
Abstract
The mollusc shell comprises a small amount of organic macromolecules, mostly proteins and polysaccharides, which, all together, constitute the skeletal organic matrix (SOM). In the recent years, the study of the SOM of about two dozens of mollusc species via transcriptomics and/or proteomics has led to the identification of hundreds of shell-associated proteins. This rapidly growing set of data allows several comparisons, shedding light on similarities and differences at the primary structure level and on some peculiar evolutionary mechanisms that may have affected SOM proteins. In addition, it constitutes a prerequisite for investigating the SOM repertoires of sub-fossils or fossil specimens, closely related to known extant species, in order to revisit diagenetic processes, i.e. how SOM proteins degrade during fossilization. These two aspects are briefly exemplified here: on the one hand, Aplysia californica, the sea hare, exhibits a vestigial internal shell that has kept a proteomic signature similar to that found in fully functional external shells. On the other hand, subfossil specimens of the giant clam Tridacna, collected in French Polynesia, precisely dated and analysed by proteomics for their SOM content, comprise several preserved proteins that can still be identified by their peptide signature, in spite of information losses likely due to diagenetic transformations.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
The mollusc shell is a remarkable composite material made of calcium carbonate at 99% and of a minor organic fraction, the skeletal organic matrix (SOM). During the mineral deposition process, the SOM is secreted by the shell-forming organ, the mantle, and remains occluded. It is considered to be the main regulator of crystallization. In addition, it exhibits an interesting potential for preservation in fossil or subfossil samples (Hare et al. 1980). Classical biochemical characterizations indicate that the SOM consists in a mixture of proteins and polysaccharides (Marin et al. 2012). For decades, the SOM was considered as a ‘black box’ and analysed biochemically in bulk. Nowadays, high-throughput screening of SOMs, via the combined use of proteomics and transcriptomics, has allowed the identification of hundreds of shell proteins that are putatively involved in shell biosynthesis, in about two dozens of model mollusc genera comprising mostly bivalves, gastropods, and, in a lesser extent, cephalopods (Marin et al. 2016). This wealth of molecular data has considerably blurred the outlines of the SOM. However, taken individually, each of these ‘shell repertoires’ represents a key component of the calcifying machinery for making a shell, and shell repertoires can be compared to each other, shedding light on the macroevolution of calcifying matrices (Kocot et al. 2016; Marie et al. 2017). In addition to giving information on the calcification process and its macroevolution, molecular data collected from shells of extant molluscs is a prerequisite for obtaining – whenever possible – similar proteins in archaeological or fossil shell samples, an emerging field defined as paleoproteomics (Demarchi et al. 2016; Wallace and Schiffbauer 2016).
In the present paper, we briefly describe two unpublished examples on the use of proteomics to identify shell proteins: the first example relates to the Californian sea hare, Aplysia californica, a heterobranch gastropod that belong to a family, the Aplysiidae, characterized by an internal atrophied shell which is weakly calcified. The second example relates to subfossil specimens of the giant clam, Tridacna sp., collected in French Polynesia and precisely dated. In both cases, proteomics was performed from their extracted SOM.
2 Materials and Methods
2.1 Materials
Fresh shells of the Californian sea hare Aplysia californica were obtained from RSMAS at the University of Miami (Ph. Gillette) after sacrifice of the living animals according to ethical rules. Six shells of the giant clam Tridacna sp., including fresh, recent and subfossil specimens, were collected by one of us (E. S) on two sites of French Polynesia: Motu Piti Aau, Motu Mute, Bora Bora (Society Islands) and Motu Tepapuri, Mangareva (Gambier archipelago). The fresh Tridacna specimens were used as reference material.
2.2 Structural and Geochemical Characterizations
Series of structural characterizations of the shells were performed by SEM observations on polished sections or on freshly broken shell pieces that were slightly etched (EDTA 1% wt/vol, 3–5 min.). Minute fragments were sampled and powdered and the powder analysed by FT-IR spectroscopy, in order to check the mineralogy (aragonite). For Tridacna samples, thick sections were made for cathodoluminescence, epifluorescence and XRD analyses. In addition, the subfossil samples were dated via 14C measurements (Beta Analytic, Miami, FL, USA).
2.3 Extraction of the Aplysia and Tridacna SOMs
All Tridacna shells were scrupulously abraded and cleaned (two or three extended bleaching treatments with sodium hypochlorite) in order to remove putative contaminants. The clean powders were decalcified overnight with acetic acid, and the soluble and insoluble fractions were fractionated by centrifugation. All the subsequent steps leading to freeze-dried matrices were performed as previously described (Ramos-Silva et al. 2014). For Aplysia californica, the thinness of shells required adapting the cleaning/extraction protocol, which consisted first in protein desorption in successive bathes of TBS buffer containing Tween 20 (0.1%), NaN3 (0.001%), pH 9.2 for 1 week. After drying and reduction into powder, the samples were decalcified and centrifuged, leading to the fractionation of the soluble and insoluble matrices. The soluble fraction was desalted by several centrifugations/resuspension in water, in Vivaspin 20 cells (3 kDa cutoff), while the insoluble was rinsed with water. Both fractions were freeze-dried.
