
Abstract
We evaluated the migration of radionuclides (131I, 129I, 134Cs, 136Cs, 137Cs, and 132Te) in the surface soil after the Fukushima nuclear accident. The radionuclides in the soil collected late March in 2011 were barely leached with ultrapure water, indicating that these are insoluble. We observed the chemical behavior of 137Cs and 129I in soil: (1) 137Cs was predominantly adsorbed within a depth of 2.5 cm from the ground surface; (2) 137Cs was hardly released from soil by the water leaching experiments that lasted for 270 days; (3) approximately, more than 90 % of 137Cs was adsorbed on organic matters and the residual fractions, while 129I was mainly fixed on the Fe-Mn oxide and organically bounded fraction. Therefore, we conclude that 137Cs and 129I in soil seldom leach into the soil water and migrate downward because of the irreversible adsorption. The shallow groundwater which residence time is short.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
A number of radionuclides (including 137Cs, 134Cs, 136Cs, 131I, 132Te) were released into the atmosphere from the Fukushima Daiichi NPP accident in March, 2011. On March 15–17 and 21–23, deposition increased in the areas surrounding Fukushima prefecture because north–easterly, easterly, and south–easterly winds under a low-pressure system transported the radionuclides from the Fukushima NPP, and subsequent precipitation associated with the same system washed radioactive materials out of the radioactivity plume, thereby effectively depositing them on land [1–5]. In addition to the radioactive plume that covered the Fukushima Prefecture, two other large plumes suffered severe radioactive contamination over north Japan. Precipitation from these plumes caused high-radioactive spots across wide areas including the Tokyo metropolitan [1].
Although the eastern parts in the Tokyo metropolitan area are located far from the Fukushima NPP, high 137Cs and 131I deposition was observed in the areas. Therefore, the Tokyo metropolitan area is one of the hot spots of radioactive fallout from the Fukushima NPP accident [4]. The Tokyo metropolitan hot-spot area has a high-density population, and many residents have been worried about the radiation exposure from the Fukushima NPP. The released 131I from the Fukushima NPP contaminated tap water with rainfall that precipitated in the Tokyo metropolitan hot-spot areas on March 23, 2011, whereas 131I was also detected in the tap water of the Kanto and Tohoku regions through mid-April [1].
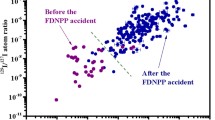
At present, 131I released from the Fukushima NPP accident in March, 2011 is not detected in the environment, because its short half-life is only 8 days. On the contrary, 129I with a half-life of 15.7 Ma is easily found. The 129I/127I ratio measured in surface soil could provide information on the local deposition of 131I released from the Fukushima NPP, if the ratio of 131I and 129I before release from the broken reactors can be estimated [6, 7]. The 131I behavior in the unsaturated zone and shallow groundwater immediately after the accident may be inferred from the 129I content.
This study has two objectives: (1) to examine the behavior of 137Cs and 131I in the surface soil of the Kanto loam in the Tokyo metropolitan hot-spot areas and Fukushima immediately after the accident and (2) to determine the 137Cs and 129I speciation in the soil to evaluate 137Cs and 131I contamination in the shallow groundwater which residence time is short.
2 Material and Methods
2.1 Soil Samples
One surface-soil sample within a depth of 0.5 cm and four surface-soil samples within a depth of 1 cm were collected on March 29, 2011 at the western Tokyo metropolitan area (WTMA, W1) and on March, 30 and 31, 2011 at the eastern Tokyo metropolitan hot-spot area (ETMA, E1-4), respectively. The soil samples collected at western Tokyo were placed in a polypropylene vessel (vessel A), 5 cm in diameter and 10 cm in height, and each of the four soil samples collected at eastern Tokyo was packed into a polypropylene vessel (vessel B), 2.2 cm in diameter and 1.2 cm in height.
