
Abstract
Mechanisms are the how of chemical reactions. Substances are individuated by their structures at the molecular scale, so a chemical reaction is just the transformation of reagent structures into product structures. Explaining a chemical reaction must therefore involve different hypotheses about how this might happen: proposing, investigating and sometimes eliminating different possible pathways from reagents to products. One distinctive aspect of mechanisms in chemistry is that they are broken down into a few basic kinds of step involving the breaking and making of bonds between atoms. This is necessary for chemical kinetics, the study of how fast reactions happen, and what affects it. It draws on G.N. Lewis’ identification of the chemical bond as involving shared electrons, which from the 1920s achieved the commensuration of chemistry and physics. The breaking or making of a bond just is the transfer of electrons, so a chemical bond on one side of an equation might be balanced on the other side by the appearance of a corresponding quantity of excess charge. A bond is understood to have been exchanged for a pair of electrons. Since reaction mechanisms rely on identities, doesn’t the establishment of a reaction mechanism explain away the chemical phenomena, showing that they are no more than the movement of charges and masses? In one sense yes: these mechanisms seem to involve a conserved-quantity conception of causation. But in another sense no: the ‘lower-level’ entities can do what they do only when embedded in higher-level organisation or structure. There need be no threat of reduction.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
7.1 Introduction
In this paper, I will defend the following claims which, taken together, amount to a detailed conception of the metaphysics of mechanisms in chemistry.
-
1.
Chemical reactions are processes individuated by the molecular-scale structures with which they begin (the reagents) and end (the products).
-
2.
A reaction mechanism is a description of how a chemical reaction might possibly happen: how the reagents are transformed into the products.
-
3.
Different pathways from reagents to products are individuated by how they are composed of a few basic kinds of step involving transfers of such conserved quantities as mass, charge and energy.
-
4.
This view of mechanisms has no tendency to support the reduction of chemistry to physics, or reductionism more generally.
My intention in this article is not to say anything complicated or controversial about reaction mechanisms in chemistry, but instead to relate some obvious and basic facts about them to the recent wave of philosophical literature on mechanisms. Even though chemistry has a long and illustrious tradition of thinking about mechanisms – in the twentieth century, multiple Nobel prizes were awarded for work on mechanisms – mechanist philosophers have almost entirely looked elsewhere for examples. Thus, for instance, the word ‘chemistry’ does not occur in the influential paper that initiated that wave (Machamer et al., 2000). In the major survey article by Craver and Tabery (2016), chemistry is mentioned only once on its own account (that is, as a science that investigates mechanisms), and then only as a subject to be addressed in future work. I think there are some distinctive and interesting things to say about reaction mechanisms in chemistry.
7.2 What Is a Chemical Reaction?
A chemical reaction is a type of process in which chemical change occurs.Footnote 1 Chemical reactions can be specified at two different scales, or levels:Footnote 2 as involving chemical substances, or as involving species at the molecular scale (atoms, ions or groups of atoms). Chemical change is not just any change: for a chemical reaction to have taken place during a process, the substances or species at the end must be chemically different from those at the beginning. The melting of ice does not count as a chemical reaction, although it does, of course, have a mechanism:
Given all the above, it seems obvious that chemical reactions are individuated by the substances or species with which they begin (the reagents) and those with which they end (the products). Chemical reactions can be specified at different levels of abstraction. Consider for instance the reaction between hydrogen chloride and sodium hydroxide to form sodium chloride and water. From there one might abstract to yield a reaction type in which an unspecified hydrogen halide reacts with an unspecified metal hydroxide to give a metal halide and water. Yet more abstractly, one might consider as a class reactions between acids and bases to give a salt and water. The same is true in organic chemistry: one might consider the oxidation of propan-2-ol (CH3CHOHCH3) to propanone (CH3COCH3, better known as acetone), or more abstractly the oxidation of secondary alcohols of the general form R1CHOHR2 to ketones of the general form R1COR2, with R1 and R2 being unspecified alkyl groups. A description of a chemical reaction always picks out a reaction type, but the level of abstraction of that description may vary.
Chemists give information about chemical reactions using representations of the relevant processes, which are often, but not always, balanced chemical equations. These involve chemical formulae, which are themselves representations of the structure of the relevant chemical substances, which again can be couched at different levels of detail (or abstraction). The simplest kind of chemical formula – the empirical formula – represents only the elemental composition of a substance. Because the information it provides about structureFootnote 3 is limited, this formula will fail to distinguish between distinct substances. For instance, acetone, mentioned earlier, has the empirical formula C3H6O, which is shared by about 10 distinct substances including cyclopropanol and ethanal (the substance formerly known as acetaldehyde). Empirical formulae are not very informative: for any purpose that requires isomers be distinguished, more detailed structural formulae will need to be used.
