
Abstract
Upon photoexcitation, molecules break apart, following different dissociation reactions. We can image these dissociation pathways using Coulomb explosion imaging (CEI) and sometimes we can control them using asymmetric laser fields. In the formaldehyde molecule, we can see fragments following the direct, conventional dissociation path, as well as fragments deviating from this minimum energy path. So-called roaming fragments or “roamers” explore the potential energy landscape in a statistical manner and could be directly captured in real-time, despite the signal’s statistical character. This is possible due to the single-molecule sensitivity of CEI and we could show that the onset of roaming occurs actually several orders of magnitude earlier than previously expected. In the polar molecule OCS we go one step further by controlling the fragmentation process using two-color asymmetric laser fields. In addition to expected direct ionization effects, we also see post-ionization contributions, which are usually not visible in heavy polar molecules. We thus show in two different examples that CEI provides the means to extract new, unexpected pathways, which would otherwise remain hidden underneath a strong background.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others
Keywords
- Realtime roaming dynamics
- Ultrafast imaging
- Coulomb explosion imaging
- Femtosecond dynamics
- Coherent quantum control
- Molecular movie
1 Introduction
How to image statistical dynamics of individual reaction pathways hidden in an overwhelming background? How to image and differentiate the dynamics of various ionization pathways, which are otherwise only visual in high-resolution spectroscopy? We are addressing these questions by making use of the capability of Coulomb explosion imaging (CEI) to detect the ion-breakup of single-molecules in real time. The beauty of CEI is, that it provides full 3D reconstruction of the molecular fragment momenta for each molecule, individually. This allows us to separate channels, which would be smeared out in other time resolved spectroscopy approaches.
Conventional time-resolved spectroscopy would often struggle to separate features that are occurring on a few-percent level from the background, since in general one would average over an ensemble of molecules. If these features relate to a coherent motion (like e.g. wavepacket dynamics showing up as a clear oscillation) one might still be able to identify this on top of a background. However, statistically occurring events like uni-molecular dissociation signatures (including roaming) would not be separable from the background [1]. The fact that CEI allows to identify specific signatures of specific pathways to separate dissociation channels in a time resolved way is what makes it unique, since it allows to directly probe the evolution of the distance between fragments during a statistical decay.
2 Roaming in Formaldehyde
We investigated the unimolecular dissociation reactions in the deuterated formaldehyde molecule (D2CO). In addition to the conventional dissociation channels – molecular and radical dissociation – a third channel termed roaming has been discovered in 2004 [2], which bypasses the transition state. Identified through spectroscopic footprints in this prototypical molecule using high-resolution spectroscopy and found later in a variety of other molecules, roaming fragments had however never been observed directly and in a time-resolved way. We succeeded in doing so by using the highly sensitive CEI method in combination with high-level (i.e. rigorous) first-principles simulations of all critical experimental steps [3, 4]. For animated trajectories of the direct dissociation path and the roaming path, see Ref. [5]. The schematic of the experimental excitation step leading to the roaming reaction is shown in Fig. 1, together with the probing step, which induces the Coulomb explosion.

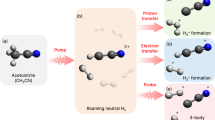
Schematic of roaming dynamics in D2CO visualized for two coordinate systems; the orange axis correspond to relevant vibrational modes, the green curved line shows the roaming reaction coordinate. A pump pulse (purple arrow, 304 nm) excites the à state at time t0 to the purple energy level. Because of vibronic coupling of the out-of-plane bending (pyramidization) and the C–O stretching mode with hot vibrational states of the ground state, a nonradiative population transfer is initiated at t1. Once on the hot ground state, three reaction types can occur: D2 + CO molecular dissociation, D + DCO radical dissociation, and roaming, each following its own reaction potential that is not explicitly shown here. Corresponding exemplary molecular structures for three subsequent times are given along the roaming reaction coordinate (green curved line). Excited molecules experience D2 vibrations (t2), which lead to one deuteron roaming around the remaining DCO fragment at large interatomic distances (t3). Eventually, the roamer D abstracts the bound deuteron and together they leave the CO fragment, which leads to D2 formation and abstraction (t4). Note that t1 to t4 do not correspond to specific absolute times but rather indicate the order of these steps within a statistical process. A strong NIR pulse (800 nm, ~8 fs duration, black arrow) probes the molecular structure at variable time delays (tpr). (Figure reprint with permission from Ref. [3])
Distinctive roaming signatures were identified in the experimental data, based on simulations (see Fig. 2 for three different time delays between the pump and the probe pulse). These roaming signatures were rendered observable by extracting rare stochastic events out of an overwhelming background. To identify the individual pathways, gates have been defined based on the individual analysis of 1500 semi-classical trajectories. These gates allow to extract an experimental observable for transient roaming features. Additionally, the identification of transient roaming features allowed to generalize the definition of roaming. While it was previously assumed that roaming results in molecular dissociation only, we observed that roaming might also be followed by radical dissociation. We further studied how the pump step and the overall dissociation depends on the employed UV pump wavelength [6]. Out of the four different wavelengths investigated (304, 314, 329, and 337 nm, corresponding to different selective populations of vibrational modes), 304 nm exhibits the fastest population transfer and dissociation times.

