
Abstract
Severe noninfectious adverse events (AEs) and transmission of pathogens by plasma-derived protein concentrates from the very beginning of their clinical use were threats for recipients (see Chap. 11 for additional information). “Standard IgG” preparations were the first available for clinical use. They were produced by the cold-ethanol fractionation methods and did not make an exception. Noninfectious severe AEs occurred while infectious AEs were rarely reported. Indeed, prior to the introduction of mass screening for infection markers of plasma donations, inadvertent transmission of HIV to recipients of factor VIII and factor IX concentrates did occur, while IgG concentrates obtained from the same plasma pool did rarely transmit HIV (Morgenthaler 2001). Rare transmissions were restricted to products not exposed to low pH. The very few incidences of HIV and some incidences of HCV transmission by IgG concentrates in the early 1990s together with many cases of coagulation factor concentrates transmitted viral disease clearly demonstrated the need to establish standardized measures to render plasma products pathogen safe. In the second half of the 1990s, authorities shifted regulatory emphasis from a scientific review of the processes to a focus on compliance to current good manufacturing practice (cGMP). The focus on cGMP compliance was applied to all aspects of plasma fractionation and the clinical use of plasma products. Court injunctions and warning letters were the consequences of this paradigm shift by authorities. This in turn resulted in a paradigm shift how the modern plasma industry operates (Steinhardt 1998).
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

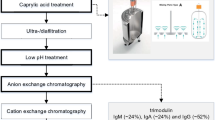
1 Introduction
From the very beginning of the clinical use of plasma-derived protin concentrates severe noninfectious adverse events (AEs) and transmission of pathogens by plasma-derived protein concentrates were threats to recipients (see Chap. 10 for additional information). First anti-infectious plasma potein concentrates were the “standard IgG” preparations. They were produced by the cold-ethanol fractionation methods and did not make an exception to the above: noninfectious severe AEs occurred while infectious AEs were rarely reported. Indeed, prior to the introduction of mass screening for infection markers of plasma donations, inadvertent transmission of HIV to recipients of factor VIII and factor IX concentrates did occur, while IgG concentrates obtained from the same plasma pool did rarely transmit HIV (Morgenthaler 2001). Rare transmissions were restricted to products not exposed to low pH. The very few incidences of HIV and some incidences of HCV transmission by IgG concentrates in the early 1990s, together with many cases of coagulation factor concentrates transmitted viral disease, clearly demonstrated the need to establish standardized measures to render plasma products pathogen safe. In the second half of the 1990s, authorities shifted regulatory emphasis from a scientific review of the processes to a focus on compliance to current good manufacturing practice (cGMP). The focus on cGMP compliance was applied to all aspects of plasma fractionation and the clinical use of plasma products. Court injunctions and warning letters were the consequences of this paradigm shift by authorities. This in turn resulted in a paradigm shift how the modern plasma industry operates (Steinhardt 1998).
The strict implementation of the recommendations by authorities resulted in today’s immunoglobulin concentrates in general being well tolerated and safe regarding the transmission of known blood-borne viruses, the agent of the transmissible spongiform encephalitis (TSE) or even emerging and reemerging zoonotic viruses. Furthermore, moden IgG concentrates have an excellent noninfectious adverse event record, particularly when used in chronic conditions.
Manufacturing of IgG concentrates is a flow of processes as outlined in Fig. 12.1. Along this flowchart, particular measures can serve to obtain clinical tolerability and to achieve reduction in pathogens potentially contaminating the starting material. Below the efforts undertaken to provide for patients IgG concentrates safe in all aspects are outlined.

Strongly simplified outline of fractionation of pooled plasma to pathogen free, well-tolerated IgG concentrates. Side fractions of the process are starting material for plasma products other than IgG. Level A of manufacturing: generating a large volume of plasma with optimal safety. Level B: plasma deprived of cryoprecipitate that contains relevant blood-clotting factors. Level C: a series of steps resulting in a crude IgG concentrate that is not tolerated intravenously. Level D: a sequence of steps rendering an IgG concentrate clinically well tolerated. Level E: lyophilized products are adjusted to the appropriate concentration, the excipient is added, the solution is filled into the vials, and the product is lyophilized. Liquid formulations are finalized similarly while leaving the freeze-drying process. Bottles with lyophilizate or IgG solutions are labeled and are ready for shipping
2 Plasma Fractionation: Regulatory Agencies’ Requests
Plasma protein concentrates, typically prepared from plasma of thousands of donors, inevitably are associated with the risk of transmitting blood-borne pathogens (see Chap. 10). Therefore, modern fractionation of human plasma for clinical use is embedded in a tight network of quality assurance (QA) put in place by recommendation of authorities. Good manufacturing practice (GMP) is part of a QA program (Slopecki et al. 2007). GMP is a guidance for industry. GMP ensures consistency in production. It describes the minimal requirement a product must fulfill for obtaining a marketing authorization and covers procedures for receipt of materials, production, packaging, labeling, quality control, release, storage, and distribution. The procedures and laws of GMP are defined by each country. GMP is a “moving target” and current GMP (cGMP) includes the most recent developments and knowledge of the field; as technology and information capabilities change, so do the requirements for maintaining good manufacturing practices. On a solid fundament of GMP, the elements for a state-of-the-art plasm product are:
-
Good clinical practice (GCP).
-
Good laboratory practice (GLP).
-
Standard operation procedures (SOPs).
-
Training of staff.
