
Abstract
Circadian clocks are biologic oscillators present in all photosensitive species that produce 24-h cycles in the transcription of rate-limiting metabolic enzymes in anticipation of the light–dark cycle. In mammals, the clock drives energetic cycles to maintain physiologic constancy during the daily switch in behavioral (sleep/wake) and nutritional (fasting/feeding) states. A molecular connection between circadian clocks and tissue metabolism was first established with the discovery that 24-h transcriptional rhythms are cell-autonomous and self-sustained in cultured fibroblasts, and that clocks are present in most tissues and comprise a robust temporal network throughout the body. A central question remains: how do circadian transcriptional programs integrate physiologic systems within individual cells of the intact animal and how does the ensemble of local clocks align temporal harmonics in the organism with the environment? Our approach to studies of metabolic regulation by the molecular clock began with analyses of metabolic pathologies in circadian mutant animals, experiments that first became possible with the cloning of the clock genes in the late 1990s. A paradox in our early studies was that the effects of circadian clock disruption were both nutrient- and time-dependent, so that, under fed conditions, animals exhibited diabetes whereas during fasting, they decompensated and died. Application of a broad range of tissue-specific genetic and biochemical approaches has now begun to provide mechanistic insight into the circadian control of metabolism.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Genetic Approaches to Dissecting Circadian Physiology
Glucose homeostasis is a dynamic process that is subjected to rhythmic variation throughout the daily light–dark cycle. Impaired glucose regulation arises from desynchrony in the integration of anabolic, catabolic, and incretin hormones across the circadian cycle and leads to metabolic syndrome and diabetes mellitus, disorders that are associated with over-nutrition, sedentary lifestyle, and sleep-wake disruption common in industrialized society. Individuals with diabetes must adjust their insulin levels differently every day and night even independently of how much they eat; however, the molecular underpinnings of circadian glucose regulation were previously not well understood. Genome-wide association and deep-sequencing studies have shown that variants of the melatonin receptor 1b and cryptochrome 2 genes correlate with glucose variation in humans, suggesting a genetic linkage between the circadian system and glucoregulatory processes in man (Bouatia-Naji et al. 2009; Mulder et al. 2009; Dupuis et al. 2010). Against this backdrop, work from our laboratory using circadian clock mutant mice first revealed an essential role for the intrinsic beta cell clock in insulin secretion, beta cell development, and diabetes mellitus (Marcheva et al. 2010). Subsequent studies in three other groups have corroborated our observation that local function of the clock transcription factor in islets is crucial for normal glucose homeostasis (Sadacca et al. 2011; Lee et al. 2013; Pulimeno et al. 2013). Importantly, work from the Dibner laboratory has been the first to manipulate and monitor clock function in isolated human islet cells, raising the possibility that future investigation into circadian cell physiology will yield new understanding of beta cell failure in man (Pulimeno et al. 2013). In recently published work, we have developed tamoxifen-inducible Cre-LoxP technology to conditionally eliminate clock gene function in pancreas (PMID 26542580). Remarkably, our results establish that acute pancreatic clock ablation in the adult is sufficient to cause diabetes mellitus in the whole animal. These new genetic studies are the first to demonstrate an essential role for the adult circadian system in beta cell glucose regulation, although a gap remains in our understanding of the cell and molecular bases for clock function in the beta cell. Using conditional gene targeting and next-generation sequencing described in the following sections, we are presently poised to dissect the genomic, biochemical, and physiologic mechanism of the clock in beta cell failure. Moreover, since clock transcription factors impact both islet cell growth and stress response, we also seek to understand the role of the islet cell clock in susceptibility to beta cell apoptosis in type 1 diabetes, in islet regenerative capacity, and in islet cell survival in insulin resistant obesity.