Surface-soil core samples (5 cm2 × 10 cm) were collected at the WTMA on August 25, 2011 and ETMA, on October 14–18, 2011, respectively. The undisturbed soil cores were sampled using a cylindrical stainless steel core sampler, 0–10 cm in depth. The soil core samples were cut into the lengths of 0–2.5, 2.5–5 cm, and 5–10 or 0–2.5 and 2.5–5.0 cm. Each sample was stored in plastic vessels (vessels A). We also collected soil in the same manner in Nagadoro, Fukushima (N1) in May, 2012. Then we measured the vertical profiles of 137Cs in soil.
2.2 Column-Infiltration Experiments Using the Rainwater from the Tokyo Metropolitan Hot-Spot Area
It rained in the southern Ibaraki Prefecture from 0:00 to 2:00 LT on March 31, 2011, resulting in a total precipitation of 5.5 mm in Tsukuba. A rainwater sample was collected during precipitation at the ETMA (Japan Meteorological Agency). The rainwater sample (101 mL) was placed in a polypropylene vessel (5 cm in diameter and 10 cm in height, vessel A).
We investigated the infiltration of 137Cs from the rainwater in the soil environment via column experiments on April 1, 2011. Sand (Toyoura Standard Sand, Toyoura Keiseki Kogyo Co., Ltd., Yamaguchi, Japan) and soil were used in the columns. The soil, classified by the World Reference Base as a haplic stagnosol, was collected from a depth of approximately 30 cm from an outcrop at the campus of Kyoto University Research Reactor Institute (KURRI), Osaka, Japan. The soil contained a small amount of organic matter (organic carbon 2.7–2.8 %, pH 5.7–5.8) and was devoid of 137Cs and 134Cs.
The sand was rinsed several times to remove clay minerals using deep groundwater that did not contain 137Cs. The sand and soil samples were packed into two columns (20 mm in diameter and approximately 1 cm in depth). After filling, columns A (sand) and B (soil) were soaked in pure water for 3 h. Next, the rainwater sample (50 mL) was passed through each column at a rate of 0.7 mL・min−1, and then, each column was rinsed with ultrapure water (84 mL). Although the flow rate used was faster than the actual rate of rainwater infiltration in the Kanto district (approximately 1.5 m・y−1), this provides a conservative estimation of radionuclide migration. After infiltration, the sand and soil samples were stored separately in polypropylene containers (vessel B). Then, these vessels were placed into a different container, and the radioactivity of the samples in each container was measured using a gamma-ray spectrometer.
2.3 Leaching of Radionuclides from Soils Using the Batch Method
Radioactivities of the 100-g soil samples collected on March 29, 2011 at WTMA were immediately measured by the following method. The soil samples were mixed with the ultrapure water and shaken by hand for 30 min. The mixture was stored for 15 min, and the solution was separated by ultra-centrifuge (Kokusan Co., Ltd, Japan), and then, radioactivity in the supernatant was measured by gamma-ray spectrometry.
Approximately 10 g of the surface-soil samples collected from the WETA were added to ultrapure and groundwater samples, and then, they were agitated at a speed of 100 rpm for 90 and 270 days. The groundwater sample was discharged from the Kanto-loam layer in the Tokyo metropolitan hot-spot area, and was sampled in March, 2010 before the Fukushima NPP accident. The concentrations of Na+, K+, Mg2+, and Ca2+ in the groundwater were measured by cation chromatography and those of Cl−, SO4 2− and NO3 − were measured by anion chromatography, whereas HCO3 − was measured by titration using HCl. Table 2.1 lists the major cation and anion of the groundwater samples.
After leaching, solid-liquid separation was conducted by centrifugation at 2000 rpm for 5 min. Then, the solution was filtered through a membrane filter with a pore size of 0.45 μm (Advantech, Co., Ltd., Japan). After storing each fraction in a U-8 vessel, radioactivities of 131I, 134Cs, 136Cs, 137Cs, and 132Te were measured by gamma-ray spectrometry.