-
Donor selection, deferral and inventory hold.
-
Mass screening of donation for infection markers by serology and nucleic acid amplification technology (NAT) testing. Recently one company added isoagglutinin titer screening in plasma donations in order to obtain plasma pools for chromatographic fractionation low in isoagglutinins anti-A and anti-B (Siani et al. 2014).
-
In-process controls (production parameters and infection markers).
-
Validation studies (manufacturing steps and dedicated pathogen reduction).
-
Polishing steps (clinical tolerability and pathogen reduction).
-
Labeling and shipment.
-
Cleaning and segregation.
-
Look back.
-
Pharmacovigilance.
-
Audits by internal and external inspectors.
A plasma fractionation, which is compliant with GMP, has undergone process validation for each manufacturing step. Process validation helps to ensure that systems are performing in the intended manner. Process validation ensures that the product has the required potency, purity, and safety. The parameters of a single process step are defined on a laboratory scale. The parameters obtained during the validation process help to define the elements of process control and provide a mechanism for ongoing quality assurance, quality control, training, competency review, and continuous improvement (Preti 1999).
Ensuring pathogen safety of a plasma product needs a tripod of measures: (1) donor selection, (2) mass screening for pathogen markers, and (3) pathogen elimination and inactivation during manufacturing. As an example how to implement pathogen safety of plasma products, the view of the European Medicines Agency is cited: “The safety of a product has to be demonstrated experimentally by validation studies on a laboratory scale. Such studies are based on the principle of deliberate contamination of the intermediate product by model viruses at given stages of the manufacturing process and monitoring the reduction in infectivity produced by the production step under consideration.” At the time when GMP was introduced, well-established fractionation procedures were in place. For validation studies, the processes had to be downscaled to a laboratory scale. In a first step, it had to be demonstrated that the scaled-down process mimics the process in production, i.e., ideally has identical process parameters. Furthermore, authorities request at least two “dedicated” virus removal/inactivation steps in sequence during the fractionation. The two methods should represent two different principles of action thereby assuring that virus reduction is complementary.
When fractionating plasma on the fundaments of cGMP and applying the “full package” of complementary recommendations a state-of-the-art immunoglobulin preparation results.
Any activity from plasma collection to delivery at the door of a customer that is not performed and documented according to the standards may have severe consequences to the noncompliant company. Deficiencies in adherence to GMP detected during inspections of companies can lead to consent decrees, and severe deviations can have the consequence of a company forced to cease distribution of its products (http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2010/09/WC500097037.pdf; https://wayback.archive-it.org/7993/20161023082812/http://www.fda.gov/mw/ucm223968.htm ; both accessed April 2017).
The leading regulatory agencies, which are enforcing the adherence to QA fractionation recommendations, are the US Food and Drug Administration (FDA; https://www.fda.gov/) and the European Medicines Agency (EMA; www.ema.europa.eu), while in all countries own agencies govern the enforcement of recommendations. A list of regional offices for Africa, for the Americas, for the Eastern Mediterranean, for Europe, for Southeast Asia, and for the Western Pacific can be found under: http://www.who.int/medicines/areas/quality_safety/regulation_legislation/ListMRAWebsites.pdf (accessed April 2017).
3 Plasma Collection: The Starting Point for Quality and Safety of a Plasma-Derived Product
IgG concentrates belong to the stable blood products. Ensuring quality of an IgG concentrate starts with collection of plasma, its correct handling, and cautious pooling.
Donor selection (level A, Fig. 12.1): Plasma fractionated to current best practice IgG concentrates all are from donors donating blood or plasma from free will. There are two types of donations: whole blood from which plasma is collected after centrifugation (recovered plasma) or plasma obtained by apheresis device (apheresis or source plasma) (Table 12.1). There is epic, for several decades’ ongoing debate which of the two ways of plasma collection is of higher ethical and better biological quality. There is no reason to step into this discussion in this book chapter. However, it has to be acknowledged that apheresis plasma collection of volumes at the upper end of what is allowed might have an influence on protein composition of the donated plasma (Laub et al. 2010). As GMP covers minimal requirements for quality, and because having been accused remunerated donations being of lesser quality and safety, the apheresis plasma collecting and fractionating industry has introduced additional voluntary regulations. These are the International Quality Plasma Program (IQPP) introducing qualified donor standard, implementation of a donor deferral registry, and a drug abuse screening and the Quality Standards of Excellence, Assurance, and Leadership (QSEAL). QSEAL covers control on incoming plasma, inventory hold, NAT testing, intermediates purchased form external suppliers, recovered plasma specification, qualified donor, and viral markers (http://www.pptaglobal.org/safety-quality/standards/qseal; accessed June 2017). With the implementation of these voluntary standards, no relevant differences are found in pathogen marker frequencies of either type of plasma donation (EMEA 2002). In summary, the main goal of donor selection is to ensure that donors with high risk are excluded form donating, or their donation is withheld from further processing (Table 12.2). The donor is informed if HIV positive.
Finally yet importantly, plasma donations require careful handling to prevent formation and accumulation of vasoactive (e.g., prekallikrein activator (PKA)) or coagulation promoting (e.g., coagulation factor FXIa) substances. Due to their physicochemical properties, their removal during the manufacturing process may remain imperfect, and they may induce severe adverse events.
