
Abstract
Virus encoded ion channels, termed viroporins, are expressed by a diverse set of viruses and have been found to target nearly every host cell membrane and compartment, including endocytic/exocytic vesicles, ER, mitochondria, Golgi, and the plasma membrane. Viroporins are generally very small (<100 amino acids) integral membrane proteins that share common structure motifs (conserved cluster of basic residues adjacent to an amphipathic alpha-helix) but only limited sequence homology between viruses. Ion channel activity of viroporins is either required for replication or greatly enhances replication and pathogenesis. Channel characteristics have been investigated using standard electrophysiological techniques, including planar lipid bilayer, liposome patch clamp or whole-cell voltage clamp. In general, viroporins are voltage-independent non-specific monovalent cation channels, with the exception of the influenza A virus M2 channel that forms a highly specific proton channel due to a conserved HXXXW motif. Viroporin channel currents range between highly variable (‘burst-like’) fluctuations to well resolved unitary (‘square-top’) transitions, and emerging data indicates the quality of channel activity is influenced by many factors, including viroporin synthesis/solubilization, the lipid environment and the ionic composition of the buffers, as well as intrinsic differences between the viroporins themselves. Compounds that block viroporin channel activity are effective antiviral drugs both in vitro and in vivo. Surprisingly distinct viroporins are inhibited by the same compounds (e.g., amantadines and amiloride derivatives), despite wide sequence divergence, raising the possibility of broadly acting antiviral drugs that target viroporins. Electrophysiology of viroporins will continue to play a critical role in elucidating the functional roles viroporins play in pathogenesis and to develop new drugs to combat viroporin-encoding pathogens.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Viral ion channel
- Amphipathic alpha-helix
- Proton channel
- Non-selective cation channels
- Planar lipid bilayer
- Xenopus oocytes
1 Introduction
Viruses are obligate cellular parasites that require host cells to support their replication cycle, which utilizes virus-encoded proteins and nucleic acids to exploit cellular processes needed from progeny virus assembly. Many viruses have evolved their own ion channels to help subvert cellular ionic gradients to their advantage. These virus-encoded ion channels are called viroporins. Research on viroporins over the last 22 years has demonstrated that most (if not all) viroporins are bona fide ion channels, in that they can display unitary single channel conductance states. However, identification and characterization of new candidate viroporins is challenging due to the lack of sequence homology between viroporins, the wide array of small molecules that can be conducted through these proteins when expressed in exogenous systems such as E. coli, and differences in ion channel characteristics (Lamb and Pinto 1997; Gonzalez and Carrasco 2003; Kelly et al. 2003). This chapter will summarize the different approaches used to identify viroporins and to demonstrate bona fide ion channel activity, with a particular emphasis on how differences in experimental details may explain differences in ion channel characteristics. The goal is to provide a guide for current and future electrophysiology analysis of viroporins and a more rational approach to understanding the electrophysiology of these truly unconventional ion channels.
1.1 Viroporins
Viroporins are a diverse class of small, hydrophobic virus encoded membrane associated proteins that oligomerize, particularly in lipid membranes or lipid-like detergents, such that an aqueous pore is formed through the membrane by an amphipathic α-helix within the viroporin domain (Gonzalez and Carrasco 2003). While the initial concept of viroporins focused on proteins that induced cell lysis or permeability to small molecule compounds, such as hygromycin B, more recent studies using electrophysiology demonstrate viroporins are not simply large pores but are bona fide viral ion channels, distinguishing them from viral fusions domains found on viral attachment and entry proteins (Gonzalez and Carrasco 2003; Nieva et al. 2012). Currently, the majority of viroporins are from mammalian viruses, with the exception of the K+-selective channel (Kcv) expressed by chlorella viruses that infect unicellular algae (Kang et al. 2004). Additional viroporins come from avian reovirus (p10), and arboviruses that replicate in both insect and mammalian hosts (orbivirus NS3, ephemerovirus α-1), and it is likely that ion channels are also encoded by bacteriophage (e.g., holins/pinholins) as well. However, identification of ion channel activity through electrophysiological assays remains to be done for these proteins (Bodelon et al. 2002; Han and Harty 2004; Joubert et al. 2014; Young 2014). Although the Chlorella virus Kcv is clearly a viral ion channel, it has structural and functional features of bacterial and eukaryotic K+ channels, suggesting a shared evolutionary origin with conventional K+ channels (Kang et al. 2004). This chapter will focus on the electrophysiology of mammalian viroporins (Table 7.1) that share only limited sequence homology with conventional host channels.
1.2 Structural Features and Classification of Viroporins
Viroporins come from a diverse set of virus families and there is little to no sequence homology among viroporins, making identification of these proteins challenging. However, all viroporins have a number of shared structural characteristics and it is by identification of these features that new viroporin candidates arise. Viroporins are relatively small (~60–275 amino acids) hydrophobic transmembrane proteins, which typically have highly basic regions of multiple lysine and arginine residues adjacent to the transmembrane domains. The basic regions are thought to aid in membrane association and invasion of the amphipathic α-helix, and the presence of the basic residues is typically well conserved (Gonzalez and Carrasco 2003). Viroporin activity is typically important for virus replication and/or pathogenesis because mutations or knockdown of viroporin expression result in reduced viral yield or attenuated disease (Nieva et al. 2012; Giorda and Hebert 2013).
In addition to primary sequence diversity, there is also considerable diversity in membrane topology among viroporins. They often have only short extra-membrane domains extending to the cytoplasm or into the intracellular membrane compartment or extracellular space, although there are examples of viroporins with long cytoplasmic domains, such as rotavirus NSP4 (Hyser et al. 2010). A classification system was recently developed based on the number of transmembrane segments (Class I or Class II) and the topology (Subclass A: N-terminus in the ER; Subclass B: N-terminus in the cytoplasm) (Nieva et al. 2012). More recent studies suggest the existence of Class III viroporins with three transmembrane segments, which includes the rotavirus NSP4 and SARS-CoV ORF3a proteins (Hyser et al. 2010; Lu et al. 2006). Thus far, no viroporin candidates with more than three transmembrane domains have been reported.
1.3 Non-electrophysiological Methods for Viroporin Characterization
The lack of sequence homology among viroporins makes genomic and meta-genomic approaches for viroporin identification nearly impossible. A common functional hallmark of viroporins is the ability to induce plasma membrane permeability of host cells and E. coli upon recombinant expression or exogenous addition to liposomes [Reviewed in (Gonzalez and Carrasco 2003; Nieva et al. 2012; Giorda and Hebert 2013)]. Traditional assays to address viroporin-induced permeabilization have been developed using E. coli cell lysis, reduction in cell viability, or growth inhibition; permeabilization of cells or liposomes to β-galactosidase substrates (OMPG), translation-blocking compounds (hygromycin B) or fluorescent molecules (e.g., FITC dextrans); and fluorescence sensors to measure conductance of protons, Ca2+, or redox state (Lama and Carrasco 1992; Taube et al. 2014; Agirre et al. 2002; Henkel et al. 2010; Hyser et al. 2012; Lin and Schroeder 2001). Many of these assays have been developed into screens for mutations that attenuate viroporin activity and chemical blockers of the channel (Agirre et al. 2002; Luscombe et al. 2010). These relatively simple assays serve as an important staging ground for understanding the viroporin’s structure and identification of mutations that provide a basis for electrophysiology studies of channel activity.
1.4 Electrophysiology Methods and Challenges for Viroporins
Since viruses utilize host cell machinery for protein synthesis and replication, the study of viroporins utilizes the same electrophysiology methodologies as do conventional ion channels. However, there are a number of challenges associated with conducting and interpreting viroporin electrophysiology studies. First, because viroporins do not share sequence motifs that provide clues about ion selectivity (except for the HXXXW motif of proton channels, see below), it can be challenging to assign an observed conductance to the viroporin. For example, voltage clamp of viroporin-expressing Xenopus leavis oocytes have successfully demonstrated ion channel function of some viroporins, but the viroporin can also activate endogenous currents, which confound the data (Shimbo et al. 1995). Similarly, while planar lipid bilayer electrophysiology (PLB) is a popular method-of-choice for initial viroporins studies, the detergents and/or solvents used for viroporin purification can cause channel-like current artifacts in PLB experiments, again making rigorous demonstration that ion channel activity can be definitively assigned to the viroporin (Kelly et al. 2003). Thus, the most successful electrophysiology studies of viroporins have utilized multiple electrophysiology techniques, comparison of wild-type and viroporin-deficient mutant proteins, and a chemical inhibitor/blocker of viroporin function, if one is known.
1.4.1 Patch Clamp
Several patch clamp techniques have been used to study viroporins, with X. leavis oocytes being the most popular system to date and the system used in the seminal paper by Pinto et al. (1992) demonstrating IAV M2 is a viral ion channel. However, in a number of cases viroporin expression induced endogenous currents that could not be attributed to the viroporin itself (Shimbo et al. 1995). One study of HIV-1 Vpu in oocytes showed induction of a slowly activating, hyperpolarization-activated nonspecific cation (HANC) current [also called hyperpolarization-dependent inward cation current (IIN) by others (Kuruma and Hartzell 1999)] that was interpreted to be a typical artifact of exogenous membrane proteins, and another study of the Sindbis virus 6K showed activation of endogenous calcium-activated chloride current (ICl) due to activation of store-operated calcium entry (SOCE) (Coady et al. 1998; Antoine et al. 2007). Similar currents were observed upon expression of HIV-1 Vpu (Schubert et al. 1996), influenza C virus M2 (CM2) (Hongo et al. 2004), and enterovirus 71 2B (Xie et al. 2011), but viroporin-defective mutants or blockers were used to demonstrate specificity. Such discrepancies are discussed individually below, but they highlight the need for cautious interpretation of data from oocytes when viroporins induce IIN and/or ICl.
Fewer electrophysiology studies of mammalian cells expressing viroporins have been performed, likely because over-expression of viroporins causes rapid cytotoxicity and damages the cell (Madan 2008). As with oocytes, mammalian cell studies of the influenza A virus (IAV) M2 proton channel represent the seminal reports, first using CV-1 cells, an African green monkey kidney cell line (Wang et al. 1994) and subsequently using mouse erythroleukemia cells (MEL) stably expressing M2 (Chizhmakov et al. 1996). More recent studies on SARS CoV E protein, HIV-1 Vpu, and RSV SH protein utilized transient expression in the HEK293 or HEK293T cells (Pervushin et al. 2009; Bodelon et al. 2002; Gan et al. 2012). The low conductance of most viroporins studied thus far makes the study of macroscopic whole-cell currents an attractive alternative to PLB, provided the viroporin localizes to the plasma membrane and interference from cellular channels can be ruled out with viroporin-defective mutants and/or inhibitors.
