
Abstract
In the oral mucosa, epithelial cells work not only as a physical barrier to pathogens, but also play a pivotal role in initiating immune responses to microbes. Interleukin (IL)-33, a member of the IL-1 family, is constitutively expressed in epithelial cells and amplifies Th2-type inflammatory immune responses. We found that IL-33 was detected in the inflamed gingival epithelium from chronic periodontitis patients, and periodontopathic Porphyromonas gingivalis strongly increased expressions of IL-33 mRNA and molecules in human gingival epithelial cells. In contrast, fimbriae, a lipopeptide and lipopolysaccharide derived from P. gingivalis were not active in this respect. Protease inhibitors specific for gingipains efficiently inhibited the induction of IL-33 mRNA by stimulation with P. gingivalis. Furthermore, P. gingivalis KDP136, a gingipains-null mutant, did not increase IL-33 mRNA expression. We also demonstrated that P. gingivalis upregulated IL-33 mRNA expression through protease-activated receptor-2, phospholipase C, mitogen-activated protein kinase p38 and NF-κB. IL-33 is suggested to negatively regulate antimicrobial peptide LL-37, resulting in attenuation of innate immune responses of gingival epithelial cells in chronic periodontitis. Possible roles of IL-33 in inflammation in the oral mucosa are discussed.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
In mucosal immune systems, including the oral mucosa, epithelial cells work not only as a physical barrier to bacterial pathogens, but also play a pivotal role in initiating and amplifying Th2-type immune responses in response to bacterial components [1, 2]. Epithelial cells produce interleukin (IL)-33, IL-25, and thymic stromal lymphopoietin, which may be involved in the development and regulation of Th2-type inflammatory responses. IL-33 is a member of the IL-1 cytokine family, and is constitutively expressed in epithelial cells, endothelial cells and fibroblasts [3]. IL-33 consists of two domains: a non-classical homeodomain-like helix-turn-helix DNA-binding domain, which consists of a chromatin-binding motif (CBM) and a nuclear localization sequence (NLS), and an IL-1-like domain [4] (Fig. 25.1a). IL-33 localizes in the nuclei of resting epithelial cells and acts as an alarmin when released from necrotic cells (Fig. 25.1b). IL-33 has a protective role in inflammatory bowel disease [7] and in the initiation of Toxoplasma infection that polarizes adaptive responses towards a Th2-biased response, which is protective in this disease [8]. In contrast, a lot of evidence suggests that IL-33 is also involved in the development of chronic inflammatory diseases such as arthritis [9]. IL-33 signals through the IL-33 receptor (IL-33R), which consists of heterodimers of ST2 and IL-1 receptor accessory protein (IL-1RAcP) [5, 6] (Fig. 25.2). IL-33 upon binding to ST2 induces the recruitment of IL-1RAcP and myeloid differentiation primary-response protein 88 (MyD88) to the Toll/IL-1R (TIR) domain in the cytoplasmic region of ST2. The MyD88 and TRAF6 complex activates NF-κB- and MAP kinase-mediated signaling pathways [5, 6].

(a) Structure of IL-33 protein. IL-33 is a 30-kDa protein that consists of 270 amino acids. IL-33 consists of two domains: a helix-turn-helix domain and an IL-1-like domain. The helix-turn-helix domain contains a chromatin-binding motif (CBM) and a nuclear localization signal (NLS). (b) Release of IL-33. Active form of IL-33 is thought to be released by necrosis. IL-33 is inactivated by cleaving with caspase-3 or caspase-7 during apoptosis. Based on Martin, Oboki et al. and Palmer and Gabay [4–6]

IL-33 signaling pathways. IL-33 signaling through the IL-33 receptor (IL-33R), which consists ST2 and IL-1 receptor accessory protein (IL-1RAcP) dimers and myeloid differentiation primary-response protein 88 (MyD88) to the Toll/IL-1R (TIR) domain in the cytoplasmic region of ST2. The MyD88 and TRAF6 complex activates NF-κB- and MAP kinase-mediated signaling pathways. Based on Martin, Oboki et al. and Palmer and Gabay [4–6]
Porphyromonas gingivalis is a periodontopathic pathogen in chronic periodontitis and has a variety of virulence factors that induce proinflammatory cytokines leading to chronic inflammation, resulting in destruction of periodontal tissues. In the dental/oral field, only limited information on IL-33 is available (see Table 25.1). TNF-α induces IL-33 expression in human gingival fibroblasts [12], although whether IL-33 expression is increased in the periodontal tissues in chronic periodontitis patients and the functions of IL-33 in the modulation of chronic periodontitis remain unelucidated. This review article shows possible regulation of IL-33 expression in human gingival epithelial cells in response to P. gingivalis based on our recent studies, and discusses possible roles of IL-33 expressed in gingival epithelial cells in relation to the pathogenesis of chronic periodontitis.