I have argued elsewhere for microstructural essentialism, the thesis that chemical substances are the substances they are in virtue of their structures at the molecular scale (see Hendry, 2023, Forthcoming, from which the following discussion is drawn). I will not argue here for full-blown microstructural essentialism but only the weaker claim that microstructuralism is the official ideology of chemistry, suffusing its approach to mechanisms. I will do that via three arguments drawing on the practice of chemistry, concerning (i) the centrality of structure at the molecular scale to chemical classification and nomenclature, and the complete absence of any other non-microstructural criteria; (ii) the role of microstructure in explaining and predicting the chemical and physical behaviour of substances, and (iii) the fact that no other systematic basis for individuating substances is consistent with chemical practice, and the epistemic interests that underlie it. I will develop those three arguments in turn.
The case for microstructuralism concerning chemical classification and nomenclature is particularly strong in the case of the chemical elements. Since 1923, the International Union of Pure and Applied Chemistry (IUPAC) has quite explicitly identified nuclear charge as what characterises the various chemical elements (for the historical background see van der Vet, 1979; Kragh, 2000). I have argued that the historical record supports realism about the elements as natural kinds, because the IUPAC change reflected a series of discoveries (Hendry, 2006, 2010a). I correspondingly disagree with LaPorte (2004, Chapter 4), who has argued that, prior to the twentieth century, it was indeterminate whether the names of the chemical elements referred to classes of atoms which are alike in respect of their nuclear charge, or to classes of atoms alike in respect of their atomic weight, or to classes of atoms alike in both respects. For LaPorte, IUPAC’s 1923 decision had the character of a stipulation. I think it is quite natural to see the extensions of the names of the elements as being determinate before 1923, and IUPAC’s identification as simply the recognition of determinate membership (see Hendry, 2006, 2010a). Silver, which has been known since ancient times, always consisted of roughly equal mixtures of two isotopes of silver (107Ag and 109Ag), differing in respect of their atomic weight. There is, of course, a remote possibility that the isotopic composition of silver changed radically over time, but that would not affect my main point, which is that the weight differences between the isotopes make very little difference to their chemical behaviour. What makes those diverse atoms count as silver is what they share, namely their nuclear charge (47), a property that explains why these diverse atoms behave chemically in very similar ways. The twentieth-century identification of nuclear charge as what individuates the elements was a discovery of this fact, rather than a stipulation or a convention.
There are good grounds for extending microstructuralism to compound substances. The rules for chemical nomenclature that IUPAC has developed over the years are based entirely on microstructural properties and relations. Consider for instance 2, 4, 6,-trinitromethylbenzene, better known as trinitrotoluene, or TNT (see Fig. 7.1).

Full structural formula for 2, 4, 6,-trinitrotoluene, or TNT
For the purposes of nomenclature this compound, which was first synthesised in the nineteenth century, is regarded as being derived from methylbenzene (toluene): counting clockwise from the methyl (–CH3) group as position 1, there are three nitro-groups (–NO2) at positions 2, 4 and 6, replacing three hydrogen atoms (conventionally the remaining hydrogen atoms at positions 3 and 5 are left out for clarity). In short, TNT is named purely on the basis of its bond structure. Now it is true that there are alternative ways of generating names for compounds (see Leigh et al., 1998), but the important point is that IUPAC’s various systems of nomenclature are all based on microstructure. Critiques of microstructuralism in chemistry never seem to mention this fact.
A second argument for microstructuralism concerns explanation. Understanding the chemical behaviour of a compound substance—its chemical reactivity—is essentially a matter of understanding how its structure transforms into the structure of other substances under various conditions. As we shall see, this involves the study of chemical reaction mechanisms. Similarly, understanding the physical behaviour of a substance, including its melting and boiling points and its spectroscopic behaviour, is a matter of understanding how, given their structure, its constituent molecules interact with each other and with radiation respectively.