One study has compared PLB to excised patches from unilamellar blisters of collapsed giant proteoliposomes to examine HCV p7 channel activity (Delcour et al. 1989; Montserret et al. 2010). Both techniques yielded similar results, including observation of multiple conductance states, which were attributed to either simultaneous insertion of multiple channels or channels composed of a different number of monomers (i.e., pentamers versus hexamers) (Montserret et al. 2010). Despite the more labor intensive preparation, liposome patch clamp has advantages over traditional PLB electrophysiology, such as direct control of the protein:lipid ratio, lack of continued channel insertion events, and the generally lower noise of patch clamp than PLB electrophysiology.
1.4.2 Planar Lipid Bilayer
The most common electrophysiology approach to study viroporins is planar lipid bilayer (PLB). The first studies of viroporins utilizing PLB were on the IAV M2 proton channel (Tosteson et al. 1994) and shortly after HIV-1 Vpr and Vpu, and the influenza B virus M2 (BM2) protein (Ewart et al. 1996; Piller et al. 1996; Sunstrom et al. 1996). While PLB studies have driven forward viroporin electrophysiology more than patch clamp studies, a disadvantage of using PLB for viroporins is that often the induced currents are highly variable without discernable conductance states, as illustrated for HCV p7 in Fig. 7.1a, but the same p7 peptide can also give rise to canonical unitary single channel states in separate experiments (Fig. 7.1b) (Premkumar et al. 2004). Further, the highly hydrophobic nature of most viroporins and the viroporin-domain peptides makes the choice of solvent important for generating high quality current recordings. Issues with solubility have been overcome by several different means, including reconstituting the viroporin into liposomes or addition of two flanking lysine resides (Torres et al. 2007). Nevertheless, clearly resolvable conductance states are observed for most viroporins, which may occur between ‘burst events’ (Henkel et al. 2010), enabling measurement of single channel conductance and ion permeability.


Representative examples of HCV p7 viroporin channel activity in planar lipid bilayer. Synthetic p7 peptide was added to the cis bath resulting in (a) variable ‘burst-like’ currents or (b) unitary single channel currents. (c) The unitary single channels were completely blocked by 100 μM hexamethylene amiloride (HMA), but HMA only partially blocked the larger ‘burst-like’ currents (not shown) (Premkumar 2004). (d) Single channel activity of HCV p7 is altered by the lipid composition of the lipid bilayer. In PC-rich membranes (left) p7 has a higher open probability than in PE-rich membranes (right). PC-rich membrane also promoted slightly higher single channel conductance of the main conductance states and the subconductance state (arrows) (Whitfield 2011)
Interestingly, most viroporins display multiple distinct single channel conductance levels and subconductance states, which are observed within single traces and are reproducible between experiments (Chien et al. 2013; Ewart et al. 2002; Henkel et al. 2010; Montserret et al. 2010; Piller et al. 1999; Verdia-Baguena et al. 2013). This phenomenon is attributed to simultaneous opening of multiple channels, and/or activity of distinct channel oligomeric species, though few studies have been conducted. More recently, the lipid composition has been shown to significantly influence viroporin conductance. Changes in the phosphatidylcholine-to-phosphatidylethanolamine (PC:PE) ratio altered HCV p7 channel open probability and conductance states (Fig. 7.1d) (Whitfield et al. 2011), and similarly changes from neutral (PC) membrane to one containing negatively charged (PS) lipids reduced coronavirus E protein conductance by 50 % and increase cation selectivity (Verdia-Baguena et al. 2013).
2 Viroporin Electrophysiology
The functional characteristics of different viroporins vary substantially, with few similarities between virus families. Nevertheless, viroporins can be generally classified into three groups based on the ions they appear to conduct: (1) proton channels (e.g., influenza M2), (2) monovalent cation channels, some of which show ion selectivity (SARS CoV ORF3a), whereas others do not have strong selectivity (HIV-1 Vpu), and (3) divalent cation channels [(e.g., rotavirus NSP4 and picornavirus 2B, for which electrophysiology studies are not yet published (see below)]. This section contains a summary of the electrophysiology data for each viroporin in these viruses.
2.1 Viral Proton Channels
2.1.1 Influenza Virus M2
Influenza A virus (IAV) M2 was the first viral ion channel recorded using electrophysiology and is therefore the prototypical viral ion channel. The rationale for testing M2 for ion channel activity was based on amantadine and ramantadine inhibiting early steps in the low pH-dependent disassembly of viral ribonucleoprotein complexes during entry, as well as protecting the hemagglutinin protein (HA) from a premature conformational change during transit through the trans Golgi (Pinto et al. 1992). Amantadine resistant mutants mapped to the transmembrane domain of M2 protein, a small (~100 amino acid) tetrameric protein that is abundant in the Golgi and plasma membrane of host cells, and a minor component of the virion (23–60 copies) (Pinto et al. 1992), suggesting a direct role for M2 in modulating pH in infected cells. Whole-cell voltage clamp of Xenopus oocytes was performed using expressed wild-type M2 (A/Udorn/72) or an amantadine-resistant M2 mutant. A large inward current was detected in the WT M2-expressing cells. This current was greater than endogenous currents, was inhibited by 10 μM amantadine, and activated ~7-fold by decreasing the pH from 7.5 to 6.2; however, while similar currents were produced by the M2 mutants, they were not blocked by 100 μM amantadine (Pinto et al. 1992). Similar currents were observed when M2 from other IAV strains were expressed in oocytes (Wang et al. 1993), and when M2 was expressed in mammalian CV-1 cells expressing WT M2 (Wang et al. 1994). Recombinant M2 (Udorn) has ion channel activity when reconstituted into phosphatidylserine:phosphatidylethanolamine (PS:PE) bilayers (Tosteson et al. 1994) and these currents were also activated by low pH and inhibited by amantadine (Wang et al. 1994). In PLB, the M2 current was highly variable, with both small (50–90 pS) and large (40–600 pS) conductances in 150 mM NH4Cl (Tosteson et al. 1994).
Initial reports suggested M2 is able to conduct protons, as well as group IA alkali cations, (Pinto et al. 1992; Shimbo et al. 1996; Tosteson et al. 1994), but more detailed investigation of ion selectivity by patch clamp of M2-expressing MEL cells (Chizhmakov et al. 1996) and oocytes and CV-1 cells (Mould et al. 2000) showed that M2 has high proton selectivity over Na+ [PH+/Na+ = 6 × 107 in MEL cells (Chizhmakov et al. 1996) and 2 × 106 in oocytes and CV-1 cells (Mould et al. 2000)]. Selective proton conductance was confirmed for bacterially expressed and purified M2 reconstituted into dimyristoyl phosphatidylcholine (DMPC) and dimyristoyl phosphatidylglycerol (DMPG) liposomes (1:5 w/w) and recorded in mixed lipid bilayers [phosphatidylethanolamine, phosphatidylcholine, phosphatidylserine, cholesterol (4:1:1:2 molar ratio)]. Single channel conductance in 1 mM HCl was 6 pS with a reversal potential (Erev) close to EH but Erev did not vary upon changes in Cl−, Na+ or tetraethylammonium (TEA) concentrations (Vijayvergiya et al. 2004). In MEL cells, the estimated single channel conductance was <10 fA, though single channel currents could not be directly measured (Chizhmakov et al. 1996). In contrast to electrophysiological measurements of M2 conductance, liposomes-based studies using the fluorescent pH indicator pyranine measured the M2 single channel conductance to be 4.1 × 10–18 A (~4.1 aA) at pH 5.7; which is approximately 4 orders of magnitude lower than the lowest patch clamp measurement (Lin and Schroeder 2001). It is unclear why such a striking difference is observed between the electrophysiology and fluorescence based assays, but it the authors argue conductance below the noise level of patch clamp may lead to inaccurate estimates of M2 single channel conductance (Lin and Schroeder 2001)
M2 channel gating and proton selectivity are determined by the conserved HXXXW motif (aa 37–41) such that pH <6.5 at the ectodomain (i.e., extracellular or ER/Golgi/vesicle lumen) protonates His-37 thereby stabilizing an interaction with Trp-41, which acts as the primary gate (Okada et al. 2001; Tang et al. 2002). Substitution of His-37 with Gly, Ala, Ser, Thr, or Glu abolishes proton selectivity but ion channel activity is retained and proton selectivity is partially restored by including imidazole in the buffer. (Wang et al. 1995; Venkataraman et al. 2005).
M2 homologs from Influenza B and C viruses, BM2 and CM2, are also tetrameric one-pass membrane proteins (Class IA viroporin), and function similarly to M2 (Stewart and Pekosz 2012; Mould et al. 2003; Betakova and Hay 2007). When expressed in Xenopus oocytes BM2 showed external pH-dependent inward currents and strong selectivity for protons but was not inhibited by amantadine (Mould et al. 2003). M2 and BM2 share little sequence homology with the exception of the HXXXW motif, which validates their shared proton selectivity (Pinto and Lamb 2006). In contrast, CM2 expression in oocytes showed a slowly developing hyperpolarization activated inward current and was only weakly pH activated. This was a Cl− current that was inhibited by the anion channel blocker DIDS but not amantadine, suggesting these were endogenous channels and not CM2 (Hongo et al. 2004; Shimbo et al. 1995). While CM2 does not contain the proton selective HXXXW motif, it was still able to protect HA from acid-induced conformational changes in the Golgi, albeit to a much lesser extent than M2 or BM2 (Betakova and Hay 2007). These biophysical studies have set the stage for an enormous scientific literature examining M2 structure, functions, and dynamics, as well as screens for new M2-blocker antiviral drugs (Loregian et al. 2014).
2.1.2 Hepatitis C Virus p7
Hepatitis C virus (HCV) is a member of the Flaviviridae family and a leading cause of cirrhosis and hepatocellular carcinoma. HCV structural and nonstructural proteins are synthesized as a single ER-associated polyprotein and cleaved by both viral and cellular proteases into the individual proteins. HCV p7 is a two-pass integral membrane protein located between the E2 envelope protein and nonstructural protein 2 (NS2) (Atoom et al. 2014). Subcellular distribution of p7 shows high levels in the ER but also localization to mitochondria or mitochondrial-adjacent membranes, and lipid droplets (Griffin et al. 2004; Vieyres et al. 2013).