2 Expression of IL-33 in Periodontal Tissues from Chronic Periodontitis Patients
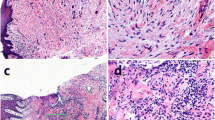
We first examined whether inflamed gingival tissues from chronic periodontitis patients expressed IL-33 by immunohistochemical studies using an anti-human IL-33 monoclonal antibody (mAb). As expected, IL-33 was strongly expressed in the cytoplasm of the inflamed gingival epithelium from chronic periodontitis patients, but only weakly detected in the normal gingival epithelium from healthy individuals. Unlike the expression of IL-33 in the gingival epithelium, IL-33 expression in the lamina propria of gingival tissues was only weakly observed. IL-33 expression is possibly upregulated in epithelial, mesenchymal, and myeloid cells in response to proinflammatory stimuli, pathogen-associated molecular patterns, and pathogens [5]. These findings suggest that gingival epithelial cells are capable of inducing IL-33 expression upon infection with periodontal pathogens.
3 Possible Induction of IL-33 by Gingipains from P. gingivalis in Gingival Epithelial Cells
3.1 P. gingivalis Induces IL-33 mRNA Expression in Human Gingival Epithelial Cells in Culture
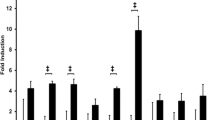
As P. gingivalis is implicated as a major pathogenic bacteria for chronic periodontitis [14], we examined the effect of P. gingivalis infection on IL-33 mRNA expression in human gingival epithelial cells in culture. The IL-33 mRNA expression was increased 20-fold at 48 h after stimulation with P. gingivalis W83 in Ca9-22 cells, which is a human gingival epithelial cell line established from squamous cell carcinoma. Pretreatment of the cells with cycloheximide, a protein synthesis inhibitor, blocked the induction of IL-33 mRNA levels, suggesting that de novo protein synthesis was required for P. gingivalis-mediated IL-33 mRNA induction. Nile et al. [10] reported that P. gingivalis LPS induced IL-33 expression in human monocytes. Therefore, we examined the possible IL-33 mRNA-inducing capacity of P. gingivalis-related specimens; fimbriae (Toll-like receptor (TLR) 2 ligand [15]) and LPS (TLR2 and 4 ligand [16]) prepared from P. gingivalis, and synthetic P. gingivalis-type lipopeptide PGTP2-RL (TLR2/1 ligand [17]). Unlike P. gingivalis whole cells, fimbriae and lipopeptide were completely inactive, and LPS specimens were only weakly active in this respect. These results suggest that gingival epithelial cells respond to P. gingivalis rather than TLR ligands for induction of IL-33 mRNA.
3.2 Involvement of Gingipains in the Induction of IL-33 mRNA Expression in Human Gingival Epithelial Cells by P. gingivalis
P. gingivalis produces two types of arginine-specific cysteine proteinases (Arg-gingipains, RgpA and RgpB) and a lysine-specific cysteine proteinase (Lys-gingipain, Kgp) [18]. Gingipains are localized to a cell-associated form, a soluble form, and released as outer membrane blebs [18]. We next examined whether enzymatic activities of gingipains are involved in the IL-33-inducing capacity in Ca9-22 cells and primary oral epithelial cells. The induction of IL-33 mRNA expression by P. gingivalis W83 in the two types was completely and significantly inhibited by FPR-cmk (Rgp inhibitor) and KYT-36 (Kgp inhibitor), respectively. Furthermore, P. gingivalis KDP136, a gingipains-null mutant, unlike P. gingivalis ATCC 33277, a wild-type parent strain of KDP136, did not induce IL-33 mRNA expression in either cell type. P. gingivalis-induced IL-33 mRNA expression was abolished by heat treatment (70 °C, 1 h) to inactivate enzymatic activities of P. gingivalis. We confirmed intact proteolytic activities for Rgps and Kgp in the whole cells of P. gingivalis W83 and ATCC 33277, but not in those of P. gingivalis KDP136. These observations suggest that the proteolytic activity of gingipains is essential for the induction of IL-33 mRNA expression by P. gingivalis in human gingival epithelial cells.