2.4 Separation of 137Cs and 129I in Soil Samples
We extracted 137Cs and 129I from three surface soil samples from the Kanto-loam layer in the ETMA and the Nagadoro in Fukushima. The soil of depths of 0–2.5 cm (E5, E6, N1) was well mixed and approximately 10 g soil sample was taken by cone and quartering method. Approximately 10 g of the samples were used in the sequential-extraction experiment [8, 9]. A ratio of solution to sample of 5 (v/w) was used for extraction in each step.
-
Fraction 1: After ultrapure water was added to the soil sample, the suspension was shaken for 24 h at room temperature, and then the suspension was stored overnight. After extraction, solution was separated from the soil residue by centrifugation at 2000 rpm for 5 min. The solution was filtered through a membrane filter with a pore size of 0.45 μm (Advantech, Co., Ltd., Japan). The fraction of the filtrate represents water-soluble species. The remaining solid on the membrane filter was combined with the residue for the next leaching step.
-
Fraction 2: 1 M of NaAc was added to the residue from Fraction 1. The suspension was shaken for 12 h at room temperature and stored overnight. The fraction of the filtrate represents exchangeable species.
-
Fraction 3: 1-M NaAc − HAc (pH 5) was added to the residue from Fraction 2, and the suspension was shaken for 12 h at room temperature. The fraction of the filtrate represents carbonate-bound species.
-
Fraction 4: 0.04-M NH2OH・HCl in 25 % (v/v) HAc (pH 2) was added to the residue from Fraction 3 and stirred in a hot-water bath at 80 °C for 4 h. The fraction of the filtrate represents species associated with solids via chemical-sorption mechanisms that can be released into the extraction solution with a weak reducing agent, and they mainly include species bound to Fe/Mn oxides.
-
Fraction 5: 30 % H2O2 was added to the residue, in which HNO3 had already been added to adjust the final pH to 2. The suspension was agitated for 2 h at 85 °C. After the suspension was cooled to room temperature, 1.8-M NH4Ac in 11 % HNO3 (v/v) was added, and the extraction continued for 30 min at room temperature. The fraction of the filtrate is associated with organic matter.
After each fraction was stored into a U-8 vessel, 137Cs was measured by gamma-ray spectrometry.
2.5 Purification of Iodine Isotopes for Accelerator Mass Spectrometry (AMS) Measurement
One-milliliter solution of nitric acid (Kanto Chemical Co., Ltd.) and 0.5 mL of H2O2 (Kanto Chemical Co., Ltd.) were added to sample solutions separated from each fraction. The dissolved iodine was oxidized to I2 and was then separated from the sample solution into 10 mL of chloroform (Wako Co., Ltd.). The chloroform was separated from the sample solution, and then 10 mL of 0.1 M of NaHSO3 (Wako Co., Ltd.) was added to the chloroform to extract I− into the NaHSO3 solution. The NaHSO3 solution was separated from the chloroform, and 1 mL of 6 M NaCl (Aldrich Co., Ltd.) was added to the solution. A 0.1 mL portion of 1 M AgNO3 (Aldrich Co., Ltd.) was then added to the solution, and the solution was agitated, causing AgI to precipitate with AgCl. This precipitation was allowed to continue for 30 min before the mixture was centrifuged for 5 min at 3000 rpm. The mixture of AgCl and AgI precipitate was separated from the solution. AgCl was separated from AgI by adding 4 mL of concentrated NH3 to dissolve AgCl only. The AgI precipitate was rinsed with 5 mL of ultrapure water to yield a pure AgI sample, which was then dried in an electric oven at 70 °C for 40 min. The AgI sample was added to Nb powder at an Nb and AgI ratio of 4 (w/w). The detail chemical separation was described by a previous paper [10]. 129I and 127I were measured by AMS (Malt, Tokyo Univ., Japan), and detailed procedure of measurement of 129I/127I atomic ratios were described by Matsuzaki et al. [11].