A third argument for microstructuralism concerns the fact that no other group of properties provides a systematic framework for naming and classifying substances, or understanding their behaviour, that is consistent with chemical practice. What are the alternatives? Paul Needham (2011) has argued that classical thermodynamics provides macroscopic relations of sameness and difference between substances, acknowledging that his macroscopic perspective is revisionary of current chemical practice, in that it divides substances more finely than chemists do. On Needham’s view, different isotopes of the same element are distinct substances even though chemists lump them together when thinking about the elements. Taking a similar stance to LaPorte, although for different reasons, Needham describes IUPAC’s identification of nuclear charge as what characterises the elements as a ‘convention’ (2008, 66). I think this is too thin a description, and historically misleading. The adoption of nuclear charge was a considered choice, which reflected the discovery that the elements occur in nature as mixtures of different isotopes. This meant that the names of the elements as they were currently being used had been discovered to refer to populations of atoms which are alike in respect of their nuclear charge, while diverse in respect of their weight. The identification of nuclear charge as what characterises the elements was therefore simply a recognition of the real basis of the periodic table (see Hendry, 2006, 2010a). Further objections to Needham’s thermodynamic criteria are provided by other examples. Orthohydrogen and parahydrogen are spin isomers of the hydrogen molecule that readily interconvert on thermal interaction: in orthohydrogen the spins are aligned, while in parahydrogen they are opposed. Orthohydrogen and parahydrogen count as different substances on the thermodynamic criteria. The same holds for populations of atoms in mutually orthogonal quantum states, such as two streams of silver atoms emerging from a Stern-Gerlach apparatus (for detailed discussion see Hendry, 2010b). A natural conclusion is that Needham’s proposed thermodynamic criteria for sameness and difference of substance track differences of physical state, without regard for whether those differences correspond to distinctions of substance.Footnote 4
These three arguments together constitute a strong positive case in favour of taking microstructuralism to be the prevailing classificatory ideology of the discipline of chemistry, and seeing chemistry’s adoption of microstructuralism as well motivated, because it is a natural ‘carving’ of chemical reality. I noted earlier that chemical reactions are individuated by the chemical substances with which they begin and end. Putting these two claims together, it follows that chemical reactions are the particular reactions they are in virtue of the structures at the molecular scale with which they begin and end.
Before we move on to considering reaction mechanisms, it is worthwhile saying something about the scope and content of microstructuralism about chemical substances. On the subject of content, I disagree with the claim, made for instance by Needham (2002, 208) and Jaap van Brakel (2000, Chapter 4) that microstructuralism entails a reductionist view of substances. It involves only a claim about what chemical substances are made of, and therefore the resources that go into forming their structures at the molecular scale. This is consistent with molecules and substances being strongly emergent, or so I shall argue in the final section. On the subject of scope, by the term ‘chemical substances’ I mean chemically homogeneous stuffs as viewed by the discipline of chemistry. Some critics of microstructuralism see the fact that some other kinds of stuff, viewed from the perspectives of scientific disciplines and other kinds of activity which may be quite different from chemistry’s, are not best understood in microstructural terms as a criticism of microstructuralism (see for instance LaPorte, 2004; Havstad, 2018). However, microstructuralism about chemical substances does not apply to milk and wool (to take two examples), because they are chemically heterogeneous stuffs. Chemists don’t decide what counts as milk or wool, although they can expertly analyse particular samples.Footnote 5 As I understand it, the same applies to protein classification as viewed from the perspective of the life sciences. Tom McLeish (2019) has argued that the physical processes underlying protein folding demonstrate many important connections with soft matter physics, a subdiscipline of condensed matter physics that studies (for instance) the mechanical, topological and thermodynamic properties of polymers, a focus that often demands that it abstracts away from their chemical composition, and engages with interactions at energy scales at which high-energy and quantum interactions are irrelevant. But the differing classification is not simply ‘higher-level’ or ‘more special’ disciplines defining their kind-terms more abstractly than ‘lower-level’, ‘less special’ or ‘less fundamental’ disciplines in a hierarchy of the sciences. Gil Santos, Gabriel Vallejos and Davide Vecchi (2020, 364) worry that microstructuralism in chemistry entails a reductionist view of processes in cells if it sees the causal powers of molecules as being determined only by the internal, or non-relational features of molecules (i.e. in abstraction from the environments in which they exert them). In my view microstructuralism about chemical substances is not a reductionist philosophy for cell biology for two kinds of reason. Firstly, proteins in vivo are rightly understood functionally, and so may not be heterogeneous chemical substances (their primary structure may vary). Given our earlier point about substances, they fall outside the scope of my earlier claim for microstructuralism. Secondly, as Santos, Vallejos and Vecchi point out, proteins participate in cellular processes which are subject to top-down constraints. It would be quite wrong to abstract away from the environment. This point is crucial, and applies outside the cellular context: the causal powers of an entity at the molecular scale depend on the environment in which they are exerted. I will return to this point in the final section.