Our analysis of the beta cell clock also opens broader insight into the role of transcription factor deregulation in beta cell failure and the unifying and distinct molecular events between tissues that culminate in diabetes mellitus. In this regard, positional cloning of genes causing Maturity Onset Diabetes of the Young (MODY) in humans has revealed that the hepatic nuclear factor (HNF) network of forkhead transcription factors plays a critical role in beta cell development and function, although knowledge of the cell-context specific determinants of HNF action in liver and pancreas remains incomplete. By analogy, an important goal in understanding how beta cell transcription defects related to the clock pathway give rise to diabetes will be to elucidate differences in the clock-controlled enhancer network in liver and pancreas and to compare the cistromes and transcriptomes for these factors in each tissue. Interestingly, there is coincidence of CLOCK/BMAL1 binding sites in liver at loci marked by monomethylated H3K4 in pancreas, although it is not known whether the enhancer state (poised, H3K4me1 vs active, H3K4me1 with H3K27Ac) in pancreas varies over the 24-h cycle. In the long term it will be necessary to evaluate the cistromes and localization of clock factors with established transcription factors involved in beta cell function over the full circadian cycle in both liver and pancreas. Such studies will further elucidate the mechanism by which the clock controls gene transcription networks involved in insulin release, namely by determining the extent to which CLOCK/BMAL1 directly binds to promoters and/or enhancers or regulates epigenetic chromatin modifiers that determine accessibility to transcription factors and RNA polymerase genome-wide. Overall, studies of the beta cell molecular clock will elucidate how glucose homeostasis is coupled to the light/dark cycle and the transcriptional determinants of circadian physiology.
Clock-NAD+-Sirtuin Pathway in Bioenergetics
A major step in understanding how the clock-NAD+ cycle impacts physiology came from the observations that circadian mutant mice become hypoglycemic and die when subject to a prolonged fast (PMID 24051248, unpublished data) and also exhibit muscle and heart failure (PMID 20956306, 21452915), all hallmarks of mitochondrial disease, which prompted us to dissect the mechanisms of clock regulation of mitochondrial function. We began our investigation into the effect of NAD+ deficiency on mitochondrial function in circadian mutant animals using a multi-faceted approach, including unbiased proteomics, which led to the identification of abnormal acetylation of enzymes involved in lipid oxidation, amino acid catabolism, tricarboxylic acid (TCA) cycle, electron transport chain (ETC), and superoxide dismutase pathways. Importantly, loss-of-function mutations in several of these oxidative enzymes have also been identified in the human metabolic myopathy syndrome and in both glioblastoma and renal cell carcinoma, indicating a broader effect of the clock-NAD+ pathway on mitochondrial metabolism in both normal and transformed cells. Using tissue- and cell-based bioenergetics assays, we discovered that abrogation of the clock impairs electron transfer from lipid to the TCA cycle, in addition to increased mitochondrial production of superoxide free radical, increasing sensitivity to genotoxic stress. Our work also showed that cells exhibit an autonomous rhythm of oxygen consumption, glucose oxidation, and mitochondrial lipid catabolism. Importantly, the oxygen consumption cycle in muscle is directly linked to metabolism of NAD+ and activity of the mitochondrial NAD+-dependent deacetylase SIRT3 (Peek et al. 2013).
Although the aforementioned work has pinpointed specific defects in clock control of mitochondrial function, several unanswered questions remain in dissecting the effect of clock-NAD+ rhythms on physiology and cell biology. First, it is not yet known how NAD+ deficiency locally within skeletal muscle contributes to respiration or exercise tolerance in circadian mutant mice or in animals subjected to environmental circadian disruption. Though skeletal muscle ablation of the clock has been achieved in our group and others, the biochemical pathways through which clock abrogation impairs oxidative capacity remain largely unknown (Dyar et al. 2014).
Second, we still do not know whether clock abrogation and NAD+ deficiency in liver or skeletal muscle impacts overall energy balance and alters the capacity to utilize carbohydrate and lipid as a fuel source. New pharmacologic (Wang et al. 2014) and genetic means to raise NAD+ both globally in the whole animal and selectively within either liver or skeletal muscle are now available and will be powerful tools in evaluating the potential to boost NAD+ as a therapeutic strategy in myopathy and liver defects of circadian mutant animals. Finally, in addition to its function as a cofactor for the class III histone deacetylases, NAD+ is a cofactor for the poly-ADP-ribosylases, critical factors in DNA repair and stress response, though the possible interaction between rhythmic regulation of NAD+ and PARP activity is not known. Lastly, NAD+ functions as an electron transport molecule and, as such, it is a direct marker of cellular redox state and the balance between glycolytic and oxidative metabolism. Whether NAD+ might participate in the bidirectional communication between metabolism and the clock system remains an area of intensive investigation. In summary, discovery of the clock as an upstream regulator of NAD+ provides a wealth of opportunity to dissect the interrelationship between circadian rhythms, physiology, and epigenetics.