2.6 Measurement of Radioactivity in Environmental Samples by Gamma-Ray Spectrometry
The radioactivity of the rainwater and soil sample was measured using a p-type high-purity germanium detector (IGC-309, Princeton Gamma-Tech) with 40 % relative efficiency and a multichannel analyzer (7600-000, Seiko EG&G) with a high-voltage circuit (7600-310) and pulse height analyzer (7600-510) in KURRI. The gamma-ray counting efficiency of the detector was estimated by constructing a relative gamma-ray-counting-efficiency curve using a certified mixed-radionuclide gamma-ray reference source containing 57Co (122.1 keV), 137Cs (661.7 keV), and 60Co (1173 and 1332 keV), which were normalized to the 1460-keV gamma-ray peak of 40K in KCl. A quadratic function was fitted to the logarithmic relationship between the relative counting efficiency and gamma-ray energy using the least-squares method and was normalized at 1460 keV. The radiation energies of 131I, 134Cs, 136Cs, 137Cs, and 132Te were 364, 796, 818, 662, and 228 keV, respectively. The sum effect of gamma rays from 134Cs was corrected by measuring the 134Cs solution. 134Cs was produced by the neutron activation of CsCl at KURRI. CsCl was dissolved in the ultrapure water and the prepared 134Cs solution. The detection limits of 131I, 134Cs, and 137Cs with a measuring time of 10,000 s were 0.11, 0.099, and 0.12 Bq for vessel B and 0.20, 0.19, and 0.23 Bq for vessel A, respectively.
The radioactivity of undisturbed core samples from the ETMA and radio-Cs samples leaching into the ultrapure water and groundwater samples was measured in the Isotopes Centre, Hokkaido University using a p-type, high-purity germanium detector (model IGC-309, Princeton Gamma-Tech) with 40 % relative efficiency. The detection limits of 134Cs and 137Cs with a measuring time of 864,000 s were 0.02 and 0.02 Bq for the U-8 vessel, respectively.
3 Results and Discussion
Table 2.2 shows the concentrations of 134Cs, 136Cs, 137Cs, 131I, and 132Te in the surface soils collected in March, 2011 from the Tokyo metropolitan hot-spot area (but only W6 was collected in August, 2011). Short half-lives for 136Cs, 131I, and 132Te were detected in the surface-soil samples except W6. The radioactivities of 131I, 134Cs, and 137Cs in the surface-soil samples except W6 collected at ETMA were measured, and W6 measured only the radioactivities of 134Cs and 137Cs. 131I was the source of the maximum radioactivity in the soil in March, 2011 in the Tokyo metropolitan hot-spot area. The concentration of 131I ranged from 9.4 to 13 k Bq・kg−1 before rain (March 30, 2011) and from 7.2 to 11 k Bq・kg−1 after rain, whereas that of 137Cs ranged from 0.72 to 3.3 k Bq・kg−1 before rain and 1.7–2.9 k Bq・kg−1 after rain (March 31). As the fallout radionuclides on the surface soils were mainly washed out from the atmospheric aerosol plume by precipitation, 137Cs concentrations being similar to 131I in the soil before rain on March 30 and after rain on March 31 suggest that the rainfall on March 21 and 22 (and the very small precipitation on March 15 and 16) was major source to most radionuclides in the soil.
The concentrations of 131I, 134Cs, and 137Cs in the rainwater collected at the ETMA toward the end of March, 2011 were 66 ± 3, 28 ± 2, and 31 ± 2 Bq・L−1, respectively. The concentration of each radionuclide was corrected at 2:00 LT, 31 March 2011.
Table 2.3 lists radioactivities trapped in both columns of sand and soil, and the 131I- and 137Cs-trapping efficiencies in both columns. In column A (sand), almost all of the 137Cs was trapped on sand with the trapping efficiency of 93 ± 6 %. In column B (soil), the 137Cs-trapping efficiency was 81 ± 5 %. Therefore, we considered almost all 137Cs in rainwater to be adsorbed on sand and the Haplic Gray Upland soil within a depth of 1 cm from the ground, while the trapping efficiency of 131I in both sand and soil columns was below the detection limit and 30 %, respectively. These data indicate that retardation of 131I in both sand and soil downward was smaller than that of 137Cs.