7.3 What Is a Reaction Mechanism?
Molecules are structured entities composed of electrons and atomic nuclei: they are ‘composed’ of electrons and nuclei in the sense that, if you take away the electrons and nuclei, there is nothing left. Chemical reactions must involve changes to molecular structures, and these must involve rearrangements of the electrons and nuclei. A proposed reaction mechanism is just a detailed proposal about how those rearrangements go. Hence a reaction mechanism will be a process involving rearrangements of nuclei and electrons that is, transfers of conserved quantities such as charge, mass and energy.
This rather high-level view is borne out by a closer look at chemists’ discussions of reaction mechanisms. In an influential textbook of theoretical organic chemistry, Edwin S. Gould defines ‘reaction mechanism’ as follows:
In the ideal case, we may consider the mechanism of a chemical reaction as a hypothetical motion picture of the participating atoms. Such a picture would presumably begin at some time before the reacting species approach each other, then go on to record the continuous paths of the atoms (and their electrons) during the reaction, and come to an end after the products have emerged. (Gould, 1959, 127)
Gould goes on to say that such a ‘hypothetical motion picture’ is not directly empirically accessible, and that in practice chemists focus on particular steps in the reaction:
Since it is not generally possible to obtain such an intimate picture, the investigation of a mechanism has come to mean obtaining information that can furnish a picture of the participating species at one or more crucial instants during the course of the reaction. (1959, 127)
These ‘crucial instants’ involve the making and breaking of chemical bonds, and other kinds of change that affect nuclear positions and electron distribution within the molecule.
Drawing on Gould and other textbook discussions, William Goodwin (2012) identifies two conceptions of a reaction mechanism. On the thick conception, a reaction mechanism is ‘roughly, a complete characterization of the dynamic process of transforming a set of reactant molecules into a set of product molecules’ (2012, 310). As Goodwin notes this is something like Gould’s motion picture (2012, 310). On the thin conception in contrast, mechanisms are ‘discrete characterizations of a transformation as a sequence of steps’ (2012, 310). This distinction raises three important issues: first, what relationship there is between thick and thin reaction mechanisms; second, how they are related to theories and models in chemistry, and third, how they relate to philosophical accounts of mechanisms and causation (see Goodwin, 2012, 310–15).
Felix Carroll argues that Gould’s motion picture ‘should be viewed as animation or simulation’ because ‘we cannot see the molecular events; we can only depict what we infer them to be’ (1998, 317). According to Goodwin, thick mechanisms are in some sense more fundamental, perhaps because they are descriptively complete, yet they are only indirectly accessible to experiment. If they have a role in chemical thinking it is as a regulative ideal of complete description. The steps in a thin mechanism, in contrast, are more accessible to chemical inference because they affect the kinetics of the reaction (how fast the reaction goes, and how rate depends on background conditions), and they leave traces on the structures of the products. I will examine some examples later. Clearly, there is a close relationship between the two: a thick mechanism is a fuller description, but the steps in a thin mechanism can be found in a thick mechanism. For that reason, and also because thick and thin conceptions represent the very same processes, I think that even if we can draw contrasts between the two conceptions of mechanism, they ought to represent the underlying processes consistently. A thin mechanism may have gaps, but we should not think of it as representing mechanisms as gappy. To get to a thin mechanism from a thick mechanism we simply focus on some crucial steps, ignoring the rest. We are abstracting rather than falsifying or removing any important features of the mechanism.
Turning to the second issue, Goodwin sees thick mechanisms as more fundamental in a second sense, that they are more directly related to fundamental theory, which he identifies with potential energy surfaces (or free energy surfaces) that figure widely in discussions of mechanisms in theoretical organic chemistry.Footnote 6 This thought further supports the idea that thick and thin mechanisms are commensurable: the steps of a thin mechanism can be identified as topological features on a PE surface. A slow, or rate-determining step will typically correspond to the traversal of a maximum.