3.3 Induction of the IL-33 Molecule by P. gingivalis in Human Gingival Epithelial Cells
Next, we examined whether P. gingivalis induced IL-33 expression in human gingival epithelial cells using an immunoblot analysis. IL-33 expression in Ca9-22 cells was increased tenfold with a peak at 4 days by stimulation with whole cell preparations of P. gingivalis. Epithelial cells constitutively express IL-33 in their nuclei under resting conditions [19]. IL-33 is a nuclear protein that is also released into the extracellular environment. To further determine whether P. gingivalis-induced IL-33 was accumulated in the nuclei or the cytoplasm, we analyzed the location of IL-33 protein in Ca9-22 cells after stimulation with P. gingivalis using immunocytochemical analysis. Although IL-33 was constitutively expressed in the resting cells, IL-33 molecules accumulated in the cytoplasm of the cells when they were stimulated with P. gingivalis for 4 days. However, the released IL-33 level was quite low (approximately 30 pg/mL), even after stimulation with P. gingivalis whole cells. IL-33 is not detected in viable human monocytic cells, even upon stimulation with P. gingivalis LPS [10]. Furthermore, IL-33 levels in gingival crevicular fluid (GCF) are not different between chronic periodontitis patients and healthy controls [11]. In fact, IL-33 is not detected in the GCF of inflamed regions from chronic periodontitis patients [13]. Further studies are required to elucidate the fate of IL-33 induced by P. gingivalis in gingival epithelial cells, which may modulate the innate immune functions of the cells in infected periodontal lesions.
4 PAR-2-p38/NF-κB-Mediated Signals in IL-33 Induction
4.1 Role of PAR-2 in the Induction of IL-33 by Gingipains
Proteinase-activated receptor-2 (PAR-2) is a seven-transmembrane domain receptor family which couples to G-proteins [20]. Rgps are capable of activating PAR-2 expressed on human gingival epithelial cells, which produce proinflammatory cytokines [21]. We examined the possible involvement of PAR-2 in P. gingivalis-induced IL-33 mRNA expression. Ca9-22 cells constitutively expressed PAR-1, −2, −3, and −4 mRNA. PAR-2 mRNA expression was increased upon stimulation with P. gingivalis W38 in Ca9-22 cells. Next, we performed inhibition of PAR-2 mRNA expression by RNA interference using a PAR-2-specific small interference RNA (siRNA). The induction of IL-33 mRNA by P. gingivalis W83 was partially inhibited in PAR-2 siRNA-transfected cells. PAR-2 is activated by a tethered ligand when cleaved by protease. A PAR-2 agonist peptide based on the tethered ligand sequences can activate PAR-2 in a proteolysis-independent manner. However, the PAR-2 agonist peptide was not able to induce IL-33 mRNA in Ca9-22 cells. Pretreatment of the cells with cytochalasin D, a particle internalization inhibitor, inhibited the IL-33 mRNA induction by P. gingivalis. These findings suggested that both proteolytic activation of PAR-2 by gingipains and the endocytosis of P. gingivalis are required for the up-regulation of IL-33 expression induced by P. gingivalis in oral epithelial cells. P. gingivalis enters gingival epithelial cells by endocytosis, which mediates binding of Rgp to the cells [22]. It must be noted that gingipains are required for maturation of P. gingivalis fimbriae [23], which is essential for internalization of the bacterium into epithelial cells [24]. However, endocytosis of both PAR-2 and P. gingivalis are probably required for the up-regulation of IL-33 in gingival epithelial cells.
