
Abstract
The deployment of progenitor cells from the second heart field (SHF) to the poles of the cardiac tube drives heart tube elongation during looping morphogenesis. Defects in this process underlie a spectrum of common forms of congenital heart defects (CHDs) affecting the cardiac poles, including conotruncal and atrioventricular septal defects. In this chapter recent findings will be reviewed concerning sequential steps in SHF development: the early emergence of the SHF lineage, the epithelial properties of SHF cells during heart tube elongation, and the importance of the transition zone as progenitor cells differentiate at the cardiac poles. Together these studies define critical regulatory nodes at successive stages of SHF deployment, providing new insights into mechanisms of heart development and the origins of CHD.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
The second heart field (SHF) contributes cardiac progenitor cells to the poles of the vertebrate heart as it elongates during looping morphogenesis [1]. This process occurs between E8 and E10.5 of mouse gestation or between weeks 3–5 of human development. The embryonic heart thus grows by a process of progenitor cell recruitment, separating proliferation of multipotent cardiac progenitor cells in the SHF from differentiated cells of the first heart field (FHF) in the early heart tube. Correct SHF deployment is essential to maximally elongate the heart tube and for subsequent alignment and septation of the future cardiac chambers [2]. Failure of SHF addition at the arterial pole results in conotruncal CHD, including ventricular septal defects, overriding aorta and tetralogy of Fallot, while defects of SHF addition at the venous pole result in primum type atrial and atrioventricular septal defects. Dissecting the mechanisms regulating SHF deployment is therefore a critical step towards understanding the origins of common forms of CHD.
In this chapter we will briefly summarize recent advances in SHF biology, focusing on three successive steps of progenitor cell deployment: (1) emergence of SHF lineages in the early mouse embryo, (2) evidence that the epithelial properties of SHF cells play an important role in controlling the progenitor cell niche during heart tube elongation, and (3) new insights into events at the cardiac poles as SHF cells transit through a transition zone to acquire a differentiated phenotype.
2 Emergence of SHF Lineages in the Early Embryo
Clonal analyses have shown that FHF and SHF cells segregate extremely early in the developing embryo, at or before the time of gastrulation, and prior to the activation of Mesp1, one of the earliest transcription factors expressed in the cardiac lineage [3, 4]. Single cell RNA sequencing data of nascent precardiac mesoderm in the Mesp1 lineage has recently revealed early segregation of anterior and posterior SHF populations, ultimately contributing to the arterial and venous poles of the heart, respectively. This study demonstrated that different cardiac progenitor cells emerge from a population of multipotent heterogeneous precardiac mesoderm, through which pseudotime trajectories to distinct emergent fates can be mapped (Fig. 49.1a) [5]. Moreover these data provide evidence of transcriptional priming of anterior and posterior SHF sub-populations, consistent with clonal analysis and vital dye labeling experiments indicating that these cells emerge from common progenitor cells in the early embryo. Progressive segregation of arterial and venous pole progenitor cells has been shown to depend on the transcription factor Tbx1, encoded by the major gene for 22q11.2 deletion syndrome (22q11.2DS, also known as Takao or DiGeorge Syndrome) [6]. CHDs affecting both conotruncal development and atrioventricular septation are observed in Tbx1 null embryos and 22q11.2DS patients, consistent with a requirement for Tbx1 in both anterior and posterior SHF sub-populations. Moreover, the SHF itself lies within a larger population of evolutionarily conserved cardiopharyngeal mesoderm giving rise to skeletal muscles of the head and neck as well as myocardium; segregation of the heart and head muscle fate within cardiopharyngeal mesoderm occurs after the segregation of FHF and SHF lineages [7].

