure of the accuracy of NMR structures and is expected to give researchers more confidence in the use and application of structural NMR. Ultimately, this should lead to a better understanding of how proteins perform their functions, with general implications for structural biology research. This work opens up enormous new research avenues in protein structure determination and the improvement of standards for protein structure refinement.
6 Conclusion
Studying the rigidity and flexibility of geometric frameworks has advanced considerably since Maxwell’s combinatorial characterization of the rigidity of mechanical frameworks in the 1800s. Mathematical advancements in rigidity theory over the last two decades have been tremendous, opening up many exciting opportunities in applied sciences and engineering. In this chapter, we have reviewed some of the latest advances in rigidity theory and its applications for the analysis of protein function at an atomistic scale. Moreover, we have shown how rigidity theory-based methods and our various algorithms and extensions can rapidly and accurately predict protein flexibility and dynamics, which can be used to decipher various aspects of protein function, including elusive issues of allostery, enzyme catalysis, GPCR signalling, or motions of intrinsically disordered proteins. Our recent development using rigidity theory in protein structure validation has led to a development of a first workable method in validation of NMR protein structures. This advance will provide confidence to users of protein structures and is expected to accelerate and improve the process of protein structure determination and aid computational drug discovery. Rigidity theory is heavily rooted in deep mathematical formulations in the area of discrete applied geometry and combinatorics, which has unfortunately remained largely inaccessible to most researchers in applied science and engineering fields. While there has been some cross-fertilization between the various scientific fields studying different aspects of rigidity and flexibility, stronger interactions and interdisciplinary training are needed between applied and theoretical scientific communities to realize the enormous potential of rigidity theory applications. We advocate that rigidity theory, through both algorithmic and mathematical progress, has significantly advanced such that it could be widely applied in the analysis of structural biological data, which can complement experimental approaches to reveal novel insights on intractable and fundamental biological enigmas of living organism. Rigidity theory exemplifies how mathematics and algorithms can make significant contributions to structural biology, biological big-data analyses, and progress in biological applications.
References
K. Roberts, B. Alberts, A. Johnson, P. Walter, T. Hunt, Molecular Biology of the Cell (Garland Science, New York, 2002)
Yip, K. M., Fischer, N., Paknia, E., Chari, A., and Stark, H. (2020). Atomic-resolution protein structure determination by cryo-EM. Nature, 587(7832), 157–161
W. Gao, S.P. Mahajan, J. Sulam, J.J. Gray, Deep learning in protein structural modeling and design. Patterns, 100142
M.Y. Galperin, X.M. Fernández-Suárez, D.J. Rigden, The 24th annual Nucleic Acids Research database issue: a look back and upcoming changes. Nucleic acids research 45(D1), D1–D11 (2017)
Henzler-Wildman, K., and Kern, D. (2007). Dynamic personalities of proteins. Nature, 450(7172), 964–972
Hartl FU, Hayer-Hartl M (2009) Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16:574–81
J.R. Lewandowski, M.E. Halse, M. Blackledge, L. Emsley, Direct observation of hierarchical protein dynamics. Science 348(6234), 578–581 (2015)