4.2 Involvement of PLC in the Induction of IL-33 by Gingipains
Because PAR-2 has been reported to be coupled to G protein, leading to activation of phospholipase C (PLC) [25, 26], we examined whether or not the PLC pathway is involved in P. gingivalis-induced IL-33 mRNA expression in Ca9-22 cells. The cells were pretreated with U-73122, a PLC inhibitor, and then stimulated P. gingivalis W83 for 48 h. The pretreatment significantly inhibited the P. gingivalis-induced IL-33 mRNA expression. In contrast, a protein kinase C (PKC) inhibitor, GF-109203X, did not inhibit either the basal level of IL-33 or the P. gingivalis-induced increase in IL-33. These findings suggest that P. gingivalis-induced up-regulation of IL-33 expression is mediated via a PAR-2-PLC-signaling pathway.
4.3 Involvement of p38 and NF-κB in the Induction of IL-33 by Gingipains
As p38 and NF-κB are implicated in PAR-2 signaling [27], we first confirmed p38 phosphorylation in Ca9-22 cells stimulated with P. gingivalis W83. Then, we demonstrated that p38 signaling is involved in P. gingivalis-induced IL-33 mRNA expression using SB203580, a p38 inhibitor. In contrast, the P. gingivalis IL-33 mRNA expression was not inhibited by PD98059, an ERK1/2 inhibitor, or SP600125, a JNK inhibitor. Further, we demonstrated that gingipains are responsible for p38 activation caused by whole cells of P. gingivalis because p38 phosphorylation induced by P. gingivalis W83 was completely inhibited when P. gingivalis was treated with Rgp inhibitor FPR-cmk plus Kgp inhibitor KYT-36. In addition, p38 phosphorylation was not observed in the cells stimulated with P. gingivalis KDP136, a gingipains-null mutant.
Next, we demonstrated NF-κB activation in Ca9-22 cells stimulated with P. gingivalis W83 using a luciferase reporter assay. We demonstrated that NF-κB signaling is involved in P. gingivalis-induced IL-33 mRNA expression because pretreatment of cells with PDTC, an NF-κB inhibitor, significantly inhibited P. gingivalis-induced IL-33 mRNA expression. The NF-κB activities induced by P. gingivalis W83 were markedly diminished by FPR-cmk plus KYT-36, and the NF-κB activation was attenuated in the cells stimulated with P. gingivalis KDP136, indicating that gingipains are responsible for NF-κB activation caused by whole cells of P. gingivalis. Taken together, we demonstrated that the P. gingivalis-derived gingipain-mediated IL-33 increase was dependent on PAR-2-PLC-p38/NF-κB signaling (Fig. 25.3).

Role of gingipains in the induction of IL-33 via PAR-2-p38/NF-κB-mediated signals
5 Future Prospects: Possible Roles of IL-33 in Gingival Epithelial Cells in Chronic Periodontitis
Gingival epithelial cells play integral roles in innate immune defense by sensing periodontal pathogens, maintaining a physical barrier, and expressing antimicrobial peptides (AMPs) [28]. AMPs constitute an important component in the innate immune response. In humans, two main classes of cationic AMPs, the cathelicidins and the defensins, are expressed in a various type of cells, such as epithelial cells, neutrophils, and macrophages. LL-37, a 37-residue mature antimicrobial peptide with two leucine residues, is a 37 amino acid peptide derived from human cathelicidin, a cationic antimicrobial peptide of 18-kDa (CAP18). The peptide is mainly produced by epithelial cells and neutrophils [29] and detected in GCF [30]. Human β-defensin-2 (hBD-2), an antimicrobial peptide of the β-defensin family, is induced by gingipains through PAR-2 in gingival epithelial cells [31]. It has been reported that hBD-2 production is down-regulated by pretreatment with IL-33 in human foreskin keratinocytes [32]. Endogenous expression of LL-37 plays an important role in intracellular killing of mycobacteria in macrophages [33].