Turning now to the last question, Goodwin (2012, 326) points out that mechanisms on the thick conception are continuous processes involving transfers of conserved quantities (for the most part, mass, charge and energy), exemplifying Wesley Salmon’s processual theory of causal explanation (Salmon, 1984). In contrast, mechanisms on the thin conception, he argues, are a better fit with Peter Machamer, Lindley Darden and Carl Craver’s definition of mechanisms as ‘entities and activities organized such that they are productive of regular changes from start to termination condition’ (2000, 3):
[M]echanisms in the thin sense are decompositions of a transformation into standardized steps, each of which is characterizable in terms of certain types of entities (nucleophiles and core atoms, for example) and their activities or capacities (such as capacities to withdraw electrons or hinder backside attack). (Goodwin, 2012, 326)
I agree with Goodwin that thin reaction mechanisms can be understood in terms of Machamer, Darden and Craver’s definition, but think that their highly abstract characterisation applies just as well to thick reaction mechanisms. Conversely, thin mechanisms focus on key steps in chemical reactions, involving the making and breaking of bonds, and relative movements of nuclei. They too must involve transfers of conserved quantities. Given my earlier constraint about consistency of philosophical characterisation between the two conceptions, it seems to me that this had better be the case. Craver and Tabery (2016, 2.3.1) present no real objections to transference theories as a way of understanding mechanisms, commenting only that references to transfers of conserved quantities are rarely explicit in the special sciences and that transference theories face traditional objections concerning absence and prevention. As we have seen, references to transfers of conserved quantities are rather explicit in chemistry, and the other issues can be set aside if we regard transference as providing an account only of reaction mechanisms in chemistry, but not necessarily of causal claims more generally.
The historical development of chemists’ theoretical understanding of reaction mechanisms supports the view I am defending here. From the 1860s, chemists developed a theory of structure for organic substances: how particular kinds of atoms are linked together in the molecules that characterise a substance. This theory was based on chemical evidence alone: that is, the details of which substances can be transformed into which other substances (see Rocke, 2010). Then G.N. Lewis proposed that the links between atoms in molecular structures were formed by the sharing of electrons in covalent bonds (Lewis, 1916). From the 1920s onwards, C.K. Ingold and others used these insights to develop a theory of reaction mechanisms, in which transformations between organic substances were understood as involving series of structural changes falling into a few basic kinds (Brock, 1992, Chapter 14; Goodwin, 2007). The key idea was that the making and breaking of chemical bonds should be understood in terms of the movement of electrons within the molecule. Since then, mechanisms have been central to explanations in organic chemistry, and have been fully integrated with theories of structure, with molecular quantum mechanics, and with kinetic, structural and spectroscopic evidence. Although Lewis’ covalent bond may look quaint since the arrival of quantum mechanics, it was highly influential in the developing understanding of reaction mechanisms and their explanatory relationship to chemical kinetics and structure. It commensurated bonds and electrons, allowing their interconversion in descriptions of reaction processes, a key insight that we will see illustrated shortly.
As mentioned, the steps in thin reaction mechanisms fall into a relatively small number of basic kinds, each of which is well understood: an atom or group of atoms leaving or joining a molecule, and a molecule, molecular fragment or ion rearranging itself. It is the thin conception that underwrites explanations within chemical kinetics, the study of how quickly chemical reactions happen, and what determines how quickly they proceed. This is because a reaction can only proceed as fast as its slowest step—the rate-determining step—and the rate will tend to depend only on the availability of species involved in this step.Footnote 7
I will illustrate these points with a couple of examples. Consider an organic compound of the form R—L in which a substituent L (e.g. a halogen atom such as chlorine, bromine or iodine, or a group of atoms) is attached to a saturated hydrocarbon group R. The ‘leaving group’ L can be replaced with a nucleophilic species Nu− which might be the hydroxyl ion OH−, or another halide ion:
This nucleophilic substitution may happen via two different mechanisms, depending on the nature of the alkyl group R, the nature of the leaving group and the reaction conditions, including the solvent. The structures of the products can be subtly different, too. During the 1920s and 1930s Ingold developed two models (SN1 and SN2) of how these reactions might occur, in order to explain the contrasting chemical behaviour of different alkyl halides. In the SN1 mechanism, the alkyl halide first dissociates (slowly) into a carbocation (or carbonium ion, in older terminology) R+ and the leaving group X−. The carbocation then combines (quickly) with the nucleophile.