Reciprocal Control of the Clock by Nutrient
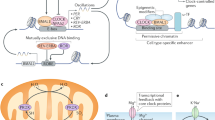
Circadian clocks are biologic oscillators that produce 24-h cycles in the transcription of rate-limiting metabolic enzymes in anticipation of the solar cycle. The molecular clock is programmed by a transcription-translation feedback loop that is comprised of activators (CLOCK/BMAL1) that induce the expression of their own repressors (CRYs/PERs) in a cycle that repeats itself every 24 h. The REV-ERB and ROR proteins form an ancillary loop that modulates Bmal1 transcription. In animals, clocks are organized hierarchically, with brain pacemaker cells synchronizing peripheral tissue clocks, leading to a classical view of the central clock as the main driver of metabolism. However, circadian oscillations within both brain and peripheral tissues have recently been shown to be sensitive to timing of nutrient availability and can become uncoupled from the light–dark cycle, as demonstrated by experimentally restricting food access to the light cycle when mice are normally resting (Damiola et al. 2000; Stokkan et al. 2001). Further, simply substituting regular with high fat chow in mice fed ad libitum lengthens periodicity of locomotor activity and alters peripheral metabolic rhythms, providing further evidence for a bidirectional relationship between clock function and metabolism (Kohsaka et al. 2007). Our discovery that diet-induced obesity reprograms both the cellular molecular clock and behavior revealed for the first time that a controlled change in nutritional environment leads to altered circadian rhythms. This idea, that circadian and metabolic systems reciprocally interact and that perturbation of the metabolic environment alters the homeostatic relationship between these systems, has been widely confirmed but still remains poorly understood at the mechanistic level. Human analyses, including genome-wide association studies, population based case–control investigation, and clinical research, have cumulatively indicated a strong interrelationship between circadian disruption, obesity, diabetes mellitus, and metabolic syndrome. Moreover, certain inflammatory and cardiovascular events, including thrombosis and nocturnal asthma, exhibit pronounced circadian variation. Surprisingly, dietary macronutrient directly impacts behavioral and molecular clock function, and circadian disruption itself exacerbates the progression of diet-induced obesity, exerting distinct effects within local metabolic organs. Moreover, limiting high-fat food to the incorrect circadian phase accelerates weight gain, whereas limiting high-fat feeding to the correct phase ameliorates hepatic steatosis, a hallmark of metabolic syndrome (Maury et al. 2010). While we previously demonstrated that diet-induced obesity reprograms the cellular molecular clock and circadian behavior, we have more recently sought to identify the macronutrient disruptor of circadian behavior by providing mice an isocaloric diet high in either saturated or unsaturated fats (SFD and UFD). Our goal is to identify the mechanism by which a macronutrient directly alters behavior and neuronal circadian pacemaker function. We propose that nutrient signaling plays a central role in inter-organ circadian communication and that circadian disruption induced by high saturated fat contributes to the rate of progression of metabolic syndrome.