Demonstration of p7 ion channel activity was performed with PLB experiments using bacterially expressed and purified GST- and His-tagged p7 (HCV J4 strain) inserted into PS:PC bilayers (Griffin et al. 2003). Addition of p7 either with or without a GST-tag induced poorly resolved bursts of current in symmetrical 0.1 M KCl and channel activity was increased by removal of the tag or measuring currents in 0.1 M CaCl2. This current was completely inhibited by addition of 1 μM amantadine to both cis and trans chambers (Griffin et al. 2003). This initial study was confirmed by two groups who used synthetic peptides of full length p7 in PE:PC (4:1 w/w) (Pavlovic et al. 2003) or PE:PS:PC (5:3:2 w/w) (Premkumar et al. 2004) bilayers. Both groups observed large variable currents (Fig. 7.1a) with conductance ranging from ~100 pS to 2 nS (Pavlovic et al. 2003); however, some apparently single channel traces were recorded (Fig. 7.1b), enabling the identification of a 10–14 pS channel in either asymmetric KCl, NaCl, or CaCl2 (Premkumar et al. 2004). Channel activity was selective for cations (Na+ or K+) over chloride; however, addition of 10 mM CaCl2 increased Cl− permeability (Premkumar et al. 2004). Long alkyl-chain iminosugar derivatives or hexamethylene amiloride (HMA), which also inhibits the HIV-1 Vpu channel, blocked p7 currents at high micromolar concentrations (Fig. 7.1c).
More consistent single channel activity was recorded using patch clamp of a full-length synthetic p7 peptide (HCV-J strain) reconstituted into asolectin liposomes. As before, variable currents were present, but two major conductance levels were discernable: 22 pS and 41 pS in 500 mM KCl (Montserret et al. 2010). Further, p7 is being used as a model ion channel to determine how lipid composition influences channel characteristics. Single channel traces in DOPC-rich [DOPC:POPE (4:1)] bilayers were compared to those in POPE-rich [DOPC:POPE (1:4)] bilayers. In the DOPC-rich bilayer, p7 exhibited a rapidly gating opening pattern with short opening times (~0.5 s), but had long (~8 s) openings with much lower frequency in the POPE-rich bilayer (Fig. 7.1d) (Whitfield et al. 2011). Despite these observed differences in channel kinetics, p7 had two main conductance states and one sub-conductance state; although the conductance levels were slightly greater in the PC-rich bilayer. However, lipid composition did not alter the voltage-independent character of p7 activity and p7 remained cation selective (Whitfield et al. 2011). Interestingly, coronavirus E protein also showed higher conductance in neutral lipid rich bilayers than in bilayers with charged lipids (Verdia-Baguena et al. 2012, 2013).
Despite p7 showing predominantly cation selective currents in PLB experiments, emerging data from cell culture studies suggest p7 to function as a proton channel, analogously to M2. First, the well characterized M2 blocker amantadine also inhibited p7 and p7 functionally complemented M2-deficient IAV to protect HA (Griffin et al. 2003; Wozniak et al. 2010). Further, p7 contains a highly conserved HXXXW/Y motif, similar to the M2 proton selectivity filter, the His-17 residue faces the pore and channel activity is inhibited by Cu2+ in a His-17 dependent manner (StGelais et al. 2007; Chew et al. 2009). However, substitutions of His-17 with Asn and Gln are present in many strains so this motif is not as strictly conserved in p7 (Meshkat et al. 2009). Thus far, proton conductance has not been demonstrated using electrophysiology but numerous drug screen studies have used PLB electrophysiology as validation for candidate p7 blockers, including the iminosugar derivatives and the drug BIT225, which are being tested for clinically relevant anti-HCV activity (Atoom et al. 2014).
2.2 Viral Monovalent Cation Channels
2.2.1 HIV-1
Human immunodeficiency virus-1 (HIV-1) encodes a series of accessory genes (nef, vif, vpr and vpu) in addition to the canonical retrovirus genes gag, pol, and env, and homologous or analogous genes are found in HIV-2 and simian immunodeficiency virus (SIV) (Strebel 2013). Two HIV-1 accessory proteins, called Viral protein regulatory (Vpr) and Viral protein unique (Vpu) have been demonstrated to have intrinsic ion channel activity, although to date the biological relevance of the observed channel function has not been determined (Strebel 2013, 2014). While channel activity of Vpr is controversial, Vpu has become a well-studied viral ion channel and used for many biophysical and structural studies (Strebel 2014). The electrophysiology studies of these viroporins are summarized below, as well as a discussion of the possible role ion channel activity may play during HIV-1 infection.
2.2.1.1 Vpr
HIV-1 Vpr is a 92 amino acid protein with a single predicted hydrophobic segment and multiple copies are incorporated into mature HIV-1 virions. Like many viral proteins, Vpr is multifunctional and assigned roles in HIV-1 replication and pathogenesis range from promotion of apoptosis, cell cycle arrest, regulation of HIV-1 reverse transcriptase, and modulation of host and viral gene transcription (Andrew and Strebel 2014).
The first evidence for intrinsic ion channel activity of Vpr utilized recombinant protein expressed in E. coli as a GST-tagged fusion protein, solubilized in 2 % CHAPS and purified by glutathione affinity chromatography, cleaved from the GST tag by thrombin and isolated by cation exchange chromatography as a detergent solubilized (0.5 % CHAPS) pure protein (Piller et al. 1996). This purified protein was added to bilayers composed of POPE:POPC (8:2 w/w) in cis chamber containing buffers of cis: 150 or 500 mM NaCl/trans: 50 mM NaCl. Purified Vpr (40–80 μL) induced poorly resolved, highly variable currents in 21 experiments, and appearance of currents required the trans chamber to be held at negative holding potentials, whereas positive holding potentials in the trans chamber rapidly inactivated currents. The reversal potential was between +30 and −40 mV, leading to a relative permeability of PNa+/PCl− of 5–12. Hyperimmune serum made against Vpr was able to neutralize the currents, which argued against current artifacts produced by CHAPS (Piller et al. 1996). A second study of Vpr truncations and mutations (also expressed in E. coli) localized the ion channel activity to the N-terminal 40 amino acids and identified Glu21 and Glu24 as potentially important for ion selectivity (Piller et al. 1999). However, other studies have focused on a downstream hydrophobic region (amino acids 52–83 or 52–96) that also has ion channel activity in PLB using either a conventional setup or the “dip-tip” method (Jacotot et al. 2001; Chen et al. 2010). This Vpr domain physically associates with the mitochondrial ANT transpoter and together these proteins cooperatively formed channels with larger conductance (190 pS) than Vpr alone (55 pS) (Jacotot et al. 2001). It is interesting to note that the channel activity of Vpr 52–96 has distinct conductance states than the N-terminal peptide or full-length protein, and it was postulated that the “burst” activity may be related to greater aggregation of monomers in aqueous environments or changes in oligomerization due to membrane curvature (Chen et al. 2010). However, the question of whether Vpr ion channel activity is biologically related to a known Vpr function remains to be determined (Andrew and Strebel 2014).
2.2.1.2 HIV-1 Vpu
HIV-1 Vpu was the third viral protein to be reported to have bona fide ion channel activity, after influenza virus M2 and HIV-1 Vpr; however, it has become one of the most intensely studied proteins, second only to IAV M2. Vpu is an ~81 amino acid, 16 kDa protein expressed from a rev-dependent bicistronic mRNA, which also encodes the env gene, and is co-translationally inserted into the ER membrane (Bour and Strebel 2003). Vpu is a type I integral membrane protein with a N-terminal hydrophobic membrane anchor comprised of residues 1–33. Like many HIV proteins, Vpu carries out multiple functions during infection, including down-regulation of CD4 expression through ERAD-mediated degradation, inhibition of NF-kB activation, sequestration and degradation of tetherin (a.k.a. BST-2) to increase HIV-1 particle release from infected cells, and ion channel activity of the N-terminal domain (Bour and Strebel 2003). While the molecular mechanisms of CD4 degradation are fairly well characterized, much work continues on how Vpu counteracts tetherin and the biological relevance of Vpu ion channel activity remains unsolved (Strebel 2014).
Vpu was initially postulated to be an ion channel due to topological similarities with the influenza A virus M2 proton channel and researchers speculated that Vpu may have an analogous role during HIV-1 replication. The first demonstration of Vpu ion channel activity utilized E. coli and PLB-based systems (Ewart et al. 1996). GST-tagged Vpu was expressed in E. coli, extracted from membranes using CHAPS, and affinity purified prior to thrombin-mediated removal of GST. Addition of purified Vpu to a bilayer composed of POPE:POPC (8:2) in asymmetric NaCl (500 mM NaCl cis and 50 mM NaCl trans) resulted in reproducible ion channel activity. Vpu showed a fivefold preference for monovalent cations (Na+ or K+) over anions, though conductivity of anions (Cl− and PO4−) were evident; however, as is often observed with viroporins, the conductivity varied drastically, from 14 to 280 pS, which was attributed to the amount of Vpu added to the chamber (Ewart et al. 1996).
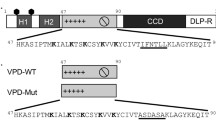
These initial results were confirmed later that year using voltage clamp of Xenopus oocytes expressing wild-type Vpu or mutants with the conserved serine phosphosphorylation sites (aa 52 and 56) mutated to arginine (these are critical for downregulation of host cell CD4). A mutant with a scrambled transmembrane domain that lacks channel activity was used as a control (Schubert et al. 1996). At 96 h post-injection of mRNA, the scrambled transmembrane mutant did not produce any current over background but WT Vpu produced a small (~130 nA), but consistent increase in inward current at −130 mV. In contrast, the Ser52Arg/Ser56Arg mutant resulted in a massively increased current (~600 nA) due to differences in expression between the wild-type and serine mutant proteins (Schubert et al. 1996). Since the serine-to-arginine mutant has the same transmembrane sequence as wild-type Vpu, the authors used this for characterization of Vpu channel activity in oocytes. The currents showed slow activation and no inactivation during the 2 s hyperpolarizing pulses. The current was primarily carried by Na+ or K+, with negligible contribution of divalent cations (Ca2+) or anions (Cl−, F−, Br−, or I−) and was not blocked by the IAV M2 blocker amantadine or Cl− channel blockers niflumic and flufenamic acids, providing evidence against activation of endogenous Cl− channels by Vpu. Whole cell studies were confirmed using synthetic peptides to the Vpu transmembrane domain for either wild-type or the scrambled TM domain mutant. The peptides were applied to POPE:POPC bilayers and distinct single channel activity was observed (Schubert et al. 1996). As with oocytes, the permeability was selective for monovalent cations. A single channel conductance was determined as 12–48 pS in 0.5 M NaCl or 17–61 pS in 0.5 M KCl, much less variable than the previous bilayer study (Ewart et al. 1996). The scrambled transmembrane domain did not form discrete currents but caused erratic current fluctuations, demonstrating specificity of the observed channel function to the Vpu sequence (Schubert et al. 1996).