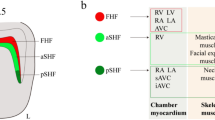
Advances in the second heart field. (a) Early segregation of SHF cells in nascent cardiac mesoderm visualized by single cell gene expression analysis (adapted from [5]). (b) Actin enriched basal filopodia (arrows) in epithelial SHF cells visualized using Utrophin-GFP (adapted from [10]). (c) Segmentation of the apical surface of cells in the dorsal pericardial wall showing an increased cell size (red) and elongation in the posterior region (adapted from [15]). (d) Cartoon showing the transition zones (arrows) between proliferating progenitor cells (green) and differentiated cardiomyocytes (red) at the arterial and venous poles. (e, f) Failure of dorsal mesenchymal protrusion (DMP) development results in atrioventricular septal defects (asterisk) in Tbx1 null hearts (adapted from [6]). aSHF anterior second heart field, pSHF posterior second heart field, FHF first heart field, End endocardium, AP arterial pole, VP venous pole, PAS primary atrial septum, RA right atrium, LA left atrium, RV right ventricle, LA left ventricle. Scale bars: b, 10 μm; e, f: 200 μm
3 Epithelial Properties of the SHF Cell Regulate the Progenitor Cell Niche
Inducible Cre recombinase alleles allowing genetic tracing of early differentiating cardiomyocytes (Smooth muscle actin) or progenitor cells (Six2) reveal the progressive contribution of SHF cells to the early heart tube [8, 9]. Whereas FHF cells differentiate in epithelial splanchnic mesoderm in the anterior lateral region of the embryo, SHF cells are situated in medial splanchnic mesoderm forming the dorsal pericardial wall (DPW). SHF cells also have an epithelial phenotype with a smooth apical surface with adherence and tight junctions facing the pericardial cavity [10]. In contrast to typical epithelia, the basolateral side of DPW cells is characterized by a mesenchymal-like convoluted cell membrane with actin-enriched filopodial structures (Fig. 49.1b). Confocal time-lapse imaging of living SHF cells in thick slice culture has revealed that these filopodia are highly dynamic and contact the underlying endodermal and mesenchymal cells [10]. Phospho-Tyrosine accumulates in the basal filopodia of SHF cells, consistent with activation of intercellular signaling receptors and a potential signaling role in maintaining the progenitor cell niche. Filopodia formation is impaired in mouse embryos lacking Tbx1, associated with elevated levels of the apical determinant atypical PKCz and abnormal SHF deployment. Exposure of embryos to a physiological activator of aPKCz phenocopies the filopodial phenotype of Tbx1 null embryos, reducing proliferation and enhancing differentiation in the SHF [10].
N-cadherin is also required in the SHF to maintain the progenitor cell niche. Conditional loss of N-cadherin results in elevated differentiation and reduced proliferation associated with decreased Wnt β-catenin signaling [11]. Non-canonical Wnt signaling also plays important roles in SHF development. In particular, loss of Wnt5a, expressed in splanchnic mesoderm in the posterior SHF, results in arterial pole defects similar to those observed in Tbx1 null embryos [12]. Furthermore, Wnt5a has been shown to be a direct target gene of Tbx1 in the SHF [13]. The regional expression of Wnt5a has been proposed to drive mesenchymal cell intercalation in the DPW, creating a pushing force promoting SHF deployment to the arterial pole [14]. Increased cell adhesion in the anterior SHF may reinforce this process by creating a pulling force, supported by missexpression experiments in transgenic mice [14] and evidence that E-cadherin accumulates in the apical membrane of cells in the anterior but not posterior SHF [10].
Ventral views of the dorsal pericardial wall epithelium have shown that cells in the posterior region are under greater epithelial tension than cells close to the arterial pole. This is observed as an increase in the cell size, cell elongation oriented towards the arterial pole and accumulation of active actomyosin complexes along the long membrane of elongated cells (Fig. 49.1c) [15]. Moreover this is associated with elevated proliferation in the posterior SHF, oriented distribution of daughter cells after cell division, and clonal growth patterns directed towards the arterial pole of the heart, likely promoting heart tube elongation. A biomechanical feedback loop between SHF deployment and tension in the DPW, driven by the process of heart tube elongation itself, may accelerate terminal stages of SHF deployment [15]. Cells in the DPW are initially attached to the heart tube along its entire length by the dorsal mesocardium. Rupture of this transient mesentery isolates the SHF from the looping heart tube with the exception of the cardiac poles. Elevated epithelial tension in the posterior SHF is observed only after addition of SHF cells is restricted to the cardiac poles and tension is dependent on heart tube elongation [15]. Interestingly, elevated nuclear accumulation of the transcriptional co-factor YAP in the posterior region of the SHF may provide a link between epithelial tension and proliferation [15]. Ongoing work aims to refine this working model by characterizing the sources of tension and the pathways by which this impacts on proliferation in the SHF.