Mass screening of donations (level A, Fig. 12.1): Mass screening is the measure to exclude donations potentially contaminated by pathogens such as virus(es). Stable blood products inherit a particular risk for transmission of viruses because the staring material being a pool of thousands of individual donations (Table 12.3). Parameters of mass screening for virus markers are outlined in Table 12.4 and include serological and nucleic acid amplification technology (NAT) testing. NAT testing in addition to serological screening was introduced in order to shorten the time period of “window donations.” During a window period, a donor already harbors the infective agent and is infective while seroconversion has not occurred yet, i.e., antibody formation has not set in, or antibodies are at a titer not detectable by validated serological methods. By introducing NAT testing, the estimated risk of an undetected infectious donation entering the blood supply dropped as given in Fig. 12.2. In addition to the minimal requirements given by GMP, most manufacturers request human parvovirus B19 and HAV NAT testing of plasma donations.

Estimated risk of an undetected donation entering the blood supply when mass screening is by serology only or NAT is performed in addition (Offergeld et al. 2005). The reciprocal of risk rate of undetected infectious donations is given; the higher the bar, the lower the risk rate
NAT testing is performed in mini-pools and largely can be automated. Aliquots from bar code labeled pilot tubes are transferred to, e.g., a micro titer plate. In such an X/Y layout, each aliquot is unequivocally associated to an individual donation. Starting NAT testing, aliquots from the aliquots are pooled, undergo validated amplification of part(s) of pathogen’s genome, and the resulting genome equivalents are quantified. The size of the mini-pool depends on amplification efficiency and minimal infectious dose of the given pathogen. In case a mini-pool is reactive, pooling is repeated, this time by rows and columns. From the row and the column pool being reactive, the X/Y layout allows the identification of the reactive donation. The contaminated donation is withheld from pooling and is destroyed safely.
Plasma fractionation starts with pooling of individual donations to an appropriate volume. Before starting the fractionation process, an in-process control is performed to ascertain absence of potential contaminating viruses (Table 12.4). This reduces the risk of pathogen transmission and reduces the financial risk of losing an entire lot of IgG concentrate.
4 Fractionation Methods in Use to Obtain From Plasma Well-Tolerated, Highly Pure IgG Concentrates
Over many decades, the development of IgG fractionation was driven to achieve intravenous tolerability and increase recovery and purity of the concentrates. Today two different main methods are in place how to fractionate plasma: the cold-ethanol fractionation and the ion-exchange chromatography process (Fig. 12.1). Before applying either method, frozen plasma is thawed at 0 °C. The part remaining insoluble at 0 °C is separated and represents the cryoprecipitate that harbors the blood coagulation factors (level A to B in Fig. 12.1). The supernatant is the cryo-poor plasma that contains a wide array of plasma proteins, including immunoglobulins and albumin.
Cold-ethanol fractionation is either according to Cohn/Oncley or to Kistler-Nitschmann (Oncley et al. 1949; Nitschmann et al. 1954) (see Chap. 10 as well). From cryo-poor plasma, a precipitate is obtained which contains the immunoglobulins (level B, Fig. 12.1). The supernatant is the starting material for albumin fractionation. Further suspension/precipitation steps of the precipitate containing the immunoglobulins finally end up what is termed “IgG bulk” (level C, Fig. 12.1). This crude IgG concentrate then undergoes various polishing steps (Table 12.5). Polishing steps and the final formulation are those differing the most among various companies (levels D and E, Fig. 12.1).
The early “standard” IgG concentrates underwent only few polishing steps of the “IgG bulk” material. They contained aggregates that rendered them not tolerable when applied intravenously. Typically, these i.m. products were of a solution strength of 16–16.5%. By introducing virus inactivation/removal steps to the original manufacturing process, some of these products survived to our days.
A “first generation” of IgG products for intravenous use (IVIGs) was deprived of unwanted Fc-effector functions by harsh enzyme treatment. The digested IgG molecules were heavily impaired in their biological functions however were well tolerated. One product still available posted on a Japanese website (see Chap. 13). For a “second generation” of IVIGs, intact IgG molecules were chemically modified in order to prevent overt spontaneous complement activation in the recipients’ circulation. Only a very few of these chemically modified products are still available in some regional markets. They are mentioned in Chap. 13 of this book. The “third generation” of IVIGs was made intravenously tolerable by gentle polishing steps (level D, Fig. 12.1).
Although in many aspects excellent, the two cold-ethanol fractionation methods reach their limits when pushing for improved recovery without loss of purity. For this reason, the plasma industry is moving toward the ion-exchange chromatography fractionation of IgG. This manufacturing method allows relative high recoveries at high purity. Before cryo-poor plasma or the intermediate fraction deprived of albumin can be fed onto ion-exchange columns, they must be free of lipids (level B, Fig. 12.1). The most widely used technique to get rid of lipids is by octanoic acid (=caprylic acid) precipitation. Caprylic acid precipitation was the first to be used in combination with DEAE cellulose columns for the isolation of IgG from plasma at good yield (Steinbuch and Audran 1969).
During chromatographic fractionation, lipid-free plasma undergoes several anion- and/or cation-exchange chromatography steps (Bertolini 1998; Dhainaut et al. 2013; Cramer et al. 2009; Trejo et al. 2003). The resulting IgG solutions again undergo polishing steps in order to achieve tolerability (Table 12.5).