The simplistic single transmembrane topology of Vpu has also made this viroporin a model system for understanding the structure-function relationship of these minimalistic ion channels. Specific residue mutations within the Vpu transmembrane domain altered ion channel activity, particularly mutation of Ser24 to alanine or leucine caused a reduction in channel activity because this residue is needed to maintain a hydrated channel within the Vpu oligomeric bundle (Mehnert et al. 2008; Padhi et al. 2014). Further, mutation of the adjacent Trp23 to leucine significantly increased channel activity and open probability, but did not change ion selectivity, which remained selective for monovalent cations and followed the Eisenman series I (Mehnert et al. 2008). Interestingly, the same Trp23Leu mutation also disrupted Vpu-mediated downregulation of CD4 due to increase Vpu oligomerization, which is inactive for targeting CD4 for degradation (Magadan and Bonifacino 2012). The interplay between Vpu’s role for targeting CD4 for degradation, sequestration of tetherin and the ion channel activity of the transmembrane domain are just beginning to be characterized (Strebel 2014).
Despite the initial studies, Vpu ion channel activity remained controversial for several years, particularly because another oocyte voltage-clamp study showed that wild-type Vpu induced the hyperpolarization-activated nonspecific cation current (HANC) often seen in oocytes, as well as it inhibited endogenous potassium currents rather than producing a de novo current (Coady et al. 1998). Further, Vpu blockers were not identified for several more years. Eventually, aliphatic modified amiloride derivatives such as hexamethylene amiloride (HMA) or dimethylamiloride (DMA)were found to block Vpu channel activity in PLB and to inhibit HIV-1 replication in cultured monocyte-derived macrophages (Ewart et al. 2004). HMA became the lead compound in a screen for novel drug-like molecules for blocking Vpu channel activity as candidate HIV-1 antiviral drugs, and from these screens the drug BIT225 was developed by Biotron Limited in Australia. BIT225 (40 μM) blocks Vpu channel activity in PLB and inhibited HIV-1 virus release for a range of strains in the low micromolar range (Khoury et al. 2010). Interestingly, BIT225 also inhibits the HCV p7 viroporin and has moved onto clinical trials against both HIV and HCV through Biotron Limited (Luscombe et al. 2010).
The biological relevance of Vpu ion channel activity remains unknown. Since the ability of Vpu to increase HIV-1 particle release mapped to the N-terminal hydrophobic domain, as does ion channel activity, it was the leading thought for many years that Vpu ion channel function was related to increasing particle release; however, more recent evidence shows that it is the hydrophobic surface of the Vpu viroporin domain that interacts with tetherin, and the BIT225 Vpu blocker and mutations that block Vpu channel function do not disrupt tetherin down-regulation by Vpu (Bolduan et al. 2011; Kuhl et al. 2011). Current studies implicate Vpu in plasma membrane depolarization through both its intrinsic ion channel activity and through impaired host potassium channel targeting to the plasma membrane, which is caused through Vpu-mediated degradation of the TASK-1 potassium channel (Hsu et al. 2010, 2004). Nevertheless, little Vpu makes it to the plasma membrane and a direct role for Vpu in altering the membrane potential of T cells or monocyte derived macrophages has not been demonstrated. Further work is needed to determine the importance, if any, Vpu ion channel activity has for HIV-1 replication (Strebel 2014).
2.2.2 Alphavirus 6K
Alphaviruses are members of the Togaviridae family and include many emerging viruses of clinical importance for humans, particularly Chikungunya virus (Morrison 2014). The alphavirus structural proteins are produced from a subgenomic mRNA as a polyprotein and cleaved by both viral and cellular proteases into the individual proteins, PE2, 6K and E1. The 6K proteins are small (~58–61 amino acid) and hydrophobic, and are critical for membrane insertion and processing of the other structural proteins (Melton et al. 2002). The ability of the 6K protein to form an ion channel was speculated about even before the viroporin field emerged, and non-electrophysiological experiments using permeability of E. coli initially identified 6K as a viroporin (Ulug et al. 1984; Sanz et al. 1994). Electrophysiology of 6K was first examined using purified bacterially expressed protein and synthetic peptides of 6K from Ross River virus (RRV) and from Barmah Forest virus (BFV) (Melton et al. 2002). For the bacterially expressed and purified protein, the authors had to reconstitute purified GST-tagged protein solubilized in 400 mg/mL CHAPS into liposomes comprised of phosphatidylethanolamine, phosphatidylcholine, and phosphatidylserine (3:1:1 ratio) and CHAPS was removed by dialysis. Interestingly, while 6K had channel activity after the GST-tag was removed, the intact GST-fusion protein had no channel activity indicating the importance of how viroporins are expressed and purified to obtain a functional protein (Melton et al. 2002).
6K PLB experiments utilized membranes with an identical composition to the liposomes and asymmetric bath solutions (cis: 10 mM TES, 500 mM NaCl; trans: 10 mM TES, 60 mM NaCl). 6K-containing liposomes were added directly to the cis chamber and spontaneously incorporated into the bilayer, resulting in variable currents with conductance between 400 and 800 pS. Currents produced from both RRV and BFV 6K were qualitatively similar to each other, as well as for either the bacterially expressed protein or synthetic peptide (Melton et al. 2002). Channel activity was voltage-dependent, with greater activity at negative membrane voltages than positive. In asymmetric solutions the reversal potential was ~48 mV, revealing a 16-fold preference for Na+ over Cl−. The permeabilities of monovalent cations (Na+ and K+) were nearly equivalent, but a three to sixfold preference of Na+ was found over Ca2+. Addition of hyperimmune sera after channel activity was observed to abolish all currents and mutation of a conserved serine residue (Ser32) to proline reduced the monovalent/divalent selectivity, indicating Ser32 may be involved in the selectivity filter structure (Melton et al. 2002).
These initial PLB results were later confirmed using voltage-clamp of Sindbis virus 6K expressing Xenopus oocytes (Antoine et al. 2007). While expression of wild-type or myc-tagged 6K in oocytes did not induce currents attributable to 6K, it did induce endogenous currents similar to those seen for HIV-1 Vpu and influenza C M2 (Coady et al. 1998; Hongo et al. 2004). A deeper investigation of the currents demonstrated 6K increased both IIN and ICl in oocytes (Antoine et al. 2007), including a 10.2-fold increase in IIN, and a 2.9-fold increase in ICl1-S, 8.4-fold increase in ICl2, and a 12.2-fold increase in ICl1-T (Kuruma and Hartzell 1999). Treating 6K-expressing oocytes with BAPTA-AM abolished the activation of both IIN and ICl, as did treating cells with low extracellular Cl−, Ba2+, Gd3+ or La3+ (Ca2+ channel blockers), niflumate (Cl− channel blocker), caffeine (InsP3 inhibitor), and thapsigargin. Finally, the authors showed that 6K caused depletion of ER Ca2+ stores and a 13-fold induction of store-operated calcium entry (SOCE) (Antoine et al. 2007). These profound alterations in ion conductance led to shrinkage of the oocytes and cytotoxicity that were partially rescued by incubation in buffers that decreased ER Ca2+ release (e.g., presence of caffeine) or reduced SOCE (e.g., low Ca2+ medium) (Antoine et al. 2007). While the patch clamp studies in oocytes could not directly study 6K ion channel activity, they still provided useful information about how 6K may function during a natural infection; however, confirmation that 6K activates SOCE in mammalian cells has not been reported.
2.2.3 Coronavirus Viroporins
2.2.3.1 Coronavirus Envelope (E) Protein
The coronavirus (CoV) envelope (E) protein is a small (~9–12 kDa) integral membrane structural protein with a short N-terminal ectodomain and single transmembrane domain and plays a central role in coronavirus replication and particle assembly. While recombinant viruses with the E protein deleted are viable in vitro, they show deficient replication and a small plaque phenotype, suggesting that E protein is important, but not essential (Wilson et al. 2004). As with most viroporins, the small size and hydrophobicity of E protein led researchers to predict that it had ion channel activity. This was confirmed using synthetic peptides of full-length SARS-CoV E and a short peptide corresponding to the N-terminal domain (aa1 −40) that were dissolved in TFE and incorporated into bilayers composed of a lipid mix of POPE/POPS/POPC (3:1:1 ratio). Addition of either full-length or truncated peptide resulted in the induction of channel currents in >60 experiments, and initiation of channel activity was enhanced with a negative holding potential relative to the cis chamber. Channel activity tested in asymmetric buffers (cis 500 mM NaCl, trans:50 mM NaCl) had a reversal potential of ~48 mV, 52 pS conductance at −88 mV and 47 pS at −48 mV, and greater current at negative potentials. Both peptides were more selective for cations over anions and were more selective for Na+ than K+. To demonstrate specificity, stable channels were inhibited by 70 % upon addition of hyperimmune serum made to the N-terminal domain into the cis chamber (Wilson et al. 2004). E protein from other CoV strains also formed cation selective channels with relatively similar conductance levels, but the selectivity for Na+ or K+ varied (Wilson et al. 2006). Like HIV-1 Vpu and HCV p7, E protein channel activity was blocked by HMA (EC50 = 10.2 μM) and reduced MHV and human CoV-229E replication in cell culture with EC50 of 3.9 and 1.3 μM, respectively (Wilson et al. 2006).
Continued work on E protein channel activity have used synthetic peptides with flanking lysine residues added to improve solubility and this resulted in higher quality single channel activity for wild-type SARS-CoV E, but channel activity was abolished for point mutants, Asn15Ala and Val25Phe. The wild-type channel characteristics showed a Na+ selective channel with 45 pS conductance in a 500 mM/5 mM asymmetric NaCl buffer system. Further, this study demonstrated channel activity was also inhibited by 100 μM amantadine (Torres et al. 2007). Finally, lipid composition influenced E protein channel activity, with negatively charged lipid bilayers (DPhPS) yielding channels with greater cation selectivity, weak selectivity of K+ over Na+, and higher conductance than those in neutral DPhPC lipid bilayers (Verdia-Baguena et al. 2012, 2013).
Despite considerable electrophysiology data on different CoV E proteins, the role ion channel activity has for CoV during infection remains unknown and while E protein can traffic to the plasma membrane, few electrophysiology studies in mammalian cells have been conducted. Whole-cell voltage clamp experiments using HEK293 cells transiently expressing SARS-CoV E found E protein induced moderate inward and large outward currents that were not observed in control cells. These currents were inhibited by HMA and did not show substantial selectivity between Na+ and K+, characteristics similar to PLB studies (Pervushin et al. 2009). However, others have found no E protein localization to the plasma membrane when expressed in HET293T cells and decreased, not increased, whole cell current (Nieto-Torres et al. 2011). Further, E protein disrupted host cell Na+/K+ ATPase α1 and ENaC channels; however, it is not known whether this is related to its ion channel function (Ji et al. 2009). Further work on the biological relevance of E protein channel activity is needed but E protein remains a potential target for antiviral drug development.