A.W. Fenton, Allostery: an illustrated definition for the second secret of life. Trends Biochem. Science 33, 420–425 (2008)
Nussinov R, CJ Tsai (2013) Allostery in disease and drug discovery, Cell, 153(2), 293–305
J. Liu, R. Nussinov, Allostery: an overview of its history, concepts, methods, and applications. PLoS Comput. Biol. 12(6) (2016)
Kim TH, Mehrabi P, Ren A, Sljoka A, Ing C, Bezginov A, Ye LB, Pomes R, Prosser RS and Pai EF (2017) The role of dimer asymmetry and protomer dynamics in enzyme catalysis, Science, 355, 262–U287
Huang, S., Pandey, A., Tran, D., Villanueva, N., Kitao, A., Sunahara, R., Sljoka, A., and Prosser, R. (2021). Delineating the conformational landscape of the adenosine A2A receptor during G protein coupling. Cell, 184(7), 1884–1894
W. Whiteley, Counting out to the fexibility of molecules. Phys. Biol. 2, S116–S126 (2005)
C. F. Mourkazel, P. M. Duxbury in rigidity theory and applications, ed. by M.F. Thorpe, P.M. Duxbury (Kluwer Academic/Plenum Publishers, 1999), p. 69
Kuhn LA, Rader DJ, Thorpe MF (2001) Protein flexibility predictions using graph theory, Proteins, 44:150–65
A. Sljoka, Algorithms in rigidity theory with applications to protein flexibility and mechanical linkages. Ph.D thesis, York University, Toronto, 2012
A.J. Rader, B.M. Hespenheide, L.A. Kuhn, M.F. Thorpe, Protein unfolding: rigidity lost. Proceedings of the National Academy of Sciences 99(6), 3540–3545 (2002)
L. Ye, C. Neale, A. Sljoka, D. Pichugin, N. Tsuchimura, R. Sunahara, S. Prosser, et al, Bidirectional regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 1(9), 1372 (2018)
S.M. Hermans, C. Pfleger, C. Nutschel, C.A. Hanke, H. Gohlke, Rigidity theory for biomolecules: concepts, software, and applications. Wiley Interdisciplinary Reviews: Computational Molecular Science 7(4), (2017)
S.A. Wells, S. Menor, B.M. Hespenheide, M.F. Thorpe, Constrained geometric simulation of diffusive motion in proteins. Phys. Biol. 2, S12736 (2005)
D.W. Farrell, K. Speranskiy, M.F. Thorpe, Generating stereochemically acceptable protein pathways. Proteins: Struct. Funct. Bioinf. 78(14), 2908–2921 (2010)
S. Zhu, A. Shala, A. Bezginov, A. Sljoka, G. Audette, D. Wilson, Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PLoS ONE 10(3) (2015)
S.L. Seyler, A. Kumar, M.F. Thorpe, O. Beckstein, Path similarity analysis: a method for quantifying macromolecular pathways. PLOS Comput. Biol. 11(10) (2015)
P. Mehrabi, C. Di Pietrantonio, T. Kim, A. Sljoka, K. Taverner, C. Ing, N. Kruglyak, R. Pomès, E. Pai, R. Prosser, Substrate-based allosteric regulation of a homodimeric enzyme. Journal of the American Chemical Society 141(29), 11540–11556 (2019)
A. Sljoka, D. Wilson, Probing Protein Ensemble Rigidity and predictions of Hydrogen-Deuterium exchange. Physical Biology 10, (2013)
B. Deng, S. Zhu, A.M. Macklin, J. Xu, C. Lento, A. Sljoka, D. Wilson, Suppressing allostery in epitope mapping experiments using millisecond hydrogen/deuterium exchange mass spectrometry. MAbs 1, 10 (2017)
G. Wieczorek, P. Zielenkiewicz, DeltaF508 mutation increases conformational flexibility of CFTR protein. J Cyst Fibros 7, 295–300 (2008)
F.R. Salsbury Jr., Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Current opinion in pharmacology 10(6), 738–744 (2010)
I.R. Kleckner, M.P. Foster, An introduction to NMR-based approaches for measuring protein dynamics. Biochimica et Biophysica Acta (BBA). Proteins Proteomics 1814(8), 942–968
E.A. Jares-Erijman, T.M. Jovin, FRET imaging. Nature biotechnology 21(11), 1387–1395 (2003)
McCammon, J. A., Gelin, B. R., and Karplus, M. (1977). Dynamics of folded proteins. Nature, 267(5612), 585–590
D.E. Shaw, et al., Anton, a special-purpose machine for molecular dynamics. Commun. ACM (ACM) 51(7), 9197 (2008)