With some chromatographically obtained preparations, an elevated frequency of hemolytic reactions emerged. These adverse events were supposed being related to the presence of higher titers of isoagglutinins than seen in products obtained with the cold-ethanol fractionation. In order to reduce levels of isoagglutinins anti-A and anti-B, a particular polishing step was introduced by some manufacturers: immunoaffinity chromatography using columns with corresponding trisaccharides coupled to the matrix (Dhainaut et al. 2013; Höfferer et al. 2015; Späth et al. 2015).
5 Widening the Pathogen Safety Margins During Fractionation and Polishing
Donor selection, mass screening, and implementation of added voluntary quality measures are important for pathogen safety of a plasma product. However, by far they are not sufficient for an optimal safety bill. “Building in” safety to the fractionation process is what finally can render a product pathogen safe.
Among the stable plasma products, the manufacturing of IgG concentrates offers the most steps effective enough to reduce or inactivate viruses without harming too much the biologic function of the molecules. Indeed, IgG concentrates largely remained free from transmitting viruses. This was remarkable as patients with impaired immune defense may require monthly administration of immunoglobulins lifelong, or patients with autoimmune/inflammatory diseases might require high to very high doses of immunoglobulin therapy sometimes over a prolonged period and nevertheless remained virus free. Beside donor selection and mass screening the third pillar on which pathogen safety is built on is the reduction of potentially present pathogens during manufacturing (levels B and C, Fig. 12.1). Either an existing fractionation step is validated accordingly, or dedicated virus inactivation/removal steps are introduced to the manufacturing process. Aspects of the validation process are outlined in Table 12.6.
There are three, in their principles different, mechanisms that contribute to the elimination or inactivation of viruses potentially present during manufacturing of plasma products (Kempf et al. 2007), namely:
-
1.
Inactivation
-
Solvent/detergent (S/D) treatment: disruption of virus envelope
-
Caprylic (octanoic) acid treatment
-
Treatment a low pH: destructive conformational changes of structural proteins
-
Heat: dry or wet (wet = pasteurization), disruption of envelope and destructive conformational change of structural proteins (e.g. capsid proteins)
-
-
2.
Elimination based on size: virus filtration (formerly termed nanofiltration). Elimination at large scale of possibly contaminating pathogens based on size is the most recent technique to free plasma products form pathogens. Virus filtration at large scale was introduced by the Central Laboratory of the Swiss Red Cross, Blood Transfusion Service in the late 1990s (virus filtration on a modest size (Stucki et al. 1997); virus filtration at large scale: submission dossier for Sandoglobulin NF in 1999). With the progress in manufacturing of filters with smaller and smaller pores, virus filtration has become a universal key process in assuring pathogen safety—size matters only.
-
3.
Partitioning, e.g., virus in the precipitate while the supernatant, is processed further. Filtration with filter aids can take advantage of the filter aids tightly binding and adsorbing viruses in precipitates and despite their small size retaining them (technical term: depth filtration). Partitioning involving filtration techniques are not to confound with virus filtration that is a dedicated virus removal principle.
The highest level of product safety is achieved when all three principles are applied in a stepwise and diligent manner during the fractionation process (Table 12.8). As an example the LRFs obtained by various methods are listed in Table 12.9. LFRs obtained on basis of varying principles are additive in defining the safety margin of an entire manufacturing process.
The results published by various manufacturers of the various virus inactivation and removal steps allow to conclude that by applying the various techniques sequentially:
-
Inactivation of enveloped viruses is usually achieved well.
-
Current inactivation methods show limited efficiency for non-enveloped viruses.
-
Virus filtration has the potential to completely remove the smallest and most robust non-enveloped viruses known.
-
Partitioning processes additionally contribute to viral safety.
The combination of these pathogens elimination methods also provides confidence that the various fractionation processes can cope with newly emerging viruses.
6 Emerging Human Pathogens
So far, no proven transmission by IgG concentrates of (re-)emerging viruses in the last two decades occurred. This might be credited to the tightly implemented QA framework. The (re-)emerging pathogens are predominantly of zoonotic nature. Hence, their reservoirs are animals and their (re-)emergence is associated with hygienic conditions. Transmission of enveloped zoonotic viruses over the species barrier with severe consequences for man have been described for Chikungunya virus, MERS coronavirus, avian influenza virus (bird flu; H5N1), SARS coronavirus, West Nile virus, Ebola virus, Zika virus, others. The presence of these viruses in blood donors were reported for West Nile virus, SARS coronavirus and Zika virus which persists in the cellular blood compartment. Validation studies have assured the safety regarding transmission by plasma products of relevant enveloped zoonotic viruses (Table 12.10). In general, enveloped viruses can relatively easily be inactivated, e.g., by solvent/detergent treatment.
For non-enveloped viruses, gene mutations are a prerequisite to cross the species barrier. Hepatitis E virus (HEV) is a non-enveloped zoonotic virus of pigs. Four genotypes are known. Genotypes 3 and 4 are recognized as zoonotic pathogens in industrialized countries. Transmissions are by pork blood (in rare cooked meat). Transmission of HEV by plasma exchange has been reported (Mallet et al. 2016). Prevention of HEV transmission is relevant as infection can have severe neurological consequences, even in immunocompetent individuals (Blasco-Perrin et al. 2015; Higuchi et al. 2015; Pérez Torre et al. 2015; Scanvion et al. 2017). Elimination or inactivation of HEV during the manufacturing of plasma products apparently is sufficient in preventing pathogen transmission (Table 12.10).