2.2.3.2 SARS-CoV ORF3a and ORF8
The SARS coronavirus (SARS-CoV) encodes two other viroporins, called ORF3a and ORF8. Like E protein, ion channel activity of these proteins has been identified, but their functional roles have not be determined. Their role is thought not to be critical for virus replication or virulence in vivo, because engineered virus or naturally occurring deletion mutants lacking ORF3a or ORF8, respectively, retain high replication and pathogenesis (DeDiego et al. 2014; Yount et al. 2005).
SARS ORF3a is the largest viroporin protein identified thus far, composed of 274 amino acids and three transmembrane domains, that is transported to the surface of virus-infected cells and is also incorporated as a minor component of mature virions. ORF3a folds into disulfide-stabilized homotetramers with a short N-terminal ectodomain and longer cytoplasmic tail. Based on topology and oligomerization, ORF3a was predicted to have ion channel activity, which was confirmed using voltage clamp in Xenopus oocytes expressing wild-type ORF3a and abrogation of channel activity for a cysteine-to-alanine mutant that failed to oligomerize (Lu et al. 2006). ORF3a increased K+ currents in oocytes that are insensitive to typical K+ channel blockers (tetraethylammonium and cesium) but was blocked by 10 mM Ba2+ in the bath buffer. Synthetic peptides of the three SARS ORF3a transmembrane domains have been tested in PLB experiments (POPE:DOPC, 1:4) to identify transmembrane segment 3 to be the main channel domain. While channel activity was influenced when the other transmembrane segments were mix in, they did not recapitulate the channel activity observed for full-length protein. Full-length ORF3a had single channel conductance levels of 3.0, 6.1 and 12.2 pS, and, in contrast with the oocyte voltage clamp data, was able to conduct Ca2+ with conductance of 10 and 58 pS in 500 mM CaCl2 (Chien et al. 2013). However, whether Ca2+ conductance is a biologically relevant function of ORF3a remains unknown.
Similar K+ channel activity was observed for ORF3 from PEDV and ORF4a from coronavirus 229E, and mutations in PEDV ORF3 found in attenuated strains correlated with decreased currents. In contrast to the K+ selective conductance observed for SARS ORF3a, ORF4a conducted Na+, K+, Rb+, Cs+ to similar levels, while Li+ partially reduced currents, indicating ORF4a is a non-selective monovalent cation channel (Zhang et al. 2014). This suggests that ORF3a and ORF4a may be functional homologs, and while they increase virion release, the mechanism by which this occurs is unknown (Wang et al. 2012; Zhang et al. 2014).
While SARS ORF3a is the largest viroporin identified, the SARS ORF8a viroporin is the smallest at only 39 amino acids (Chen et al. 2011). Channel recordings of an ORF8a synthetic peptide were performed in POPC bilayers at 38 °C (to simulate body temperature) in the presence of DTT because the protein contains four conserved cysteine residues that align along one face of the amphipathic alpha-helix. Currents were variable but with a primary conductance state of 8.8 pS with weak cation selectivity. Using computational modeling, ORF8a is expected to form a pentameric channel assembly; however, both the structure and ion channel function remain to be confirmed, as well as the possible importance of the channel function for virus replication (Chen et al. 2011).
2.2.4 Respiratory Syncytial Virus SH
Paramyxoviruses are nonsegmented negative stranded RNA viruses that include a number of key human pathogens, including Mumps virus (MuV) and human respiratory syncytial virus (hRSV) (Parks 2013, 24056173). Three membrane proteins are encoded by paramyxoviruses, the fusion protein (F), the glycoprotein (G) and small hydrophobic protein (SH) (McLellan et al. 2013). The SH protein is a 64–65 amino acid protein with a single transmembrane domain with the N-terminus remaining in the cytoplasm and the C-terminus forming the ectodomain. RSV SH homologs are found in parainfluenza virus 5, mumps virus and J paramyxovirus and human metapneumovirus; and while deletion of SH does not affect virus replication in vitro, it results in attenuation in vivo (Gan et al. 2008). Therefore the role of the SH protein remains unknown; however, SH is implicated in blocking TNF-alpha-mediated apoptosis and activation of the inflammasome (Triantafilou et al. 2013). Due to its short length, high hydrophobic nature and the small amounts found in virions, SH was predicted to be a viroporin and initially studied in the E. coli-based functional assays and shown to have viroporin-like characteristics (Perez et al. 1997).
Channel activity of a SH synthetic peptide was tested using PLB [POPE:POPS:POPC (5:3:2 v/v/v)] in asymmetric buffer (cis, 500 mM NaCl; trans, 50 mM NaCl) and found to have a cation-selective channel activity with a conductance of ~35 pS and reversal potential at −68.1 mV, near the sodium Nernst potential of 59 mV. Unlike the influenza A M2 proton channel, SH was not low-pH activated, but was in fact inhibited by decreasing the pH to 4.0 (Gan et al. 2008). Structural studies of E. coli expressed SH show formation of channel-like homopentameric rings and solved the NMR structure of SH in detergent micelles (Carter et al. 2010; Gan et al. 2012). Finally, using a liposome dye release-based drug screen, a potential RSV SH blocker called pyronin B was identified (Li et al. 2014). Pyronin B caused a 60 % inhibition of SH channel activity of purified full-length SH in PLBs, with a Kd of 6.8 μM. NMR and docking studies determined that pyronin B interacted with SH residues 39–44, and mutation of Ala39 to serine resulted in nearly complete resistance to pyronin B-mediated inhibition of channel activity (Li et al. 2014). Pyronin B was also able to block RSV replication in Vero cells, a monkey kidney cell line, making this a promising lead compound for a RSV antiviral drug.
In attempts to understand the biological role and molecular regulation of SH channel activity, whole cell patch clamp of SH-expressing HEK293T cells was performed, using artificial cerebrospinal fluid bath buffer and a potassium gluconate-based pipette buffer to mimic physiological conditions found in vivo (Gan et al. 2012). SH induced a fivefold higher current at 70 mV in low pH (5.5) than at neutral pH (7.4), and the reversal potential was close to 0 mV, indicating no selectivity between Na+ and K+. To examine the mechanism of the low-pH activation, mutants of two conserved histidine residues (His22 and His51) to alanine or phenylalanine were tested and had similar or increased (His22) currents at pH 5.5 relative to wild-type SH. Double mutations eliminated SH channel activity, suggesting the histidine residues are functionally important for SH but not whether they are involved in pH-dependent activation (Gan et al. 2012). However, the HEK293 patch clamp studies are not conclusive because the same group was unable to reproduce the low-pH activation of SH currents in a later study (Li et al. 2014). Thus, the biological role for SH channel activity during RSV infection, and whether channel activity is linked to dysregulation of cytokine signaling (e.g., TNF-alpha or inflammosomes) remains unknown (Triantafilou et al. 2013).
2.2.5 Influenza A Virus PB1-F2
While the influenza A virus M2 proton channel is the most well studied and well defined viral ion channel, another influenza A protein called PB1-F2 is gaining notoriety as a virulence factor in highly pathogenic influenza strains. PB1-F2 is encoded by the second genome segment by a +1 alternate open reading frame in many influenza A strains, but is absent from influenza B virus. The functional role(s) of PB1-F2 are still being elucidated; however, at least one of its primary roles is the induction of cellular apoptosis by activation of the intrinsic apoptotic pathway (Chanturiya et al. 2004). Structural studies indicate PB1-F2 forms a positively charged amphipathic alpha-helix and has a high propensity for oligomerization. A mitochondrial localization sequence was first defined to be between amino acids 65–87 (Gibbs et al. 2003) and then refined to amino acids 46–75, with two conserved basic residues (Lys73 & Arg75) to be required for trafficking to mitochondria and association with the inner and outer mitochondrial membranes (Yamada et al. 2004). Localization of PB1-F2 to mitochondria resulted in a loss of mitochondrial membrane potential, release of cytochrome c and induction of apoptosis (Gibbs et al. 2003). The first study to examine the potential ion channel function of PB1-F2 used planar lipid bilayers of various compositions (Chanturiya et al. 2004). Addition of PB1-F2 to PLB chambers induced currents without typical unitary fluctuations of channels and the lack of transitions was not influenced by different lipid or buffer compositions; however, the observed currents were greatest with neutral lipids in monovalent cation-based solutions. PB1-F2 is selective for cations over anions, but addition of Ca2+ increased Cl− permeability. Based on the membrane destabilization and lack of discrete channel-like behavior, PB1-F2 was thought to form lipidic pores rather than proteinaceous channels (Chanturiya et al. 2004). However, a recent study of a synthetic PB1-F2 peptide in phosphatidylcholine bilayers found that typical channel events were observed among the unresolved fast fluctuations. Traces containing unitary channel activity were occasionally interrupted by strong bursts of unresolved current and there appeared to be two distinct open levels (O1 and O2). In contrast with the previous study, PB1-F2 conducted anions better than cations, but did not discriminate between K+ and Na+ or Cl− and gluconate, and was able to conduct Ca2+ (Henkel et al. 2010). More work is needed to determine whether the channel activity of PB1-F2 is directly responsible for induction of apoptosis through disruption of the mitochondria membrane potential, possibly by directly patching mitochondria from influenza A-infected cells or cells expressing PB1-F2.
2.2.6 Dengue Virus M
Dengue virus is a member of the Flaviviruses in the Togaviridae family. Dengue viruses cause up to 528 million infections per year, with approximately 20 % of these infections resulting in clinically significant disease, making it an emerging virus of significance to public health (Salazar et al. 2014). The Dengue virus membrane protein (M) is a transmembrane protein that is co-translationally inserted into the ER membrane and the mature 75-aa protein is produced from the preM precursor by furin cleavage (Hsieh et al. 2014). Dengue virus entry requires endosomal acidification, suggesting preM or M may have ion channel activity (Premkumar et al. 2005). PLB studies (PE:PS:PC, 5:3:2 ratio) of a 40 amino acid synthetic C-terminal peptide in asymmetric NaCl or KCl buffers (cis, 500 mM; trans, 50 mM) showed large and variable currents, with few single channel transitions (~6 pS) (Premkumar et al. 2005). The observed reversal potential of +38 mV suggested K+ selectivity over Cl−. Both HMA and amantadine inhibited M channel activity (Premkumar et al. 2005). However, a recent study refuted the PLB data, as no significant conductance from preM/M was detected by voltage clamp of Xenopus oocytes (Wong et al. 2011). Given the few studies on preM/M, future studies are needed to determine whether preM/M forms a bona fide ion channel.