To address whether development of chronic periodontits is attributable to IL-33, we examined the influence of P. gingivalis-mediated IL-33 production on the induction of LL-37 in human gingival epithelial cells. Indeed, we found that P. gingivalis-induced CAMP (LL-37 gene) mRNA expression was up-regulated in IL-33 siRNA-transfected Ca9-22 cells. LL-37 is capable of proteolytically degradation by gingipains secreted by P. gingivalis; however, the antibacterial activity of LL-37 is still intact in the presence of P. gingivalis proteases [34]. These findings indicate that IL-33 may attenuate antimicrobial immune responses by epithelial cells against bacterial mucosal infections.
In this study, we revealed that P. gingivalis induced IL-33 via PAR-2-PLC-p38/NF-κB signaling pathways and that the IL-33 down-regulated LL-37 expression in human gingival epithelial cells. These findings suggest evasion of innate immune responses by P. gingivalis is due to inhibition of antimicrobial peptide expression (Fig. 25.4). Further studies are necessary to elucidate the role of intracellular IL-33 in maintaining host defense mechanisms in gingival epithelial cells against periodontal diseases.

A hypothetical model for the role of IL-33 induced by P. gingivalis in the down-regulation of LL-37 expression in human gingival epithelial cells
References
Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–90.
Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–35.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90.
Martin MU. Special aspects of interleukin-33 and the IL-33 receptor complex. Semin Immunol. 2013;25(6):449–57. doi:10.1016/j.smim.2013.10.006.
Oboki K, Ohno T, Kajiwara N, Saito H, Nakae S. IL-33 and IL-33 receptors in host defense and diseases. Allergol Int. 2010;59:143–60.
Palmer G, Gabay C. Interleukin-33 biology with potential insights into human diseases. Nat Rev Rheumatol. 2011;7(6):321–9. doi:10.1038/nrrheum.2011.53.
Pastorelli L, Garg RR, Hoang SB, Spina L, Mattioli B, Scarpa M, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A. 2010;107(17):8017–22. doi:10.1073/pnas.0912678107.
Jones LA, Roberts F, Nickdel MB, Brombacher F, McKenzie AN, Henriquez FL, et al. IL-33 receptor (T1/ST2) signalling is necessary to prevent the development of encephalitis in mice infected with Toxoplasma gondii. Eur J Immunol. 2010;40(2):426–36. doi:10.1002/eji.200939705.
Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci U S A. 2008;105(31):10913–8. doi:10.1073/pnas.0801898105.
Nile CJ, Barksby E, Jitprasertwong P, Preshaw PM, Taylor JJ. Expression and regulation of interleukin-33 in human monocytes. Immunology. 2010;130:172–80.
Buduneli N, Ozcaka O, Nalbantsoy A. Interleukin-33 levels in gingival crevicular fluid, saliva, or plasma do not differentiate chronic periodontitis. J Periodontol. 2012;83(3):362–8. doi:10.1902/jop.2011.110239.
Beklen A, Tsaous Memet G. Interleukin-1 superfamily member, interleukin-33, in periodontal diseases. Biotech Histochem. 2013; doi:10.3109/10520295.2013.832800.
Papathanasiou E, Teles F, Griffin T, Arguello E, Finkelman M, Hanley J, et al. Gingival crevicular fluid levels of interferon-gamma, but not interleukin-4 or −33 or thymic stromal lymphopoietin, are increased in inflamed sites in patients with periodontal disease. J Periodontal Res. 2014;49(1):55–61. doi:10.1111/jre.12078.
Pathirana RD, O’Brien-Simpson NM, Reynolds EC. Host immune responses to Porphyromonas gingivalis antigens. Periodontol 2000. 2010;52(1):218–37. doi:10.1111/j.1600-0757.2009.00330.x.
Ogawa T, Asai Y, Hashimoto M, Uchida H. Bacterial fimbriae activate human peripheral blood monocytes utilizing TLR2, CD14 and CD11a/CD18 as cellular receptors. Eur J Immunol. 2002;32:2543–50.
Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72(9):5041–51. doi:10.1128/IAI.72.9.5041-5051.2004.
Hashimoto M, Asai Y, Ogawa T. Separation and structural analysis of lipoprotein in a lipopolysaccharide preparation from Porphyromonas gingivalis. Int Immunol. 2004;16:1431–7.