2.4 Proteomics on SOMs
All lyophilized samples were submitted to proteomic analysis (3P5 platform, Institut Cochin, Université Paris Descartes, Paris), after trypsic digestion, as previously described (Immel et al. 2016). For Aplysia californica, assigning identified peptides to known proteins was performed by using Mascot program (version 2.5, MatrixScience, London, UK) against the nonredundant NCBInr database. The search was restricted to ‘Other Metazoa’ dataset, which comprises a large collection of transcriptomic and genomic sequences from Aplysia californica, publicly accessible at NCBI (www.ncbi.nlm.nih.gov/). For Tridacna sp., an unpublished EST database provided by one of us (Dr. T. Takeuchi) from mantle tissues of the coral reef-associated crocus giant clam Tridacna crocea was used for protein identification.
3 Results
3.1 Proteomics on Aplysia californica Shell Matrices
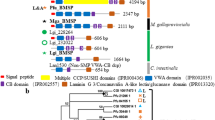
The internal shell of Aplysia californica is lightly calcified, chitinous, made of aragonite of the crossed-lamellar type (see Fig. 34.1a, b). It was submitted to a complete structural, chemical, biochemical and proteomic characterization that will be the subject of an extended publication in preparation. In the present paper, we simply summarize few of the outcomes obtained by proteomics. Our investigations generated several hits with proteins – known or unknown – from Aplysia californica. In total, we obtained 40 hits with proteins identified by more than two peptides and several additional hits with proteins identified with one peptide. We classified the protein hits according to the similarity of each of their primary structure to known functional domains or domains with a peculiar signature in terms of amino acid composition. As shown in Fig. 34.1c, we obtained six categories: enzymes, protease inhibitors, ECM/ECM-like (extracellular matrix), cation-interacting proteins, proteins containing LCDs/RLCDs (repetitive low complexity domains). The last category (others) comprises proteins that cannot be included in the five others. It is to note that the heterogeneous class of proteins containing LCDs/RLCDs represents the biggest group of shell proteins. It contains hydrophobic proteins in addition to P-rich, D-rich and S-rich proteins.

(a) The internal shell of Aplysia californica; the shell is lightly calcified. Top, apex (posterior); bottom, anterior part, which corresponds to the non-calcified growing shell margin. (b) Microstructure of the shell, observed in cross section; the dorsal side is on the right, the ventral, on the left. (c) Abridged list of proteins identified in the SOM of A. californica by proteomics. LCDs/RLCDs stands for low complexity domains/repetitive low complexity domains, ECM for extracellular matrix. The columns on the right indicate their theoretical molecular weight (MW) in kDaltons, their calculated isoelectric point (pI) and the presence (Y) or absence (N) of signal peptides. Signal peptides were identified by SignalP 4.1, while molecular weights and isoelectric points were calculated by ProtParam (after removal of the signal peptides when present). Both tools are accessible at http://www.expasy.ch/tools/. The fact that several secreted proteins do not exhibit a signal peptide could indicate that some of the sequences are not complete
3.2 Proteomics on Subfossil Tridacna Samples from French Polynesia
The subfossil samples of the giant clam Tridacna sp. were carefully checked for their preservation state, taking the fresh shells as reference. In particular, micro-samplings made across the thickness of the shell to identify the mineralogy via FT-IR spectroscopy showed that all shells were fully aragonitic and not recrystallized (data not shown). All of them exhibited the classical crossed-lamellar microstructure. In the subfossil shells, we however saw important alterations and perforations in their outermost and innermost layers (which were subsequently discarded) while the core layer was intact.
Proteomic investigations performed on the fresh shells generated up to 134 protein hits, 46 of which corresponding to proteins identified by at least two peptides. For subfossil shells, these numbers decreased drastically. For example, the GAM-14 sample, the age of which was precisely determined at 2880 ± 30 BP (before present), exhibited a total of 40 hits, but only 4 of them correspond to proteins identified by at least 2 peptides. Figure 34.2 shows an example of a new LCD-containing protein (P-rich), unambiguously identified in the fresh Tridacna sample, owing to good protein sequence coverage by peptides (38%, 17 peptides) all along the sequence. In the subfossil sample, the percentage of coverage of this protein sequence by peptides drops to 4% only (5 peptides), which suggests that the other non-covered parts of the sequence may be submitted to diagenetic transformation and/or hydrolysis. A complete view of all the results will be resumed in a publication in preparation.