Snapshots of dissociating D2CO molecules for different time delays in experiment, pumped by 304 nm (a–c), with the total kinetic energy released in the Coulomb explosion plotted against the angle between the two deuteron momentum vectors (see inset on the bottom left). Geometries that correspond to different molecular roads, as obtained from the simulations, are marked by the colored curves: gray, equilibrium (d); red–yellow dash, roaming (e); green, radical dissociation (f); blue, molecular dissociation (g)
3 Post-Ionization Dynamics in OCS
In another experiment, we have tailored the laser fields used for excitation to investigate and control the dissociation mechanisms of the prototypical heavy polar molecule OCS into the two break-up channels of the dication, OCS2+ → O+ + CS+ and OC+ + S+, in phase-locked two-color intense laser fields.
Two obtain these asymmetric laser fields, a fundamental beam is superimposed with its second harmonic while controlling the relative time delay between both of them. The electric field of the resulting two-color laser field, composed of the fundamental (carrier frequency of ω) and parallel polarized second harmonic (2ω) pulses, can be expressed as:
where Fω and F2ω are the electric field amplitudes of the fundamental and second harmonic fields, respectively, and ϕ is the relative phase between both fields.
In these control experiments, the O+ + CS+ channel accounts for about 10% of the cases and the OC+ + S+ channel for about 90% (averaged over all phases). The branching ratio of the breaking of the C–O and C–S bonds followed a pronounced 2π-oscillation with a modulation depth of 11%, depending on the relative phase of the two-color laser fields. The fragment ejection direction of both break-up channels reflects the anisotropy of the tunneling ionization rate, following a 2π-periodicity, as well. The two dissociation pathways in the C–S bond breaking channel show different phase dependencies of the fragment ejection direction, which are assigned to post-ionization dynamics. These observations, resulting from the excitation with asymmetric two-color intense laser fields, supported by state-of-the-art theoretical simulations, reveal the importance of post-ionization population dynamics in addition to tunneling ionization in the molecular fragmentation processes, even for heavy polar molecules [7].
To evaluate the fragment ejection direction quantitatively, the asymmetry parameter A is defined as a function of Ekin and ϕ,
where Y+ and Y− are the fragment yields with positive and negative momenta along the laser polarization direction (x-axis), respectively.
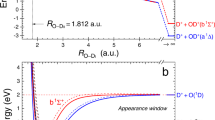
The results of this evaluation for both breakup channels are shown in Fig. 3. Both channels peak at around 5.7 eV, as shown in green and purple. This main contribution is assigned to the phase dependence of the direct tunnel ionization step. However, in addition, we observe a shoulder peak at around 4 eV in the O+ + CS+ channel, which we prove to be related to post-ionization dynamics. For this, we combine experimental analysis and state of the art theory simulations based on real-time time dependent density functional theory (rt-TDDFT) for the ionization step, as well as semi-classical surface hopping simulations to consider the involved nuclear dynamics.

Phase dependent total kinetic energy release Ekin spectrum of the OC+ + S+ channel (left) and the O+ + CS+ channel (right). (a + d) averaged spectrum over the relative phase ϕ between the ω and 2ω laser fields. (b + e) Two-dimensional plot of the asymmetry parameter as a function of Ekin and ϕ. (c) Asymmetry parameter of the OC+ fragments integrated over 5–10 eV (green circles) and 2.5–4.5 eV (orange squares) vs. ϕ. The dotted lines are sine fits. (f) Same as (c), but the O+ fragments are integrated over 3.5–7 eV (purple triangles). (Figure reprint with permission from Ref. [7])
While post-ionization dynamics has been previously identified in light polar molecules, as well as in non-polar molecules [8, 9], it is in general not visible in heavy polar molecules, due to the dominant nature of the direct tunnel ionization contributions. Here, we have been able to separate these two contributions.