4 SHF Cells at the Cardiac Poles Pass Through a Transition Zone
As SHF cells contribute to the cardiac poles they pass through a transition zone (TZ), or zone of commitment (Fig. 49.1d). The TZ is characterized by a cardiomyoblast phenotype, altered cell shape and actin cytoskeleton remodeling associated with sarcomere assembly and the onset of differentiation [16,17,18]. Cells in the TZ are distinguished by reduced FlnA and Hopx expression, the latter associated with modulating intercellular signals controlling the switch from proliferation to differentiation [17, 18]. Downregulation of the SHF transcriptional program is mediated by the cardiac transcription factor Nkx2-5 that competes with the SHF activator Isl1 to repress transcription at the Fgf10 locus [19]. Similarly, binding of Nkx2-5 to sites in the Popdc2 gene initially bound by Meis1 is associated with spatiotemporal progression of cells through the TZ [20]. Defects in TZ development result in failure to fully extend the outflow tract and subsequent ventriculoarterial alignment defects. For example, the planar cell polarity gene Vangl2 controls the epithelial properties of cells in the TZ, and outflow tract alignment defects including double outlet right ventricle are observed in conditional mutant embryos [16]. At the venous pole, SHF cells give rise to the dorsal mesenchymal protrusion (DMP) contributing to the muscular base of the primary atrial septum [21]. Multiple transcription factors, including Tbx5, Osr1, Gata, and Fox factors, as well as several intercellular signaling pathways, have been implicated in DMP development [21, 22]. Defects in the development of the DMP underlie atrial primum type CHD, a class of atrioventricular septal defect. Such defects, in addition to failure of sub-pulmonary myocardium development at the arterial pole, are observed in Tbx1 null hearts and constitute part of the CHD spectrum in TBX1 haploinsufficent 22q11.2DS patients (Fig. 49.1e, f) [6, 23].
5 Future Directions and Clinical Implications
The studies discussed above identify critical regulatory nodes at successive stages of SHF deployment, providing new insights into the mechanisms of heart morphogenesis. Further analysis using dynamic approaches, single cell transcriptomic and epigenetic studies and lineage analysis will refine our understanding of these steps and generate a more complete blueprint of SHF deployment with which to dissect the origins of CHD and develop future preventive and, potentially, therapeutic approaches.
References
Kelly RG. The second heart field. Curr Top Dev Biol. 2012;100:33–65. https://doi.org/10.1016/B978-0-12-387786-4.00002-6.
Epstein JA. Franklin H. Epstein Lecture. Cardiac development and implications for heart disease. N Engl J Med. 2010;363(17):1638–47. https://doi.org/10.1056/NEJMra1003941.
Devine WP, Wythe JD, George M, Koshiba-Takeuchi K, Bruneau BG. Early patterning and specification of cardiac progenitors in gastrulating mesoderm. Elife. 2014;3 https://doi.org/10.7554/eLife.03848.
Lescroart F, Chabab S, Lin X, Rulands S, Paulissen C, Rodolosse A, Auer H, Achouri Y, Dubois C, Bondue A, Simons BD, Blanpain C. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat Cell Biol. 2014;16(9):829–40. https://doi.org/10.1038/ncb3024.
Lescroart F, Wang X, Lin X, Swedlund B, Gargouri S, Sanchez-Danes A, Moignard V, Dubois C, Paulissen C, Kinston S, Gottgens B, Blanpain C. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science. 2018;359(6380):1177–81. https://doi.org/10.1126/science.aao4174.
Rana MS, Theveniau-Ruissy M, De Bono C, Mesbah K, Francou A, Rammah M, Dominguez JN, Roux M, Laforest B, Anderson RH, Mohun T, Zaffran S, Christoffels VM, Kelly RG. Tbx1 coordinates addition of posterior second heart field progenitor cells to the arterial and venous poles of the heart. Circ Res. 2014;115(9):790–9. https://doi.org/10.1161/CIRCRESAHA.115.305020.
Diogo R, Kelly RG, Christiaen L, Levine M, Ziermann JM, Molnar JL, Noden DM, Tzahor E. A new heart for a new head in vertebrate cardiopharyngeal evolution. Nature. 2015;520(7548):466–73. https://doi.org/10.1038/nature14435.
Choquet C, Marcadet L, Beyer S, Kelly RG, Miquerol L. Segregation of central ventricular conduction system lineages in early SMA+ cardiomyocytes occurs prior to heart tube formation. J Cardiovasc Dev Dis. 2016;3(1) https://doi.org/10.3390/jcdd3010002.
Zhou L, Liu J, Xiang M, Olson P, Guzzetta A, Zhang K, Moskowitz IP, Xie L. Gata4 potentiates second heart field proliferation and Hedgehog signaling for cardiac septation. Proc Natl Acad Sci U S A. 2017;114(8):E1422–31. https://doi.org/10.1073/pnas.1605137114.
Francou A, Saint-Michel E, Mesbah K, Kelly RG. TBX1 regulates epithelial polarity and dynamic basal filopodia in the second heart field. Development. 2014;141(22):4320–31. https://doi.org/10.1242/dev.115022.