2.3 Viral Divalent Cation Conductance
A number of viruses and viroporins disrupt host cell Ca2+ homeostasis during infection; however, thus far, none of the viroporins that have been studied by electrophysiology has shown selectivity for divalent cations over monovalent cations (Zhou et al. 2009). Nevertheless, some degree of Ca2+ conductivity has been observed for some viroporins. PLB recordings of bacterially expressed p7 in 0.1 M CaCl2 resulted in significantly larger currents than in KCl; however, whether p7 antagonists (e.g., HMA or amantadine) or channel-disrupting mutations block Ca2+ conductance was not studied (Griffin et al. 2003). A similar experimental set-up (cis 500 mM KCl, trans 500 mM CaCl2) demonstrated that influenza A virus PB1-F2 was also able to conduct Ca2+ and a liposome-based assay using Fluo-3 loaded vesicles confirmed the ability of PB1-F2 to conduct Ca2+ into the liposome (Henkel et al. 2010).
In general, Ca2+ conductance by viroporins is demonstrated through indirect means, usually through fluorescent Ca2+ imaging to measure disruption in host cell Ca2+ homeostasis by examining the resulting activation of host cell channels due to viroporin-mediated Ca2+ currents. As detailed above, whole-cell voltage clamp experiments of Sindbis virus 6K in oocytes showed activation of SOCE and calcium-activated chloride channels, but current attributable to 6K was not detected (Antoine et al. 2007). Activation of SOCE by viroporin-mediated release of ER Ca2+ stores and activation of STIM1 may be a function shared by other ER-localized viroporins, as demonstrated for the rotavirus viroporin nonstructural protein 4 (NSP4) and predicted for the picornavirus 2B (van Kuppeveld et al. 1997; Hyser et al. 2013). The resulting Ca2+ fluxes affect multiple cell functions that facilitate virus replication and/or are involved in disease (Zhou et al. 2009).
For rotavirus, NSP4 viroporin activity is sufficient to disrupt Ca2+ homeostasis and therefore an excellent model to study the electrophysiology of Ca2+-conducting viroporins (Hyser et al. 2013). Using both liposome patch clamp and PLB electrophysiology of a 44-amino acid synthetic NSP4 viroporin domain peptide (aa47-90), we have observed ion channel activity, including both the highly variable “burst-like” activity and unitary single channel conductance. In liposome patch clamp of NSP4 in asymmetric buffers (pipette 150 mM KCl; bath 75 mM CaCl2), single channels were observed with both positive and negative pipette voltages, indicating NSP4 can conduct both K+ and Ca2+, although contribution of Cl− cannot be ruled out (Hyser and Delcour, unpublished data, 2015). Of particular interest is how viroporins such as NSP4 and 2B, but not other ER-localized viroporins (e.g., HIV-1 Vpu), release ER Ca2+. A highly conserved Ca2+ binding site in NSP4 may represent a regulatory motif, as it is found immediately adjacent to the NSP4 viroporin domain in the cytoplasmic coiled-coil domain (Chacko et al. 2011; Sastri et al. 2014). This motif is selective for Ca2+ and Ba2+, but not Mg2+, and binding is lost at pH <5.6, which corresponds with a conversion of the protein from a Ca2+-bound tetramer to a Ca2+-free pentamer with an open central channel (Sastri et al. 2014). Further electrophysiological studies are needed to establish whether these channel show any Ca2+ selectivity or whether they are non-selective channels that exploit the natural Ca2+ gradient across the ER membrane to provide the driving force for Ca2+ over other ions.
2.4 Viral Anion Channels
All currently identified viroporins preferentially conduct cations over anions; although high concentration of CaCl2 or some transmembrane domain mutants alter the channel selectivity, but not to the extent that the viroporin becomes an anion channel (Griffin et al. 2003; Henkel et al. 2010). The primary exception to this have been recombinant viroporins from HIV-1 Vpu and alphavirus 6K expressed in Xenopus oocytes, where strong Cl− currents were detected by whole cell voltage clamp (Coady et al. 1998; Melton et al. 2002; Antoine et al. 2007). Such was also the case for enterovirus 71 (EV71) 2B. Recombinant EV71 2B was expressed in Xenopus oocytes and large hyperpolarization-dependent currents were detected, but not for a transmembrane deletion mutant. Replacing Cl− with gluconate reduced currents and the current was blocked by the well-characterized chloride channel blocker DIDs, while replacing Na+ with NMDG had little effect, which suggested the invoked currents were carried by Cl−. The authors postulated that the 2B anion channel activity may cause elevated Ca2+ indirectly by reducing ATP synthesis or by disrupting anion balance across the Golgi membrane, causing leakiness to Ca2+ (Xie et al. 2011). However, this study did not address the possible indirect activation of oocyte Ca2+-activated chloride channels (CaCC), similar to what was determined for the Sindbis virus 6K protein expressed in oocytes (Antoine et al. 2007). These conflicting results underline the need for more studies of viroporins that disrupt host Ca2+ homeostasis. Further, if 2B is indirectly activating CaCC channels through induction of SOCE, then there are no candidate viroporin anion channels yet identified and this represents an interesting niche to be filled.
3 Summary and Future Directions
The first identification of viral ion channels occurred 23 years ago and the electrophysiological study of these proteins has accelerated rapidly ever since. However, it is safe to say that the viroporin field is still in its infancy, with many more questions remaining about viroporin biology to answer. Of primary importance to understanding viroporin electrophysiology is continued efforts to develop robust methods for expression, purification, and analysis of viroporins. This is particularly important because of the tendency of viroporins to induce highly variable “burst-like” currents rather than the canonical (and sought after) clear unitary conductance states (i.e., square-topped channel activity). While the burst-like activity is still likely true ion channel activity, it makes it more difficult to quantify the channel characteristics and therefore inpairs direct comparisons between different studies. This is further compounded by the fact that viroporins are able to adopt multiple oligomeric states, which is particularly true for peptides of the isolated transmembrane domains. Work to design and test peptides that adopt specific oligomeric states, such as with the Vpu tetramer versus pentamer, demonstrated these different oligomers may have different channel characteristics (Becker et al. 2004). Further, the evidence that the lipid environment and ionic concentration impact viroporin channel characteristics suggests these variables are important considerations for optimization of clear channel recordings (Whitfield et al. 2011; Verdia-Baguena et al. 2012, 2013). Establishing a systematic methodology for the reconstitution and testing of viroporins is needed to facilitate comparison between currently known viroporins, and to provide an experimentally robust framework for the testing of newly discovered viroporins, such as the papillomavirus E5 and ephemeral fever rhabdovirus α1 proteins, as well as many others (Wetherill et al. 2012; Joubert et al. 2014).
The majority of viroporins identified were tested due to similarities between a protein of interest and a known viroporin, typically based on transmembrane domain location and topology, and general sequence similarities in the viroporin signature motifs (polybasic domain and amphipathic α-helix). Using this methodology, the viroporins identified to date all have relatively non-specific cation channel activity, with the notable exception of IAV M2, which is a very specific proton channel (Chizhmakov et al. 1996). This raises the question of whether current methods to identify viroporins are too narrow and therefore only identifying those proteins that have similar functions, i.e., non-specific cation channels. While many classically defined viroporins have yet to be examined using electrophysiological techniques [reviewed in (Nieva et al. 2012; Giorda and Hebert 2013)], it is likely that we have only scratched the surface for identifying and characterizing viral ion channels. Given the fundamental importance ion channels play in cellular homeostasis, it is inconceivable to think viroporins are not found among viruses from all of the kingdoms of life, including among bacterial and archeal phages. A likely example in bacteriophage are pinholins, which induce cell lysis by depolarization of the host membrane but are too small to allow passage of endolysins like their larger cousins the holins (Young 2014). No candidate ion channels have been reported for archeal phages, and with the exception of the Kcv potassium channel in Phycodnaviridae, ion channels in protozoan and plate viruses remain unexplored. Since viroporins of mammalian viruses have proven to be a key aspect of pathogenesis, as well as exceptional models for minimalistic ion channel structure/function experiments, the search for viroporins from other groups of viruses represents a new challenge for collaboration between microbiologists and physiologists, particularly because electrophysiology equipment and expertise are not a typical part of the virologist’s experimental armamentarium. Further, because there is little primary amino acid sequence similarity among viroporins, protein sequence alignments are unlikely to yield identification of promising candidates across kingdoms. Therefore, new ways to identify viroporin motifs, perhaps capitalizing on the biophysical requirements needed for ion channel formation (e.g., clustered basic residues and high amphipathicity), are needed to search through the huge amount of sequence data becoming available from next-generation genomic and metagenomic sequencing of environmental, animal and human samples.
In conclusion, the current electrophysiological knowledge of viroporins demonstrates that they are truly unconventional ion channels. This underlines the importance of studying viroporins in terms of basic biology and for biomedical research. Much can be learned about ion channel biophysics by studying viroporins, because while few viroporins are ion selective, they do appear to discriminate between cations and anions, which is remarkable given the simple structure of viroporins relative to conventional ion channels. Further, viroporins invariably play critical functional roles for virus replication and pathogenesis and in many cases, specific blockers make effective antiviral drugs. Therefore, continued work is needed to establish robust methods for viroporin identification, characterization of the ion channel activity, and screening for blockers that may be useful therapeutics to combat disease.