We do observe the influence of post-ionization dynamics in various aspects of our joint experiment – theory work. Not all of the effects are discussed in the here presented text, for a detailed discussion we refer to Ref. [7]. Above table summarizes two observations, pointing out that the main effect occurring after the ionization (and controlled by the two-color laser field) is a population transfer between electronic states of the dication (Table 1):
In summary, the two example cases of roaming in formaldehyde and fragmentation control in the OCS molecule show that the CEI method, sensitive to single molecules, is a powerful tool to identify contributions conventionally hidden under a huge background, even if these contributions relate to a statistically occurring signal.
References
Ding, X., Forbes, R., Kübel, M., Lee, K.F., Spanner, M., Naumov, A.Y., Villeneuve, D.M., Stolow, A., Corkum, P.B., Staudte, A.: Threshold photodissociation dynamics of NO2 studied by time-resolved cold target recoil ion momentum spectroscopy. J. Chem. Phys. 151, 174301 (2019)
Townsend, D., Lahankar, S.A., Lee, S.K., Chambreau, S.D., Suits, A.G., Zhang, X., Rheinecker, J., Harding, L.B., Bowman, J.M.: The roaming atom: straying from the reaction path in formaldehyde decomposition. Science. 306, 1158 (2004)
Endo, T., Simon, V., Neville, P., Wanie, S., Beaulieu, C., Qu, J., Dechamps, P., Lassonde, B.E., Schmidt, H., Fujise, M., Fushitani, A., Hishikawa, P.L., Houston, J.M., Bowman, M.S., Schuurman, F.L., Ibrahim, H.: Capturing roaming molecular fragments in real-time. Science. 370(80), 1072 (2020)
Endo, T., Qu, C., Ibrahim, H.: A molecular road movie. Phys. Today. 74, 62 (2021)
H. Ibrahim, The Molecular Road Movie, 2020
Endo, T., Neville, S.P., Lassonde, P., Qu, C., Fujise, H., Fushitani, M., Hishikawa, A., Houston, P.L., Bowman, J.M., Légaré, F., Schuurman, M.S., Ibrahim, H.: Electronic relaxation and dissociation dynamics in formaldehyde: pump wavelength dependence. Phys. Chem. Chem. Phys. 24, 1779 (2022)
Endo, T., Ziems, K.M., Richter, M., Fröbel, F.G., Hishikawa, A., Gräfe, S., Légaré, F., Ibrahim, H.: Post-ionization dynamics of the polar molecule OCS in asymmetric laser fields. Front. Chem. 10, 859750 (2022)
Wanie, V., Ibrahim, H., Beaulieu, S., Thiré, N., Schmidt, B.E., Deng, Y., Alnaser, A.S., Litvinyuk, I.V., Tong, X.-M., Légaré, F.: Coherent control of D2/H2 dissociative ionization by a mid-infrared two-color laser field. J. Phys. B Atomic Mol. Phys. 49, 025601 (2016)
Ibrahim, H., Lefebvre, C., Bandrauk, A.D., Staudte, A., Légaré, F.: H2: the benchmark molecule for ultrafast science and technologies. J. Phys. B Atomic Mol. Phys. 51, 42002 (2018)
Acknowledgments
We thank A. Larameé for technical support and B. Wales and J. Sanderson from the university of Waterloo for providing the analysis software for the CEI measurement files; Funding: This work was supported by the Canada Foundation for Innovation, NSERC, FRQNT, the JSPS Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (grant no. S2602 to T. E. and A. H.), and the World Research Unit (B-1) of Reaction Infography (R-ing) at Nagoya University, Japan (A. H. and F. L.). J. M. B. acknowledges financial support from NASA (grant no. 80NSSC20K0360).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2024 The Author(s)
About this paper

Cite this paper
Endo, T. et al. (2024). Imaging Ultrafast Dissociation Dynamics: OCS & Roaming in Formaldehyde. In: Argenti, L., Chini, M., Fang, L. (eds) Proceedings of the 8th International Conference on Attosecond Science and Technology. ATTO 2023. Springer Proceedings in Physics, vol 300. Springer, Cham. https://doi.org/10.1007/978-3-031-47938-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-031-47938-0_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-47937-3
Online ISBN: 978-3-031-47938-0
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)