Soh BS, Buac K, Xu H, Li E, Ng SY, Wu H, Chmielowiec J, Jiang X, Bu L, Li RA, Cowan C, Chien KR. N-cadherin prevents the premature differentiation of anterior heart field progenitors in the pharyngeal mesodermal microenvironment. Cell Res. 2014;24(12):1420–32. https://doi.org/10.1038/cr.2014.142.
Sinha T, Li D, Theveniau-Ruissy M, Hutson MR, Kelly RG, Wang J. Loss of Wnt5a disrupts second heart field cell deployment and may contribute to OFT malformations in DiGeorge syndrome. Hum Mol Genet. 2015;24(6):1704–16. https://doi.org/10.1093/hmg/ddu584.
Chen L, Fulcoli FG, Ferrentino R, Martucciello S, Illingworth EA, Baldini A. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 2012;8(3):e1002571. https://doi.org/10.1371/journal.pgen.1002571.
Li D, Sinha T, Ajima R, Seo HS, Yamaguchi TP, Wang J. Spatial regulation of cell cohesion by Wnt5a during second heart field progenitor deployment. Dev Biol. 2016;412(1):18–31. https://doi.org/10.1016/j.ydbio.2016.02.017.
Francou A, De Bono C, Kelly RG. Epithelial tension in the second heart field promotes mouse heart tube elongation. Nat Commun. 2017;8:14770. https://doi.org/10.1038/ncomms14770.
Ramsbottom SA, Sharma V, Rhee HJ, Eley L, Phillips HM, Rigby HF, Dean C, Chaudhry B, Henderson DJ. Vangl2-regulated polarisation of second heart field-derived cells is required for outflow tract lengthening during cardiac development. PLoS Genet. 2014;10(12):e1004871. https://doi.org/10.1371/journal.pgen.1004871.
Jain R, Li D, Gupta M, Manderfield LJ, Ifkovits JL, Wang Q, Liu F, Liu Y, Poleshko A, Padmanabhan A, Raum JC, Li L, Morrisey EE, Lu MM, Won KJ, Epstein JA. HEART DEVELOPMENT. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science. 2015;348(6242):aaa6071. https://doi.org/10.1126/science.aaa6071.
Metais A, Lamsoul I, Melet A, Uttenweiler-Joseph S, Poincloux R, Stefanovic S, Valiere A, Gonzalez de Peredo A, Stella A, Burlet-Schiltz O, Zaffran S, Lutz PG, Moog-Lutz C. Asb2alpha-filamin A axis is essential for actin cytoskeleton remodeling during heart development. Circ Res. 2018;122:e34. https://doi.org/10.1161/CIRCRESAHA.117.312015.
Watanabe Y, Zaffran S, Kuroiwa A, Higuchi H, Ogura T, Harvey RP, Kelly RG, Buckingham M. Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nkx2-5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proc Natl Acad Sci U S A. 2012;109(45):18273–80. https://doi.org/10.1073/pnas.1215360109.
Dupays L, Shang C, Wilson R, Kotecha S, Wood S, Towers N, Mohun T. Sequential binding of MEIS1 and NKX2-5 on the Popdc2 gene: a mechanism for spatiotemporal regulation of enhancers during cardiogenesis. Cell Rep. 2015;13(1):183–95. https://doi.org/10.1016/j.celrep.2015.08.065.
Briggs LE, Kakarla J, Wessels A. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation. 2012;84(1):117–30. https://doi.org/10.1016/j.diff.2012.05.006.
Xie L, Hoffmann AD, Burnicka-Turek O, Friedland-Little JM, Zhang K, Moskowitz IP. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev Cell. 2012;23(2):280–91. https://doi.org/10.1016/j.devcel.2012.06.006.
Momma K. Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol. 2010;105(11):1617–24. https://doi.org/10.1016/j.amjcard.2010.01.333.
Acknowledgements
Research in the author’s laboratory is supported by the Fondation pour la Recherche Médicale (DEQ20150331717), the Association Française contre les Myopathies, a grant from the Fondation Leducq (Transatlantic Network of Excellence 15CVD01) and the Agence National pour la Recherche (Heartbox project).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Kelly, R.G. (2020). Advances in the Second Heart Field. In: Nakanishi, T., Baldwin, H., Fineman, J., Yamagishi, H. (eds) Molecular Mechanism of Congenital Heart Disease and Pulmonary Hypertension. Springer, Singapore. https://doi.org/10.1007/978-981-15-1185-1_49
Download citation
DOI: https://doi.org/10.1007/978-981-15-1185-1_49
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-1184-4
Online ISBN: 978-981-15-1185-1
eBook Packages: MedicineMedicine (R0)