Potempa J, Sroka A, Imamura T, Travis J. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: structure, function and assembly of multidomain protein complexes. Curr Protein Pept Sci. 2003;4(6):397–407.
Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One. 2008;3:e3331.
Rothmeier AS, Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol. 2012;34:133–49.
Lourbakos A, Potempa J, Travis J, D'Andrea MR, Andrade-Gordon P, Santulli R, et al. Arginine-specific protease from Porphyromonas gingivalis activates protease-activated receptors on human oral epithelial cells and induces interleukin-6 secretion. Infect Immun. 2001;69:5121–30.
Boisvert H, Duncan MJ. Clathrin-dependent entry of a gingipain adhesin peptide and Porphyromonas gingivalis into host cells. Cell Microbiol. 2008;10(12):2538–52. doi:10.1111/j.1462-5822.2008.01228.x.
Nakayama K, Yoshimura F, Kadowaki T, Yamamoto K. Involvement of arginine-specific cysteine proteinase (Arg-gingipain) in fimbriation of Porphyromonas gingivalis. J Bacteriol. 1996;178(10):2818–24.
Amano A, Nakagawa I, Okahashi N, Hamada N. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J Periodontal Res. 2004;39(2):136–42.
Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, et al. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J. 1996;314(Pt 3):1009–16.
Böhm SK, Khitin LM, Grady EF, Aponte G, Payan DG, Bunnett NW. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J Biol Chem. 1996;271:22003–16.
Belham CM, Tate RJ, Scott PH, Pemberton AD, Miller HR, Wadsworth RM, et al. Trypsin stimulates proteinase-activated receptor-2-dependent and -independent activation of mitogen-activated protein kinases. Biochem J. 1996;320(Pt 3):939–46.
Gorr SU. Antimicrobial peptides in periodontal innate defense. Front Oral Biol. 2012;15:84–98. doi:10.1159/000329673.
Vandamme D, Landuyt B, Luyten W, Schoofs L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol. 2012;280(1):22–35. doi:10.1016/j.cellimm.2012.11.009.
Puklo M, Guentsch A, Hiemstra PS, Eick S, Potempa J. Analysis of neutrophil-derived antimicrobial peptides in gingival crevicular fluid suggests importance of cathelicidin LL-37 in the innate immune response against periodontogenic bacteria. Oral Microbiol Immunol. 2008;23(4):328–35. doi:10.1111/j.1399-302X.2008.00433.x.
Chung WO, Hansen SR, Rao D, Dale BA. Protease-activated receptor signaling increases epithelial antimicrobial peptide expression. J Immunol. 2004;173(8):5165–70.
Alase A, Seltmann J, Werfel T, Wittmann M. Interleukin-33 modulates the expression of human beta-defensin 2 in human primary keratinocytes and may influence the susceptibility to bacterial superinfection in acute atopic dermatitis. Br J Dermatol. 2012;167(6):1386–9. doi:10.1111/j.1365-2133.2012.11140.x.
Sonawane A, Santos JC, Mishra BB, Jena P, Progida C, Sorensen OE, et al. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell Microbiol. 2011;13(10):1601–17. doi:10.1111/j.1462-5822.2011.01644.x.
Gutner M, Chaushu S, Balter D, Bachrach G. Saliva enables the antimicrobial activity of LL-37 in the presence of proteases of Porphyromonas gingivalis. Infect Immun. 2009;77(12):5558–63. doi:10.1128/IAI.00648-09.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this paper
Cite this paper
Tada, H., Shimauchi, H., Takada, H., Matsushita, K. (2015). Possible Roles of IL-33 in Periodontal Diseases: Porphyromonas gingivalis Induced IL-33 in Human Gingival Epithelial Cells. In: Sasaki, K., Suzuki, O., Takahashi, N. (eds) Interface Oral Health Science 2014. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55192-8_25
Download citation
DOI: https://doi.org/10.1007/978-4-431-55192-8_25
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55125-6
Online ISBN: 978-4-431-55192-8
eBook Packages: MedicineMedicine (R0)