In general non-enveloped viruses do mutate slowly, with one known exception, the DNA of Parvoviridae which shows a similar mutation frequency to RNA viruses (Boschetti et al. 2005; Shackelton and Holmes 2006). Under distinct immune and adaptive pressures, specific amino acid changes in parvoviruses endowed tropism shifts in the animal world (Lopez-Bueno et al. 2006). Hence, new variants/species tropisms have to be expected. Theoretically, small non-enveloped viruses represent the biggest threat for transfusion incidences in the future, and hopefully the technique of virus filtration is sufficient or will be improved to eliminate eventually emerging Parvoviridae with human tropism.
7 Transmissible Spongiform Encephalopathies (TSEs)
Also of animal origin is the agent of the variant Creutzfeldt-Jakob disease (vCJD), a TSE. vCJD with great certainty was transmitted from cow (mad cow disease or bovine spongiform encephalitis = BSE) to humans (Bruce et al. 1997). BSE emerged due to an “optimized” production process of animal carcass-derived material fed to cows (Wilesmith et al. 1991). Other forms of human spongiform encephalitides exist and can be iatrogenic/sporadic, inherited/genetic, or acquired/infectious (Table 12.11).
Spongiform encephalitides are mediated by the misfolded isoform “scrapie” of a normal cellular prion protein, PrPC which is ubiquitous in cells and is a protein highly conserved over the evolution. The misfolded “scrapie” prion protein (PrPSc) results in fatal neurodegenerative diseases of mammals. Although a protein-only agent, PrPSc is “infectious” in a sense that the misfolded protein might induce conformation changes of the normal PrPC resulting in sticky rod-like and fibrillary particles accumulating in the brain.
Serum, plasma, and leukocytes from vCJD patients might harbor infectious PrPSc. Indeed, transmission of PrPSc has been reported in three symptomatic and one non-symptomatic cases of non-leukocyte-depleted red blood cell concentrates (Hewitt et al. 2006; Peden et al. 2004). Measures to eliminate possibly contaminating PrPSc during plasma fractionation became mandatory. It is accepted that inactivation of PrPSc is not possible without destruction of the biological activity of a plasma product. Hence, elimination by partitioning or exclusion by size remains the two possibilities (Cai et al. 2002). Validation studies were performed at different manufacturers’ site with different spikes and different detection systems. The manufacturing steps studied for removal capacity of PrPSc included precipitation, adsorption, chromatography, and filtration, as well as combined steps. They proved to be highly potent in removal of the TSE agents possibly contaminating plasma (Cai et al. 2013). Reduction by individual steps in sequence was additive (Table 12.12). Furthermore, the declining incidence of vCJD, the stringent donor selection due to geographic donor deferral policy, and the additive nature of removal steps render the agent of vCJD unlikely to be transmitted by plasma products. Indeed, no proven transmission of vCJD by IgG concentrates was ever reported (Ritchie et al. 2016), even not when the administration of a possibly PrPSc contaminated IgG concentrate occurred (El-Shanawany et al. 2009). Ascertained transmission by other plasma products has not been described neither.
The sporadic/iatrogenic CJD (sCJD) is not the same as vCJD (Table 12.11). sCJD represents about 85% of all spongiform encephalitides of humans and affects elderly people. There exist several surveillance studies on the transmission of sCJD by blood and blood products. These are conducted by the American Red Cross, in the UK and in France (Crowder et al. 2017; Martin and Trouvin 2013; Urwin et al. 2016). These studies did not report sCJD in patient populations treated with blood and plasma products. This was the basis not to consider sCJD transmissible by blood transfusion or by plasma products. However, the Emerging Infectious Disease Journal in May 2017 posted in electronic form a report on two patients receiving coagulation factor concentrates and dying from sCJD. Authors conclude (citation): “A causal link between the treatment with plasma products and the development of sCJD has not been established, and the occurrence of these cases may simply reflect a chance event in the context of systematic surveillance for CJD in large populations.” It will be of outmost importance whether and how many new cases will be reported in future.
8 Completing the Full Package for a Safe and Efficacious Immunoglobulin Concentrate
Final formulation: To ensure stability/tolerability of a product over its shelf life, the right selection of stabilizers is of particular importance. Products can by lyophilized (=freeze-dried). The basic physicochemical process of freeze-drying is the replacement of the layer of water surrounding the IgG molecules. This layer, which is also termed “hydration shell” or “hydration sphere,” keeps the IgG molecules in solution (Makarov et al. 2002). In dehydrated products, e.g., oxidation of the preparation is largely prevented, and aggregation or dimer formation is suppressed. Hence, the excipient only to a part serves stabilization of a freeze-dried product, while its major role is to make sure the lyophilized protein can be reconstituted entirely within reasonable time (no aggregates remaining). The compounds that can be injected intravenously with the best physicochemical properties to simulate water are sugars followed by some amino acids. Although associated with osmotic nephrosis in patients at risk, several lyophilized products containing sucrose are still on some markets (see also Chap. 13).
In response for the request of more convenient handling, the liquid preparations were developed. In liquid preparations, aging and continuous interaction of molecules are inherent. The stabilizer has to preserve the characteristics without hampering their quality by preventing oxidation and IgG-IgG interactions, e.g., limiting IgG dimer formation. The most widely used stabilizers for liquid preparations are glycine, L-proline, maltose, and sorbitol.