References
Agirre A, Barco A, Carrasco L, Nieva JL (2002) Viroporin-mediated membrane permeabilization. Pore formation by nonstructural poliovirus 2B protein. J Biol Chem 277:40434–40441
Andrew A, Strebel K (2014) HIV-1 accessory proteins: Vpu and Vif. Methods Mol Biol 1087:135–158
Antoine AF, Montpellier C, Cailliau K, Browaeys-Poly E, Vilain JP, Dubuisson J (2007) The alphavirus 6K protein activates endogenous ionic conductances when expressed in Xenopus oocytes. J Membr Biol 215:37–48
Atoom AM, Taylor NG, Russell RS (2014) The elusive function of the hepatitis C virus p7 protein. Virology 462–463:377–387
Becker CF, Oblatt-Montal M, Kochendoerfer GG, Montal M (2004) Chemical synthesis and single channel properties of tetrameric and pentameric TASPs (template-assembled synthetic proteins) derived from the transmembrane domain of HIV virus protein u (Vpu). J Biol Chem 279:17483–17489
Betakova T, Hay AJ (2007) Evidence that the CM2 protein of influenza C virus can modify the pH of the exocytic pathway of transfected cells. J Gen Virol 88:2291–2296
Bodelon G, Labrada L, Martinez-Costas J, Benavente J (2002) Modification of late membrane permeability in avian reovirus-infected cells: viroporin activity of the S1-encoded nonstructural p10 protein. J Biol Chem 277:17789–17796
Bolduan S, Votteler J, Lodermeyer V, Greiner T, Koppensteiner H, Schindler M, Thiel G, Schubert U (2011) Ion channel activity of HIV-1 Vpu is dispensable for counteraction of CD317. Virology 416:75–85
Bour S, Strebel K (2003) The HIV-1 Vpu protein: a multifunctional enhancer of viral particle release. Microbes Infect 5:1029–1039
Carter SD, Dent KC, Atkins E, Foster TL, Verow M, Gorny P, Harris M, Hiscox JA, Ranson NA, Griffin S, Barr JN (2010) Direct visualization of the small hydrophobic protein of human respiratory syncytial virus reveals the structural basis for membrane permeability. FEBS Lett 584:2786–2790
Chacko AR, Arifullah M, Sastri NP, Jeyakanthan J, Ueno G, Sekar K, Read RJ, Dodson EJ, Rao DC, Suguna K (2011) Novel pentameric structure of the diarrhea-inducing region of the rotavirus enterotoxigenic protein NSP4. J Virol 85:12721–12732
Chanturiya AN, Basanez G, Schubert U, Henklein P, Yewdell JW, Zimmerberg J (2004) p B1–F2, an influenza A virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J Virol 78:6304–6312
Chen CP, Kremer C, Henklein P, Schubert U, Fink RH, Fischer WB (2010) Modulating the activity of the channel-forming segment of Vpr protein from HIV-1. Eur Biophys J 39:1089–1095
Chen CC, Kruger J, Sramala I, Hsu HJ, Henklein P, Chen YM, Fischer WB (2011) ORF8a of SARS-CoV forms an ion channel: experiments and molecular dynamics simulations. Biochim Biophys Acta 1808:572–579
Chew CF, Vijayan R, Chang J, Zitzmann N, Biggin PC (2009) Determination of pore-lining residues in the hepatitis C virus p7 protein. Biophys J 96:L10–L12
Chien TH, Chiang YL, Chen CP, Henklein P, Hanel K, Hwang IS, Willbold D, Fischer WB (2013) Assembling an ion channel: ORF 3a from SARS-CoV. Biopolymers 99:628–635
Chizhmakov IV, Geraghty FM, Ogden DC, Hayhurst A, Antoniou M, Hay AJ (1996) Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J Physiol 494(Pt 2):329–336
Coady MJ, Daniel NG, Tiganos E, Allain B, Friborg J, Lapointe JY, Cohen EA (1998) Effects of Vpu expression on Xenopus oocyte membrane conductance. Virology 244:39–49
DeDiego ML, Nieto-Torres JL, Jimenez-Guardeno JM, Regla-Nava JA, Castano-Rodriguez C, Fernandez-Delgado R, Usera F, Enjuanes L (2014) Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res 194:124–137
Delcour AH, Martinac B, Adler J, Kung C (1989) Modified reconstitution method used in patch-clamp studies of Escherichia coli ion channels. Biophys J 56:631–636
Ewart GD, Sutherland T, Gage PW, Cox GB (1996) The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J Virol 70:7108–7115
Ewart GD, Mills K, Cox GB, Gage PW (2002) Amiloride derivatives block ion channel activity and enhancement of virus-like particle budding caused by HIV-1 protein Vpu. Eur Biophys J 31:26–35
Ewart GD, Nasr N, Naif H, Cox GB, Cunningham AL, Gage PW (2004) Potential new anti-human immunodeficiency virus type 1 compounds depress virus replication in cultured human macrophages. Antimicrob Agents Chemother 48:2325–2330
Gan SW, Ng L, Lin X, Gong X, Torres J (2008) Structure and ion channel activity of the human respiratory syncytial virus (hRSV) small hydrophobic protein transmembrane domain. Protein Sci 17:813–820
Gan SW, Tan E, Lin X, Yu D, Wang J, Tan GM, Vararattanavech A, Yeo CY, Soon CH, Soong TW, Pervushin K, Torres J (2012) The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J Biol Chem 287:24671–24689
Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW (2003) The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol 77:7214–7224
Giorda KM, Hebert DN (2013) Viroporins customize host cells for efficient viral propagation. DNA Cell Biol 32:557–564
Gonzalez ME, Carrasco L (2003) Viroporins. FEBS Lett 552:28–34
Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ (2003) The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett 535:34–38
Griffin SD, Harvey R, Clarke DS, Barclay WS, Harris M, Rowlands DJ (2004) A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J Gen Virol 85:451–461
Han Z, Harty RN (2004) The NS3 protein of bluetongue virus exhibits viroporin-like properties. J Biol Chem 279:43092–43097
Henkel M, Mitzner D, Henklein P, Meyer-Almes FJ, Moroni A, Difrancesco ML, Henkes LM, Kreim M, Kast SM, Schubert U, Thiel G (2010) The proapoptotic influenza A virus protein PB1-F2 forms a nonselective ion channel. PLoS One 5:e11112
Hongo S, Ishii K, Mori K, Takashita E, Muraki Y, Matsuzaki Y, Sugawara K (2004) Detection of ion channel activity in Xenopus laevis oocytes expressing Influenza C virus CM2 protein. Arch Virol 149:35–50
Hsieh SC, Wu YC, Zou G, Nerurkar VR, Shi PY, Wang WK (2014) Highly conserved residues in the helical domain of dengue virus type 1 precursor membrane protein are involved in assembly, precursor membrane (prM) protein cleavage, and entry. J Biol Chem 289:33149–33160
Hsu K, Seharaseyon J, Dong P, Bour S, Marban E (2004) Mutual functional destruction of HIV-1 Vpu and host TASK-1 channel. Mol Cell 14:259–267
Hsu K, Han J, Shinlapawittayatorn K, Deschenes I, Marban E (2010) Membrane potential depolarization as a triggering mechanism for Vpu-mediated HIV-1 release. Biophys J 99:1718–1725
Hyser JM, Collinson-Pautz MR, Utama B, Estes MK (2010) Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. MBio 1:e00265-10
Hyser JM, Utama B, Crawford SE, Estes MK (2012) Genetic divergence of rotavirus nonstructural protein 4 results in distinct serogroup-specific viroporin activity and intracellular punctate structure morphologies. J Virol 86:4921–4934
Hyser JM, Utama B, Crawford SE, Broughman JR, Estes MK (2013) Activation of the endoplasmic reticulum calcium sensor STIM1 and store-operated calcium entry by rotavirus requires NSP4 viroporin activity. J Virol 87:13579–13588
Jacotot E, Ferri KF, El HC, Brenner C, Druillennec S, Hoebeke J, Rustin P, Metivier D, Lenoir C, Geuskens M, Vieira HL, Loeffler M, Belzacq AS, Briand JP, Zamzami N, Edelman L, Xie ZH, Reed JC, Roques BP, Kroemer G (2001) Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J Exp Med 193:509–519
Ji HL, Song W, Gao Z, Su XF, Nie HG, Jiang Y, Peng JB, He YX, Liao Y, Zhou YJ, Tousson A, Matalon S (2009) SARS-CoV proteins decrease levels and activity of human ENaC via activation of distinct PKC isoforms. Am J Physiol Lung Cell Mol Physiol 296:L372–L383
Joubert DA, Blasdell KR, Audsley MD, Trinidad L, Monaghan P, Dave KA, Lieu KG, Amos-Ritchie R, Jans DA, Moseley GW, Gorman JJ, Walker PJ (2014) Bovine ephemeral fever rhabdovirus alpha1 protein has viroporin-like properties and binds importin beta1 and importin 7. J Virol 88:1591–1603
Kang M, Moroni A, Gazzarrini S, DiFrancesco D, Thiel G, Severino M, Van Etten JL (2004) Small potassium ion channel proteins encoded by chlorella viruses. Proc Natl Acad Sci U S A 101:5318–5324
Kelly ML, Cook JA, Brown-Augsburger P, Heinz BA, Smith MC, Pinto LH (2003) Demonstrating the intrinsic ion channel activity of virally encoded proteins. FEBS Lett 552:61–67
Khoury G, Ewart G, Luscombe C, Miller M, Wilkinson J (2010) Antiviral efficacy of the novel compound BIT225 against HIV-1 release from human macrophages. Antimicrob Agents Chemother 54:835–845
Kuhl BD, Cheng V, Donahue DA, Sloan RD, Liang C, Wilkinson J, Wainberg MA (2011) The HIV-1 Vpu viroporin inhibitor BIT225 does not affect Vpu-mediated tetherin antagonism. PLoS One 6:e27660
Kuruma A, Hartzell HC (1999) Dynamics of calcium regulation of chloride currents in Xenopus oocytes. Am J Physiol 276:C161–C175
Lama J, Carrasco L (1992) Expression of poliovirus nonstructural proteins in Escherichia coli cells. Modification of membrane permeability induced by 2B and 3A. J Biol Chem 267:15932–15937
Lamb RA, Pinto LH (1997) Do Vpu and Vpr of human immunodeficiency virus type 1 and NB of influenza B virus have ion channel activities in the viral life cycles? Virology 229:1–11
Li Y, To J, Verdia-Baguena C, Dossena S, Surya W, Huang M, Paulmichl M, Liu DX, Aguilella VM, Torres J (2014) Inhibition of the human respiratory syncytial virus small hydrophobic protein and structural variations in a bicelle environment. J Virol 88:11899–11914
Lin TI, Schroeder C (2001) Definitive assignment of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J Virol 75:3647–3656
Loregian A, Mercorelli B, Nannetti G, Compagnin C, Palu G (2014) Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell Mol Life Sci 71:3659–3683
Lu W, Zheng BJ, Xu K, Schwarz W, Du L, Wong CK, Chen J, Duan S, Deubel V, Sun B (2006) Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc Natl Acad Sci U S A 103:12540–12545
Luscombe CA, Huang Z, Murray MG, Miller M, Wilkinson J, Ewart GD (2010) A novel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viral diarrhea virus in vitro and shows synergism with recombinant interferon-alpha-2b and nucleoside analogues. Antiviral Res 86:144–153
Madan, V., Castelló, A., & Carrasco, L. (2008). Viroporins from RNA viruses induce caspase-dependent apoptosis. Cellular Microbiology, 10(2), 437–451. doi:10.1111/j.1462-5822.2007.01057.x
Magadan JG, Bonifacino JS (2012) Transmembrane domain determinants of CD4 Downregulation by HIV-1 Vpu. J Virol 86:757–772