Q. Cui, I. Bahar, Normal Mode Analysis: Theory and Applications to Biological and Chemical Systems (CRC press)
M. Sitharam, A.S. John, J. Sidman, Handbook of Geometric Constraint Systems Principles (CRC Press, 2018)
W. Whiteley, Some matroids from discrete applied geometry, in Matroid Theory, ed. by J. Bonin, J. Oxley, B. Servatius (Amer. Math. Soc., Providence, 1996), vol. 197 pp. 171–311
B. Schulze, A. Sljoka, W. Whiteley, How does symmetry impact the flexibility of proteins? Philos. Trans. Roy. Soc. A 372, 20120041 (2014)
T.S. Tay, Rigidity of multigraphs i: linking rigid bodies in n-space. J. Comb. Theory Ser. B 26, 95–112 (1984)
N. Katoh, S. Tanigawa, A Proof of the Molecular Conjecture. Discrete Comput. Geom. 45, 647–700 (2011)
A. Lee, I. Streinu, Pebble game algorithms and sparse graphs. Discrete Math. 308(1425), 1437 (2008)
B.M. Hespenheide, D.J. Jacobs, M.F. Thorpe, Structural rigidity in the capsid assembly of cowpea chlorotic mottle virus. J. Phys.: Condens. Matter 16, S5055–S5064 (2004)
J.R. Jeliazkov, A. Sljoka, D. Kuroda, N. Tsuchimura, N. Katoh, K. Tsumoto, J.J. Gray, Repertoire analysis of antibody CDR-H3 loops suggests affinity maturation does not typically result in rigidification. Front. Immunol. 9, 413 (2018)
K. Gunasekaran, M. Ma, R. Nussinov, Is allostery an intrinsic property of all dynamic proteins? Proteins: Struct. Funct. Bioinf. 57, 433443 (2004)
J.P. Changeux, F. Jacob, J. Monod, Allosteric proteins and cellular control systems. J Mol Biol 6, 306–329 (1963)
S. Bera, M. Rashid, A. Medvinsky, G.Q. Sun, B.L. Li, C. Acquisti, A. Sljoka, A. Chakraborty, Allosteric regulation of Glutamate dehydrogenase deamination activity. Scientific reports 10(1), 1–15 (2020)
N.R. Latorraca, A.J. Venkatakrishnan, R.O. Dror, GPCR dynamics: structures in motion. Chemical reviews 117(1), 139–155 (2017)
H. Gohlke, M.F. Thorpe, A natural coarse graining for simulating large biomolecular motion. Biophysical Journal 91(6), 2115–2120 (2006)
N. Kumawat, A. Tucs, S. Bera, G. Chuev, M. Fedotova, K. Tsuda, S. Kruchinin, A. Sljoka, A. Chakraborty, Prefusion conformation of SARS-CoV-2 receptor-binding domain favors interactions with human receptor ACE2. bioRxiv (2021)
N. Fowler, A. Sljoka, M. Williamson, A method for validating the accuracy of NMR protein structures. Nat. Commun. 11(1), 6321 (2020)
N. Fowler, A. Sljoka, M. Williamson, The accuracy of NMR protein structures in the Protein Data Bank. bioRxiv (2021)
H.M. Berman et al., The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000)
Gore, S. et al. Validation of structures in the Protein Data Bank. Structure 25, 1916–1927 (2017)
A.T. Brunger, Free R-value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 (1992)
D.A. Snyder, A. Bhattacharya, Y.P.J. Huang, G.T. Montelione, Assessing precision and accuracy of protein structures derived from NMR data. Proteins: Struct. Funct. Bioinf. 59, 655-661 (2005)
G.W. Vuister, R.H. Fogh, P.M.S. Hendrickx, J.F. Doreleijers, A. Gutmanas, An overview of tools for the validation of protein NMR structures. Journal of Biomolecular NMR 58, 259–285 (2014)
Spronk, C.A.E.M., Nabuurs, S.B., Krieger, E., Vriend, G. and Vuister, G.W. Validation of protein structures derived by NMR spectroscopy. Progr. NMR Spectrosc. 45, 315–337 (2004)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Sljoka, A. (2022). Structural and Functional Analysis of Proteins Using Rigidity Theory. In: Katoh, N., et al. Sublinear Computation Paradigm. Springer, Singapore. https://doi.org/10.1007/978-981-16-4095-7_14
Download citation
DOI: https://doi.org/10.1007/978-981-16-4095-7_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4094-0
Online ISBN: 978-981-16-4095-7
eBook Packages: Computer ScienceComputer Science (R0)