Example of a novel protein identified in the fresh (a) and in the subfossil (b) Tridacna sample. This protein is the translation of the sequenced transcript TRINITY_DN253411_c2_g2_i3|m.459507. This protein is rich in proline (17.2%), glycine (10.1) and alanine (9.6%) residues and its theoretical calculated pI is basic (10.63). Its function in biomineralization is unknown. In grey italic, signal peptide. The peptides identified by proteomics are underlined. Note that the full protein sequence is well covered (38%) in the case of the fresh Tridacna shell while the coverage is poor (4%) for the subfossil shell. This drop may be explained by information losses at the peptide level due to diagenetic transformations (hydrolysis, modification of chemical groups on amino acids)
4 Discussion
This paper illustrates how proteomics contributes to answer questions related to the functions, evolution and diagenesis of SOMs in mollusc shell. In the first case, we explored the protein composition of the internal shell of the Californian sea hare, Aplysia californica. Aplysiidae are usually considered as a very derived gastropod family that has emerged only 25 million years ago (Klussmann-Kolb 2004) during the Oligocene epoch. This family is characterized by the presence of an atrophied and weakly calcified internal shell, which has completely lost its primary function, the protection of the soft tissues. In spite of this regressive evolution, it is remarkable to observe that the shell of A. californica has conserved a protein repertoire that exhibits a similar signature as the ones from fully functional external shells, in terms of diversity of protein families present in the matrix.
The second example deals with the diagenetic processes that affect organic matrices associated to calcium carbonate biominerals. In a previous paper, we showed that artificial diagenesis experiments performed on fresh nacre powder samples resulted in two phenomena recorded by proteomic analyses: a decrease of the number of identified proteins correlated to harsh diagenetic conditions and, in parallel, a decrease of the number of peptides identified per protein (Parker et al. 2015). Our analyses performed on the SOMs of subfossil Tridacna tend to correlate this finding. This example gives interesting perspectives for the coming time, i.e. the possibility to track the diagenetic pathway of each shell protein, taken individually, in sub-fossil/fossil of increasing age.
References
Demarchi B, Hall S, Roncal-Herrero T, Freeman C et al (2016) Protein sequences bound to mineral surfaces persist into deep time. eLife 5:e17092
Hare PE, Hoering TC, King K (eds) (1980) Biogeochemistry of amino acids. Wiley, New York
Immel F, Broussard C, Catherinet B, Plasseraud L, Alcaraz G, Bundeleva I, Marin F (2016) The shell of the invasive bivalve species Dreissena polymorpha: biochemical, elemental and textural investigations. PlosOne 11:e0154264
Klussmann-Kolb A (2004) Phylogeny of the Aplysiidae (Gastropoda, Opisthobranchia) with new aspects of the evolution of seahares. Zool Scr 33:439–462
Kocot KM, Aguilera F, McDougall C, Jackson DJ, Degnan BM (2016) Sea shell diversity and rapidly evolving secretomes: insights into the evolution of biomineralization. Front Zool 13:23
Marie B, Arivalagan J, Mathéron L, Bolbach G, Berland S, Marie A, Marin F (2017) Deep conservation of bivalve nacre proteins highlighted by shell matrix proteomics of the Unionoida Elliptio complanata and Villosa lienosa. J R Soc Interface 14:pii20160846
Marin F, Le Roy N, Marie B (2012) The formation and mineralization of mollusk shell. Front Biosci S4:1099–1125
Marin F., Bundeleva I, Takeuchi T, Immel F, Medakovic D (2016) Organic matrices in metazoan calcium carbonate skeletons: composition, functions, evolution
Parker A, Immel F, Guichard N, Broussard C, Marin F (2015) Thermal stability of nacre proteins of the Polynesian pearl oyster: a proteomic study. Key Eng Mater 672:222–233
Ramos-Silva P, Kaandorp J, Herbst F, Plasseraud L, Alcaraz G, Stern C, Corneillat M, Guichard N, Durlet C, Luquet G, Marin F (2014) The skeleton of the staghorn coral Acropora millepora: molecular and structural characterization. Plos One 9:e97454
Wallace AF, Schiffbauer JD (2016) Paleoproteomics: proteins from the past. elife 5:e20877
Acknowledgements
This work was supported by NEWFELPRO programme (Dr. D. Medakovic), by funds from OIST (T. Takeuchi), by EC2CO project (I. Bundeleva, F. Marin) and by annual funds from UMR Biogeosciences (F. Marin).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2018 The Author(s)
About this paper

Cite this paper
Marin, F. et al. (2018). Skeletal Organic Matrices in Molluscs: Origin, Evolution, Diagenesis. In: Endo, K., Kogure, T., Nagasawa, H. (eds) Biomineralization. Springer, Singapore. https://doi.org/10.1007/978-981-13-1002-7_34
Download citation
DOI: https://doi.org/10.1007/978-981-13-1002-7_34
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1001-0
Online ISBN: 978-981-13-1002-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)