McLellan JS, Ray WC, Peeples ME (2013) Structure and function of respiratory syncytial virus surface glycoproteins. Curr Top Microbiol Immunol 372:83–104
Mehnert T, Routh A, Judge PJ, Lam YH, Fischer D, Watts A, Fischer WB (2008) Biophysical characterization of Vpu from HIV-1 suggests a channel-pore dualism. Proteins 70:1488–1497
Melton JV, Ewart GD, Weir RC, Board PG, Lee E, Gage PW (2002) Alphavirus 6K proteins form ion channels. J Biol Chem 277:46923–46931
Meshkat Z, Audsley M, Beyer C, Gowans EJ, Haqshenas G (2009) Reverse genetic analysis of a putative, influenza virus M2 HXXXW-like motif in the p7 protein of hepatitis C virus. J Viral Hepat 16:187–194
Montserret R, Saint N, Vanbelle C, Salvay AG, Simorre JP, Ebel C, Sapay N, Renisio JG, Bockmann A, Steinmann E, Pietschmann T, Dubuisson J, Chipot C, Penin F (2010) NMR structure and ion channel activity of the p7 protein from hepatitis C virus. J Biol Chem 285:31446–31461
Morrison TE (2014) Reemergence of chikungunya virus. J Virol 88:11644–11647
Mould JA, Drury JE, Frings SM, Kaupp UB, Pekosz A, Lamb RA, Pinto LH (2000) Permeation and activation of the M2 ion channel of influenza A virus. J Biol Chem 275:31038–31050
Mould JA, Paterson RG, Takeda M, Ohigashi Y, Venkataraman P, Lamb RA, Pinto LH (2003) Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes. Dev Cell 5:175–184
Nieto-Torres JL, DeDiego ML, Alvarez E, Jimenez-Guardeno JM, Regla-Nava JA, Llorente M, Kremer L, Shuo S, Enjuanes L (2011) Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 415:69–82
Nieva JL, Madan V, Carrasco L (2012) Viroporins: structure and biological functions. Nat Rev Microbiol 10:563–574
Okada A, Miura T, Takeuchi H (2001) Protonation of histidine and histidine-tryptophan interaction in the activation of the M2 ion channel from influenza a virus. Biochemistry 40:6053–6060
Padhi S, Burri RR, Jameel S, Priyakumar UD (2014) Atomistic detailed mechanism and weak cation-conducting activity of HIV-1 Vpu revealed by free energy calculations. PLoS One 9:e112983
Pavlovic D, Neville DC, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N (2003) The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc Natl Acad Sci U S A 100:6104–6108
Perez M, Garcia-Barreno B, Melero JA, Carrasco L, Guinea R (1997) Membrane permeability changes induced in Escherichia coli by the SH protein of human respiratory syncytial virus. Virology 235:342–351
Pervushin K, Tan E, Parthasarathy K, Lin X, Jiang FL, Yu D, Vararattanavech A, Soong TW, Liu DX, Torres J (2009) Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog 5:e1000511
Piller SC, Ewart GD, Premkumar A, Cox GB, Gage PW (1996) Vpr protein of human immunodeficiency virus type 1 forms cation-selective channels in planar lipid bilayers. Proc Natl Acad Sci U S A 93:111–115
Piller SC, Ewart GD, Jans DA, Gage PW, Cox GB (1999) The amino-terminal region of Vpr from human immunodeficiency virus type 1 forms ion channels and kills neurons. J Virol 73:4230–4238
Pinto LH, Lamb RA (2006) The M2 proton channels of influenza A and B viruses. J Biol Chem 281:8997–9000
Pinto LH, Holsinger LJ, Lamb RA (1992) Influenza virus M2 protein has ion channel activity. Cell 69:517–528
Premkumar A, Wilson L, Ewart GD, Gage PW (2004) Cation-selective ion channels formed by p7 of hepatitis C virus are blocked by hexamethylene amiloride. FEBS Lett 557:99–103
Premkumar A, Horan CR, Gage PW (2005) Dengue virus M protein C-terminal peptide (DVM-C) forms ion channels. J Membr Biol 204:33–38
Salazar MI, del Angel RM, Lanz-Mendoza H, Ludert JE, Pando-Robles V (2014) The role of cell proteins in dengue virus infection. J Proteomics 111:6–15
Sanz MA, Perez L, Carrasco L (1994) Semliki Forest virus 6K protein modifies membrane permeability after inducible expression in Escherichia coli cells. J Biol Chem 269:12106–12110
Sastri NP, Viskovska M, Hyser JM, Tanner MR, Horton LB, Sankaran B, Prasad BV, Estes MK (2014) Structural plasticity of the coiled-coil domain of rotavirus NSP4. J Virol 88:13602–13612
Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M (1996) Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett 398:12–18
Shimbo K, Brassard DL, Lamb RA, Pinto LH (1995) Viral and cellular small integral membrane proteins can modify ion channels endogenous to Xenopus oocytes. Biophys J 69:1819–1829
Shimbo K, Brassard DL, Lamb RA, Pinto LH (1996) Ion selectivity and activation of the M2 ion channel of influenza virus. Biophys J 70:1335–1346
Stewart SM, Pekosz A (2012) The influenza C virus CM2 protein can alter intracellular pH, and its transmembrane domain can substitute for that of the influenza A virus M2 protein and support infectious virus production. J Virol 86:1277–1281
StGelais C, Tuthill TJ, Clarke DS, Rowlands DJ, Harris M, Griffin S (2007) Inhibition of hepatitis C virus p7 membrane channels in a liposome-based assay system. Antiviral Res 76:48–58
Strebel K (2013a) HIV accessory proteins versus host restriction factors. Curr Opin Virol 3:692–699
Strebel K (2014) HIV-1 Vpu — an ion channel in search of a job, Biochimica et Biophysica Acta (BBA) - Biomembranes, 1838(4): 1074–1081, ISSN 0005-2736, http://dx.doi.org/10.1016/j.bbamem.2013.06.029.
Strebel K (2014) HIV-1 Vpu – an ion channel in search of a job. Biochim Biophys Acta 1838:1074–1081
Sunstrom NA, Premkumar LS, Premkumar A, Ewart G, Cox GB, Gage PW (1996) Ion channels formed by NB, an influenza B virus protein. J Membr Biol 150:127–132
Tang Y, Zaitseva F, Lamb RA, Pinto LH (2002) The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J Biol Chem 277:39880–39886
Taube R, Alhadeff R, Assa D, Krugliak M, Arkin IT (2014) Bacteria-based analysis of HIV-1 Vpu channel activity. PLoS One 9:e105387
Torres J, Maheswari U, Parthasarathy K, Ng L, Liu DX, Gong X (2007) Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci 16:2065–2071
Tosteson MT, Pinto LH, Holsinger LJ, Lamb RA (1994) Reconstitution of the influenza virus M2 ion channel in lipid bilayers. J Membr Biol 142:117–126
Triantafilou K, Kar S, Vakakis E, Kotecha S, Triantafilou M (2013) Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to signal inflammasome activation. Thorax 68:66–75
Ulug ET, Garry RF, Waite MR, Bose HR Jr (1984) Alterations in monovalent cation transport in Sindbis virus-infected chick cells. Virology 132:118–130
van Kuppeveld FJ, Melchers WJ, Kirkegaard K, Doedens JR (1997) Structure-function analysis of coxsackie B3 virus protein 2B. Virology 227:111–118
Venkataraman P, Lamb RA, Pinto LH (2005) Chemical rescue of histidine selectivity filter mutants of the M2 ion channel of influenza A virus. J Biol Chem 280:21463–21472
Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, DeDiego ML, Torres J, Aguilella VM, Enjuanes L (2012) Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 432:485–494
Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, DeDiego ML, Enjuanes L, Aguilella VM (2013) Analysis of SARS-CoV E protein ion channel activity by tuning the protein and lipid charge. Biochim Biophys Acta 1828:2026–2031
Vieyres G, Brohm C, Friesland M, Gentzsch J, Wolk B, Roingeard P, Steinmann E, Pietschmann T (2013) Subcellular localization and function of an epitope-tagged p7 viroporin in hepatitis C virus-producing cells. J Virol 87:1664–1678
Vijayvergiya V, Wilson R, Chorak A, Gao PF, Cross TA, Busath DD (2004) Proton conductance of influenza virus M2 protein in planar lipid bilayers. Biophys J 87:1697–1704
Wang C, Takeuchi K, Pinto LH, Lamb RA (1993) Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol 67:5585–5594
Wang C, Lamb RA, Pinto LH (1994) Direct measurement of the influenza A virus M2 protein ion channel activity in mammalian cells. Virology 205:133–140
Wang C, Lamb RA, Pinto LH (1995) Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys J 69:1363–1371
Wang K, Lu W, Chen J, Xie S, Shi H, Hsu H, Yu W, Xu K, Bian C, Fischer WB, Schwarz W, Feng L, Sun B (2012) PEDV ORF3 encodes an ion channel protein and regulates virus production. FEBS Lett 586:384–391
Wetherill LF, Holmes KK, Verow M, Muller M, Howell G, Harris M, Fishwick C, Stonehouse N, Foster R, Blair GE, Griffin S, Macdonald A (2012) High-risk human papillomavirus E5 oncoprotein displays channel-forming activity sensitive to small-molecule inhibitors. J Virol 86:5341–5351
Whitfield T, Miles AJ, Scheinost JC, Offer J, Wentworth Jr,. P, Dwek RA, Wallace BA, Biggin PC, Zitzmann N (2011) The influence of different lipid environments on the structure and function of the hepatitis C virus p7 ion channel protein. Mol Membr Biol 28(5):254–264
Wilson L, McKinlay C, Gage P, Ewart G (2004) SARS coronavirus E protein forms cation-selective ion channels. Virology 330:322–331
Wilson L, Gage P, Ewart G (2006) Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 353:294–306
Wong SS, Chebib M, Haqshenas G, Loveland B, Gowans EJ (2011) Dengue virus PrM/M proteins fail to show pH-dependent ion channel activity in Xenopus oocytes. Virology 412:83–90
Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA (2010) Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog 6:e1001087
Xie S, Wang K, Yu W, Lu W, Xu K, Wang J, Ye B, Schwarz W, Jin Q, Sun B (2011) DIDS blocks a chloride-dependent current that is mediated by the 2B protein of enterovirus 71. Cell Res 21:1271–1275
Yamada H, Chounan R, Higashi Y, Kurihara N, Kido H (2004) Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett 578:331–336
Young R (2014) Phage lysis: three steps, three choices, one outcome. J Microbiol 52:243–258
Yount B, Roberts RS, Sims AC, Deming D, Frieman MB, Sparks J, Denison MR, Davis N, Baric RS (2005) Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J Virol 79:14909–14922
Zhang R, Wang K, Lv W, Yu W, Xie S, Xu K, Schwarz W, Xiong S, Sun B (2014) The ORF4a protein of human coronavirus 229E functions as a viroporin that regulates viral production. Biochim Biophys Acta 1838:1088–1095
Zhou Y, Frey TK, Yang JJ (2009) Viral calciomics: interplays between Ca2+ and virus. Cell Calcium 46:1–17
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hyser, J.M. (2015). Viroporins. In: Delcour, A.H. (eds) Electrophysiology of Unconventional Channels and Pores. Springer Series in Biophysics, vol 18. Springer, Cham. https://doi.org/10.1007/978-3-319-20149-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-20149-8_7
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-20148-1
Online ISBN: 978-3-319-20149-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)