Summary and Future Directions
A major window to understanding how the clock is coupled to metabolism was opened with discovery of metabolic syndrome pathologies in multi-tissue circadian mutant mice, including susceptibility to diet-induced obesity, mis-timed feeding rhythms, hypoinsulinemia, and energetic collapse upon fasting. Using Cre-LoxP conditional transgenesis and dynamic endocrine testing, we have pinpointed the tissue-specific role of the clock in energy and glucose homeostasis, with our most detailed understanding of this process in liver, muscle, and endocrine pancreas. In the post-prandial condition, the beta cell clock is essential for nutrient and adenyl cyclase-induced insulin exocytosis. In contrast, the hepatocyte and myocyte clocks are required for oxidative metabolism. Circadian mutant mice die upon prolonged fasting due to mitochondrial failure, a defect that we have tied to the bioavailability of NAD+, a cofactor of the class III histone deacetylases and poly-ADP ribosylase enzymes involved in adjusting metabolic and gene regulation in response to environmental change, including glucose deprivation, oxidative damage, and cell stress. Indeed, we have found that liver and myoblasts exhibit an autonomous rhythm of oxygen consumption, glucose oxidation, and mitochondrial lipid catabolism that is directly linked to an autonomous rhythm of NAD+ metabolism and, consequently, to cyclic activity of the mitochondrial NAD+-dependent deacetylase SIRT3. NAD+ supplementation using the pro-drug NMN improves respiration in live animals, indicating that circadian control of NAD+ metabolism plays a key role in cellular and organismal respiration. A future challenge will be to determine the cell and molecular basis for the interplay between nutritional and circadian processes important in metabolic health and disease states.
References
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M, Delplanque J, Lobbens S, Rocheleau G, Durand E, De Graeve F, Chevre JC, Borch-Johnsen K, Hartikainen AL, Ruokonen A, Tichet J, Marre M, Weill J, Heude B, Tauber M, Lemaire K, Schuit F, Elliott P, Jorgensen T, Charpentier G, Hadjadj S, Cauchi S, Vaxillaire M, Sladek R, Visvikis-Siest S, Balkau B, Levy-Marchal C, Pattou F, Meyre D, Blakemore AI, Jarvelin MR, Walley AJ, Hansen T, Dina C, Pedersen O, Froguel P (2009) A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet 41:89–94
Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U (2000) Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev 14:2950–2961
Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, Lindgren CM, Magi R, Morris AP, Randall J, Johnson T, Elliott P, Rybin D, Thorleifsson G, Steinthorsdottir V, Henneman P, Grallert H, Dehghan A, Hottenga JJ, Franklin CS, Navarro P, Song K, Goel A, Perry JR, Egan JM, Lajunen T, Grarup N, Sparso T, Doney A, Voight BF, Stringham HM, Li M, Kanoni S, Shrader P, Cavalcanti-Proenca C, Kumari M, Qi L, Timpson NJ, Gieger C, Zabena C, Rocheleau G, Ingelsson E, An P, O’Connell J, Luan J, Elliott A, McCarroll SA, Payne F, Roccasecca RM, Pattou F, Sethupathy P, Ardlie K, Ariyurek Y, Balkau B, Barter P, Beilby JP, Ben-Shlomo Y, Benediktsson R, Bennett AJ, Bergmann S, Bochud M, Boerwinkle E, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Bottcher Y, Brunner E, Bumpstead SJ, Charpentier G, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Cornelis M, Crawford G, Crisponi L, Day IN, de Geus EJ, Delplanque J, Dina C, Erdos MR, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Fox CS, Frants R, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Groves CJ, Grundy S, Gwilliam R, Gyllensten U, Hadjadj S, Hallmans G, Hammond N, Han X, Hartikainen AL, Hassanali N, Hayward C, Heath SC, Hercberg S, Herder C, Hicks AA, Hillman DR, Hingorani AD, Hofman A, Hui J, Hung J, Isomaa B, Johnson PR, Jorgensen T, Jula A, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, Le Bacquer O, Lecoeur C, Li Y, Lyssenko V, Mahley R, Mangino M, Manning AK, Martinez-Larrad MT, McAteer JB, McCulloch LJ, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Morken MA, Mukherjee S, Naitza S, Narisu N, Neville MJ, Oostra BA, Orru M, Pakyz R, Palmer CN, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Perola M, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Psaty BM, Rathmann W, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Roden M, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Scott LJ, Seedorf U, Sharp SJ, Shields B, Sigurethsson G, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvanen AC, Tanaka T, Thorand B, Tichet J, Tonjes A, Tuomi T, Uitterlinden AG, van Dijk KW, van Hoek M, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Walters GB, Ward KL, Watkins H, Weedon MN, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zeggini E, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, Borecki IB, Loos RJ, Meneton P, Magnusson PK, Nathan DM, Williams GH, Hattersley AT, Silander K, Salomaa V, Smith GD, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Dedoussis GV, Serrano-Rios M, Morris AD, Lind L, Palmer LJ, Hu FB, Franks PW, Ebrahim S, Marmot M, Kao WH, Pankow JS, Sampson MJ, Kuusisto J, Laakso M, Hansen T, Pedersen O, Pramstaller PP, Wichmann HE, Illig T, Rudan I, Wright AF, Stumvoll M, Campbell H, Wilson JF, Bergman RN, Buchanan TA, Collins FS, Mohlke KL, Tuomilehto J, Valle TT, Altshuler D, Rotter JI, Siscovick DS, Penninx BW, Boomsma DI, Deloukas P, Spector TD, Frayling TM, Ferrucci L, Kong A, Thorsteinsdottir U, Stefansson K, van Duijn CM, Aulchenko YS, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Waterworth DM, Vollenweider P, Peltonen L, Mooser V, Abecasis GR, Wareham NJ, Sladek R, Froguel P, Watanabe RM, Meigs JB, Groop L, Boehnke M, McCarthy MI, Florez JC, Barroso I (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42:105–116
Dyar KA, Ciciliot S, Wright LE, Bienso RS, Tagliazucchi GM, Patel VR, Forcato M, Paz MI, Gudiksen A, Solagna F, Albiero M, Moretti I, Eckel-Mahan KL, Baldi P, Sassone-Corsi P, Rizzuto R, Bicciato S, Pilegaard H, Blaauw B, Schiaffino S (2014) Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol Metab 3:29–41
Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, Turek FW, Bass J (2007) High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab 6:414–421
Lee J, Moulik M, Fang Z, Saha P, Zou F, Xu Y, Nelson DL, Ma K, Moore DD, Yechoor VK (2013) Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Mol Cell Biol 33:2327–2338
Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, Lopez JP, Philipson LH, Bradfield CA, Crosby SD, Jebailey L, Wang X, Takahashi JS, Bass J (2010) Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 466:571–572
Maury E, Ramsey KM, Bass J (2010) Circadian rhythms and metabolic syndrome: from experimental genetics to human disease. Circ Res 106:447–462
Mulder H, Nagorny CL, Lyssenko V, Groop L (2009) Melatonin receptors in pancreatic islets: good morning to a novel type 2 diabetes gene. Diabetologia 52:1240–1249
Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J (2013) Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342:1243417
Pulimeno P, Mannic T, Sage D, Giovannoni L, Salmon P, Lemeille S, Giry-Laterriere M, Unser M, Bosco D, Bauer C, Morf J, Halban P, Philippe J, Dibner C (2013) Autonomous and self-sustained circadian oscillators displayed in human islet cells. Diabetologia 56:497–507
Sadacca LA, Lamia KA, deLemos AS, Blum B, Weitz CJ (2011) An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia 54:120–124
Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M (2001) Entrainment of the circadian clock in the liver by feeding. Science 291:490–493
Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL (2014) P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell 158:1324–1334
Acknowledgments
Funding Sources
P01AG011412 (National Institute on Aging), R01DK090625 (National Institute of Diabetes and Digestive and Kidney Disease), R01DK100814 (National Institute of Diabetes and Digestive and Kidney Disease), 17-2013-511 (Juvenile Diabetes Research Foundation with Helmsley Charitable Trust), 1-INO-2014-178-A-V (Juvenile Diabetes Research Foundation with Helmsley Charitable Trust). I thank the lab members who participated in our cited research and to KM Ramsey for helpful comments and suggestions on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/) which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
The images or other third party material in this chapter are included in the work’s Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work’s Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Bass, J. (2016). Circadian Mechanisms in Bioenergetics and Cell Metabolism. In: Sassone-Corsi, P., Christen, Y. (eds) A Time for Metabolism and Hormones. Research and Perspectives in Endocrine Interactions. Springer, Cham. https://doi.org/10.1007/978-3-319-27069-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-27069-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-27068-5
Online ISBN: 978-3-319-27069-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)