Table 2.4 shows the concentrations of 137Cs and 134Cs in the surface soil collected at the ETMA in October, 2011, 7 months after the accident. Almost all 134Cs and 137Cs in the Kanto-loam soil were within a depth of 2.5 cm from the ground. The column test and the distribution of 137Cs obtained from the core sample in the field indicated that 134Cs and 137Cs were strongly adsorbed on the surface soil within a depth of several centimetres from the surface. Vertical profiles of radionuclides in soil in Koriyama, Fukushima, showed that more than 90 % of 131I was found to be within about 5 cm depth from the surface in soil layer after the accident [12].
Following the leaching tests from the surface soil collected at WTMA into the pure water immediately after sampling in March, 2011, the leachate was separated from soil. 134Cs, 137Cs, 131I, 136Cs, and 132Te in soil did not leach into the water (Table 2.5). Table 2.5 lists the amount of radioactive Cs in the leachate when the surface soil of the ETMA or WTMA was mixed with the ultrapure water and groundwater during 90 and 270 days. Both 137Cs and 134Cs in each soil of the Kanto-loam layer and the Nagadoro were not released by leaching with both the ultrapure water and groundwater after mixing during 90 and 270 days.
Table 2.6 shows the leaching fractions of 137Cs from each soil in the Kanto-loam layer of the ETMA and in the Nagadoro, Fukushima according to the sequential-extraction procedure described previously. The amount of 137Cs leached into water (F1) from the Kanto-loam soil and Fukushima soil was below the detection limit. This has been demonstrated by the leaching results of radioactive Cs from the soil by the ultrapure water and groundwater, as shown in Table 2.5. The ratio of exchangeable 137Cs from the two soil samples (E5 and E6) was less than 1 %, with 50–60 % of 137Cs remaining in the residue. Approximately 98 % of 137Cs was in the fraction of Fe–Mn oxide, organic matter, and the residue. The 137Cs in the water fraction (F1) is undetectable and this is the same results of the extraction test obtained in Table 2.5. The small fraction of exchangeable 137Cs suggests that immediately after the 137Cs in rainwater dropping on the surface soil, 137Cs and 134Cs were strongly adsorbed onto the soil of the Kanto loam, and they were not readily leached into the soil water. The amount of 129I in fraction of F1 and F2 were 0.7–9.5 % and 1–3.8 %, respectively; therefore, 131I in unsaturated soil moved downward faster than 137Cs. More than 90 % of 137Cs was in the fraction of organic matter and the residue, while 129I was mainly fixed by Fe-Mn oxidation and organically bound.
The estimated 137Cs migration rate for the Nishiyama loam soil, which was obtained in situ at Nishiyama (Nagasaki), is 1.0 mm・y−1 [13]. This is considerably less than the rainwater infiltration rate of 2.5 m・y−1. Furthermore, 137Cs was not detected in the groundwater of the Nishiyama area, suggesting that 137Cs has not yet migrated to the groundwater table [14]. We collected 4 L of shallow groundwater in the Kashiwa city in the ETMA on August 17, 2012. The groundwater was filtered through membrane filter with a pore size of 80, 3, and 0.45 μm. The groundwater after the filtration was gradually reduced to 5 mL by a mantle heater. Both 137Cs and 134Cs were not detected in the shallow groundwater and the suspended matters collected from the filter (Table 2.7).
Although the 131I moving velocity deduced from that of 129I was greater than that of 137Cs, the 129I migration rate is lower than the water infiltration rate, due to the 129I absorption on soil in the unsaturated zone.