Cleaning and sanitation of a production line: Prevention of cross-contamination through surface-adsorbed infectious agent is a measure to be taken under cGMP. Cleaning validations involving infectious agents are problematic, e.g., because the cleaning process has to be scaled down. This in most cases is impossible, e.g., because the rheological properties are different on a small scale. Typically NaOH or sodium hypochlorite (NaClO) are used for plant sanitation after a run and at appropriate concentrations are sufficient to destroy virus infectivity. However, how about the TSE agent of vCJD? It needed a particular effort to show that there are sanitation measures that can destroy the TSE agent PrPSc. PrPres served this purpose (Table 12.13). The validation strategy was to show that PrPres after NaOH/NaClO exposure becomes on one hand noninfectious and on the other hand sensitive to proteinase K digestion. NaOH and NaClO treatment both destroy PrPres even at low NaOH and NaClO concentrations. Regulatory agencies consider current cleaning regimes adequate to assure batch-to-batch segregation.
Traceability: GMP requirements for blood and plasma derivatives include the traceability of batches from donor to recipient. The industry is responsible for interlinking a given batch of plasma product with information such as responses of the donor has given on the medical questionnaire, results of mass screening and pooling information. Traceability also has to interconnect batches and the hospital where the product was delivered. It is the responsibility of the hospital to complete traceability by documenting which IVIG batch has been given to which individual patient(s). This enables “look backs” in case any problem should be identified at the donor or recipient end. The basis of the traceability is a bar code identification system. A proper traceability system ensures that each lot can be recalled, should this be necessary. It further allows the handling of post-donation information and pharmacovigilance in an efficient and safe manner.
Obtaining the final proof of clinical efficacy and pathogen safety of an IgG concentrate: Quality assurance through surveillance programs extends product quality after its distribution. Pharmacovigilance, post-marketing studies and surveillance programs are the pillars on which the final proof for the quality of an IgG concentrate stands. Pharmacovigilance ensures the continued safety of medicinal products in use by collecting, monitoring, and assessing any type of adverse drug reactions related to the product (Ball et al. 2016), http://ec.europa.eu/health/human-use/pharmacovigilance_en. For some infections, incubation time might be long. Surveillance programs are currently the only means to conclusively obtain long-ranging safety information. Clinicians are encouraged to report adverse events, especially those that are unexpected or unusual.
References
Ball R, Robb M, Anderson SA, Dal PG. The FDA’s sentinel initiative—a comprehensive approach to medical product surveillance. Clin Pharmacol Ther. 2016;99:265–8.
Barjas-Castro ML, Angerami RN, Cunha MS, Suzuki A, Nogueira JS, Rocco IM, Maeda AY, Vasami FG, Katz G, Boin IF, Stucchi RS, Resende MR, Esposito DL, de Souza RP, da Fonseca BA, Addas-Carvalho M. Probable transfusion-transmitted Zika virus in Brazil. Transfusion. 2016;56:1684–8.
Bertolini J. Chromatographic purification of immunoglobulins. Downstream. 1998;31:21–2.
Blasco-Perrin H, Cintas P, Abravanel F, Gérolami R, D’Alteroche L, Raynal J-N, Alric L, Dupuis E, Prudhomme L, Vaucher E, Couzigou P, Liversain J-M, Buscail L, Bureau C, Vinel J-P, Kamar N, Izopet J, Péron JM. Neurologic disorders in immunocompetent patients with autochthonous acute hepatitis E. Emerg Infect Dis. 2015;21:1928–34.
Blümel J, Musso D, Teitz S, Miyabayashi T, Boller K, Schnierle BS, Baylis SA. Inactivation and removal of Zika virus during manufacture of plasma-derived medicinal products. Transfusion. 2017;57:790–6.
Boschetti N, Stucki M, Späth PJ, Kempf C. Virus safety of intravenous immunoglobulin: future challenges. Clin Rev Allergy Immunol. 2005;29:333–44.
Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501.
Cai K, Gröner A, Dichtelmüller HO, Fabbrizzi F, Flechsig E, Gajardo R, von Hoegen I, Jorquera JI, Kempf C, Kreil TR, Lee DC, Moscardini M, Polsler G, Roth NJ. Prion removal capacity of plasma protein manufacturing processes: a data collection from PPTA member companies. Transfusion. 2013;53:1894–905.
Cai K, Miller JL, Stenland CJ, Gilligan KJ, Hartwell RC, Terry JC, Evans-Storms RB, Rubenstein R, Petteway SR, Lee DC. Solvent-dependent precipitation of prion protein. Biochim Biophys Acta. 2002;1597:28–35.
Cramer M, Frei R, Sebald A, Mazzoletti P, Maeder W. Stability over 36 months of a new liquid 10% polyclonal immunoglobulin product (IgPro10, Privigen©) stabilized with L-proline. Vox Sang. 2009;96:219–25.
Crowder LA, Schonberger LB, Dodd RY, Steele WR. Creutzfeldt-Jakob disease lookback study: 21 years of surveillance for transfusion transmission risk. Transfusion. 2017;57(8):1875–8. [Epub ahead of print].
D’Aignaux JH, Costagliola D, Maccario J, De Villemeur TB, Brandel JP, Deslys JP, Hauw JJ, Chaussain JL, Agid Y, Dormont D, Alpérovitch A. Incubation period of Creutzfeldt-Jakob disease in human growth hormone recipients in France. Neurology. 1999;53:1197–201.
Dhainaut F, Guillaumat PO, Dib H, Perret G, Sauger A, De Coupade C, Beaudet M, Elzaabi M, Mouthon L. In vitro and in vivo properties differ among liquid intravenous immunoglobulin preparations. Vox Sang. 2013;104:115–26.