In Kanto loam, soil water reaches depths of 20–30 cm, considering a 1–1.5 m・y−1 infiltration rate, after 80 days corresponding to the time length of 10 times of the 131I half-lives. Mainly 137Cs was detected in litters in forest [5] but 137Cs was detected to depth of 10 cm in soil without litter [15]. Because rainy force is buffered in litters when the surface of soils has litters, rain is hard to directly enter to the deep part of the soil. Water-soluble 131I would merely move downward to depths of 30–40 cm, even if 131I penetrated a depth of 10 cm in the soil without litters and/or grass immediately after the accident. Therefore, 131I could never reach the depths of 50 cm in the groundwater table.
4 Conclusion
The sequentially chemical fractionations of 129I and 137Cs in soil indicate that most part of 137Cs and 129I were insoluble. Traces of 131I in the soil water did not reach the 50-cm depth by late June, 2011, corresponding to the time length of 10 times of 131I half-lives after the Fukushima NPP accident. Therefore, shallow groundwater could be safely useful water resource after the accident.
References
Amano H, Akiyama M, Chunlei B, Kishimoto T, Kuroda T, Muroi T, Odaira T, Ohta Y, Takeda K, Watanabe Y, Morimoto T (2012) J Environ Radioact 111:42–52
Hirose K (2012) J Environ Radioact 111:13–17
Ohta T, Mahara Y, Kubota T, Fukutani S, Fujiwara K, Takamiya K, Yosinaga H, Mizuochi H, Igarashi T (2012) J Environ Radioact 111:38–41
Ohta T, Mahara Y, Kubota T, Igarashi T (2013) Ana Sci 29:941–947
Mahara Y, Ohta T, Ogawa H, Kumata A (2014) Sci Rep 4:Article number 7121. doi:10.1038/srep07121
Muramatsu Y, Matsuzaki H, Toyama C, Ohno T (2015) J Environ Radioact 139:344–350
Miyake Y, Matsuazaki H, Fujiwara H, Saito T, Yamagata H, Honda M, Muramatsu Y (2012) Geochem J 46:327
Oughton DH, Salbu B, Riise G, Lien HN, Østby GA, Nøren A (1992) Analyst 117:481–486
Riise G, Bjørnstad HE, Lien HN, Oughton DH, Salbu B (1990) J Radioanal Nucl Chem 142:531–538
Ohta T, Mahara Y, Kubota T, Abe T, Matsueda H, Tokunaga T, Sekimot S, Takamiya K, Fukutani S, Matsuzaki H (2013) Nucl Instrum Methods Phys Res B 294:559–562
Matsuzaki H, Muramatsu Y, Kato K, Yasumoto M, Nakano C (2007) Nucl Instrum Methods Phys Res B 259:721–726
Ohno T, Muramatsu Y, Miura Y, Oda K, Inagawa N, Ogawa H, Yamazaki A, Toyama C, Saito M (2012) Geochem J 46:287–295
Mahara Y (1993) J Environ Qual 22:722–730
Mahara Y, Miyahara S (1984) J Geophy Res 89:7931–7936
Multidisciplinary investigation on radiocesium fate and transport for safety assessment for interim storage and disposal of heterogeneous waste. The initiatives for atomic energy basic and generic strategic Research, JST, 2013, 240407 (in Japaneses). Initiatives for Atomic Energy Basic and Generic Strategic Research by the Ministry of Education, Culture, Sports, Science and Technology of Japan
Acknowledgment
This study is partially supported by JST Initiatives for Atomic Energy Basic and Generic Strategic Research. We are deeply appreciative of Dr. Iimoto for scientific support and advice for the groundwater sampling.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Ohta, T. et al. (2016). Speciation of 137Cs and 129I in Soil After the Fukushima NPP Accident. In: Takahashi, T. (eds) Radiological Issues for Fukushima’s Revitalized Future. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55848-4_2
Download citation
DOI: https://doi.org/10.1007/978-4-431-55848-4_2
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55847-7
Online ISBN: 978-4-431-55848-4
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)