El-Shanawany T, Jolles S, Unsworth DJ, Williams P. A recipient of immunoglobulin from a donor who developed vCJD. Vox Sang. 2009;96:270.
EMEA. CPMP Position Statement: Non-remunerated and remunerated donors: Safety and supply of plasma-derived medicinal products. European Agency for the Evaluation of Medical Products EMEA/CPMP/BWP/1818/02; 2002. p. 1–2.
Farcet MR, Lackner C, Antoine G, Rabel PO, Wieser A, Flicker A, Unger U, Modrof J, Kreil TR. Hepatitis E virus and the safety of plasma products: investigations into the reduction capacity of manufacturing processes. Transfusion. 2016;56:383–91.
Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Three reported cases of variant Creutzfeldt-Jakob disease transmission following transfusion of labile blood components. Vox Sang. 2006;91:348.
Higuchi M-A, Fukae J, Tsugawa J, Ouma S, Takahashi K, Mishiro S, Tsuboi Y. Dysgeusia in a patient with Guillain-Barre syndrome associated with acute hepatitis E: a case report and literature review. Intern Med. 2015;54:1543–6.
Höfferer L, Glauser I, Gaida A, Willimann K, Marques Anuntes A, Siani B, Wymann S, Widmer E, El Menyawi I, Bolli R, Spycher M, Imboden M. Isoagglutinin reduction by a dedicated immunoaffinity chromatography step in the manufacturing process of human immunoglobulin products. Transfusion. 2015;55(Suppl 2):S117–21.
Jackson GS, Burk-Rafel J, Edgeworth JA, Sicilia A, Abdilahi S, Korteweg J, Mackey J, Thomas C, Wang G, Schott JM, Mummery C, Chinnery PF, Mead S, Collinge J. Population screening for variant Creutzfeldt-Jakob disease using a novel blood test: diagnostic accuracy and feasibility study. JAMA Neurol. 2014;71:421–8.
Kempf C, Stucki M, Boschetti N. Pathogen inactivation and removal procedures used in the production of intravenous immunoglobulins. Biologicals. 2007;35:35–42.
Kreil TR, Berting A, Kistner O, Kindermann J. West Nile virus and the safety of plasma derivatives: verification of high safety margins, and the validity of predictions based on model virus data. Transfusion. 2003;43:1023–8.
Kühnel D, Müller S, Pichotta A, Radomski KU, Volk A, Schmidt T. Inactivation of Zika virus by solvent/detergent treatment of human plasma and other plasma-derived products and pasteurization of human serum albumin. Transfusion. 2017;57:802–10.
Laub R, Baurin S, Timmerman D, Branckaert T, Strengers P. Specific protein content of pools of plasma for fractionation from different sources: impact of frequency of donations. Vox Sang. 2010;99:220–31.
Leydold SM, Farcet MR, Kindermann J, Modrof J, Polsler G, Berting A, Howard MK, Barrett PN, Kreil TR. Chikungunya virus and the safety of plasma products. Transfusion. 2012;52:2122–30.
Lopez-Bueno A, Villarreal LP, Almendral JM. Parvovirus variation for disease: a difference with RNA viruses? Curr Top Microbiol Immunol. 2006;299:349–70.
Makarov V, Pettitt BM, Feig M. Solvation and hydration of proteins and nucleic acids: a theoretical view of simulation and experiment. Acc Chem Res. 2002;35:376–84.
Mallet V, Sberro-Soussan R, Vallet-Pichard A, Roque-Afonso AM, Pol S. Transmission of hepatitis E virus by plasma exchange: a case report. Ann Intern Med. 2016;164:851–2.
Martin M, Trouvin JH. Risk of transmission of Creutzfeldt-Jakob disease via blood and blood products. The French risk-analysis over the last 15 years. Transfus Clin Biol. 2013;20:398–404.
Morgenthaler JJ. Securing viral safety for plasma derivatives. Transfus Med Rev. 2001;15:224–33.
Motte A, Roquelaure B, Galambrun C, Bernard F, Zandotti C, Colson P. Hepatitis E in three immunocompromized children in southeastern France. J Clin Virol. 2012;53:162–6.
Nitschmann H, Kistler P, Lergier W. Vereinfachtes Verfahren zur Gewinnung von humanem Albumin und γ-Globulin aus Blutplasma mittels Alkoholfällung - [Simplified method for isolation of human albimin and γ-globulin from plasma using the ethnol precipitation method]. Helv Chim Acta. 1954;37:866–73.
Nogueira ML, Estofolete CF, Terzian ACB, Mascarin do Vale EPB, da Silva RCMA, da Silva RF, Ramalho HJ, Fernandes Charpiot IMM, Vasilakis N, Abbud-Filho M. Zika virus infection and solid organ transplantation: a new challenge. Am J Transplant. 2017;17:791–5.
Offergeld R, Faensen D, Ritter S, Hamouda O. Human immunodeficiency virus, hepatitis C and hepatitis B infections among blood donors in Germany 2000–2002: risk of virus transmission and the impact of nucleic acid amplification testing. Euro Surveill. 2005;10:8–11.
Oncley JL, Melin M, Richert DA, Cameron JW, Gross PM. The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and β1-lipoprotein into subfractions of human plasma. J Am Chem Soc. 1949;71:541–50.
Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527–9.
Pérez Torre P, Acebrón F, Monreal E, Viedma GE, Martinez UP, FC E+®v, Alonso CA, Avil+®s O, I, Buis+ín CJ. Guillain-Barré syndrome following acute hepatitis E in a Western European man. Eur J Neurol. 2015;22:291.
Preti RA. Process validation. Cytotherapy. 1999;1:481–3.
Rabenau HF, Biesert L, Schmidt T, Bauer G, Cinatl J, Doerr HW. SARS-coronavirus (SARS-CoV) and the safety of a solvent/detergent (S/D) treated immunoglobulin preparation. Biologicals. 2005;33:95–9.
Ritchie DL, Gibson SV, Abee CR, Kreil TR, Ironside JW, Brown P. Blood transmission studies of prion infectivity in the squirrel monkey (Saimiri sciureus): the Baxter study. Transfusion. 2016;56:712–21.
Scanvion Q, Perez T, Cassim F, Outteryck O, Lanteri A, Hatron PY, Lambert M, Morell-Dubois S. Neuralgic amyotrophy triggered by hepatitis E virus: a particular phenotype. J Neurol. 2017;264:770–80.
Shackelton LA, Holmes EC. Phylogenetic evidence for the rapid evolution of human B19 erythrovirus. J Virol. 2006;80:3666–9.
Siani B, Willimann K, Wymann S, Marques AA, Widmer E. Isoagglutinin reduction in human immunoglobulin products by donor screening. Biol Ther. 2014;4:15–26.
Slopecki A, Smith K, Moore S. The value of good manufacturing practice to a blood service in managing the delivery of quality. Vox Sang. 2007;92:187–96.
Späth PJ, Granata G, La Marra F, Kuijpers TW, Quinti I. On the dark side of therapies with immunoglobulin concentrates. The adverse events. Front Immunol. 2015;6:11.
Steinbuch M, Audran R. The isolation of IgG from mammalian sera with the aid of caprylic acid. Arch Biochem Biophys. 1969;134:279–84.
Steinhardt B. Blood plasma safety. Plasma product risks are low if good manufacturing practices are followed. B-278739. Washington: GAO/HEHS; 1998. p. 1–47. Ref Type: Report.
Stucki M, Moudry R, Kempf C, Omar A, Schlegel A, Lerch PG. Characterisation of a chromatographically produced anti-D immunoglobulin product. J Chromatogr B Biomed Sci Appl. 1997;700:241–8.
Tamura A, Shimizu YK, Tanaka T, Kuroda K, Arakawa Y, Takahashi K, Mishiro S, Shimizu K, Moriyama M. Persistent infection of hepatitis E virus transmitted by blood transfusion in a patient with T-cell lymphoma. Hepatol Res. 2007;37:113–20.
Trejo SR, Hotta JA, Lebing W, Stenland C, Storms RE, Lee DC, Li H, Petteway S, Remington KM. Evaluation of virus and prion reduction in a new intravenous immunoglobulin manufacturing process. Vox Sang. 2003;84:176–87.
Urwin P, Thanigaikumar K, Ironside JW, Molesworth A, Knight RS, Hewitt PE, Llewelyn C, Mackenzie J, Will RG. Sporadic Creutzfeldt-Jakob disease in 2 plasma product recipients, United Kingdom. Emerg Infect Dis. 2017;23:893.
Urwin PJ, Mackenzie JM, Llewelyn CA, Will RG, Hewitt PE. Creutzfeldt-Jakob disease and blood transfusion: updated results of the UK Transfusion Medicine Epidemiology Review Study. Vox Sang. 2016;110:310–6.
Venturi G, Zammarchi L, Fortuna C, Remoli ME, Benedetti E, Fiorentini C, Trotta M, Rizzo C, Mantella A, Rezza G, Bartoloni A. An autochthonous case of zika due to possible sexual transmission, Florence, Italy, 2014. Eurosurveillance. 2016;21:1–4.
Wilesmith JW, Ryan JB, Atkinson MJ. Bovine spongiform encephalopathy: epidemiological studies on the origin. Vet Rec. 1991;128:199–203.
Yang H, Huang Y, Gregori L, Asher DM, Bui T, Forshee RA, Anderson SA. Geographic exposure risk of variant Creutzfeldt-Jakob disease in US blood donors: a risk-ranking model to evaluate alternative donor-deferral policies. Transfusion. 2017;57:924–32.
Yunoki M, Urayama T, Yamamoto I, Abe S, Ikuta K. Heat sensitivity of a SARS-associated coronavirus introduced into plasma products. Vox Sang. 2004;87:302–3.
Acknowledgments
The help of Christoph Kempf, University of Berne, is greatly appreciated, as well as information kindly provided by Roland Hubner, The Federal Public Service (FPS) Health, Food Chain and Environment, Belgium.
Some web sites for additional information:
http://www.ukhcdo.org/patient-information
http://case.edu/med/pathology/centers/npdpsc
http://www.who.int/medicines/areas/quality_safety/regulation_legislation/ListMRAWebsites.pdf
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Späth, P.J. (2018). Essentials of the Production of Safe and Efficacious State-of-the-Art Polyclonal IgG Concentrates. In: Imbach, P. (eds) Antibody Therapy. Springer, Cham. https://doi.org/10.1007/978-3-319-68038-5_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-68038-5_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-68037-8
Online ISBN: 978-3-319-68038-5
eBook Packages: MedicineMedicine (R0)