-
Step 1 (slow): R—L ⇌ R+ + L−
-
Step 2 (fast): R+ + Nu− → R—Nu
The slowest (and therefore rate-determining) step involves just one type of molecule, RL, and SN1 means ‘unimolecular nucleophilic substitution’. The rate of the reaction can be expected to depend on the concentration of RL (written [RL]) and be independent of the nucleophile concentration [Nu−].
In the SN2 mechanism, substitution occurs when, as described by Gould (1959, 252), the leaving group is ‘pushed off’ the molecule by the incoming nucleophile Nu−:
The reaction requires a bimolecular collision between the nucleophile and the target molecule (hence ‘SN2’), so the reaction rate can be expected to be proportional to both [RL] and [Nu−].
Aside from the kinetics of these reactions, the SN1 and SN2 models also explain the differing stereochemical effects of nucleophilic substitution on different kinds of alkyl halide. Consider again the SN1 mechanism. The molecular geometry of a saturated carbon atom is tetrahedral, and if it is bonded to four different functional groups of atoms it is asymmetrical: like a left or right hand it will not be superimposable on its mirror image (its enantiomer). In contrast the carbocation intermediate produced by step 1 has a trigonal planar geometry. So, when the nucleophile Nu− approaches, it may do so in either of two directions, yielding an equal (racemic) mixture of two enantiomers (see Fig. 7.2).

Two possible reactions of a trigonal planar carbonium ion with nucleophile Nu−
The SN2 mechanism, in contrast, requires a bimolecular reaction between the nucleophile and the alkyl halide. One might think that the nucleophile could approach from either the same side as the leaving group, or from the opposite side (see Fig. 7.3). In practice, ‘SN2 reactions invariably proceed with inversion of configuration via back-side attack.’ (Roberts & Caserio, 1965, 297). Thus in a chiral molecule the nucleophile is not simply substituted for the leaving group: the product will correspond to the enantiomer of the reagent, rather than the reagent itself.Footnote 8

Nucleophilic attack via the SN2 mechanism, showing inversion of configuration
Groups of atoms leaving or joining a molecule necessarily involve the breaking or formation of bonds, and the breaking or formation of bonds involves transfer of electronic charge. This is quite explicit when curly arrows are added to the chemical equations constituting a mechanism.Footnote 9 The SN1 mechanism, for instance, might be represented as follows:
-
Step 1 (slow):
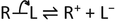
-
Step 2 (fast):
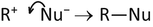
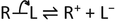
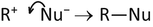
The curly arrow in step 1 represents the net transfer of the electron pair constituting a bond to the leaving group L. Since the bond was constituted by a shared electron pair, one of which had been contributed by L itself, the result is a single excess negative charge on L. Hence it is a negative ion L− that leaves. The curly arrow in step 2 represents the net transfer of two electrons from the nucleophile Nu− to a bond between it and the central carbon atom in the carbocation R+.
The mechanism therefore implements the relationship of constitution between electron pairs and chemical bonds introduced by Lewis (1916). Realisation is also an appropriate way to characterise the relationship, however. The concept of a chemical bond was introduced into organic chemistry during the 1860s to account for the sameness and difference of various organic substances, in terms of a bonding relation holding between atoms (Rocke, 2010). In a 1936 presidential address to the Chemical Society (later to become the Royal Society of Chemistry), Nevil Sidgwick pointed out that in this structural theory ‘No assumption whatever is made as to the mechanism of this linkage.’ (Sidgwick, 1936, 533) In short, in the 1860s the organic chemists identified a theoretical role for which, in the 1910s, Lewis identified shared electron pairs as the realiser, although he did not have a detailed account (and certainly no mathematical theory) of how electrons did the realising. The relationship remains complicated: the central explanatory theory is quantum mechanics, but that theory itself, applied to ensembles of electrons and nuclei, has nothing to say on the subject of bonds. Chemists need to find the bonds in quantum mechanics: the theory can, with the help of the Born-Oppenheimer approximation (localisation of the nuclei, and instantaneous neglect of their motion) provide an excellent theory of the electron-density distribution and how it interacts with changing nuclear positions. Mathematical analysis of a molecule’s electron-density distribution yields bond paths between atoms which resemble the familiar bond topologies of chemistry (see Bader, 1990; Popelier, 2000).
Reaction mechanisms are possible pathways from reagents to products in two distinct senses of possibility. In the case of SN1 and SN2, both pathways might be physically possible even if one will typically be favoured. Both might be followed by different molecules in the same reaction vessel. Both, in fact, are actual pathways. However, Roberts and Caserio discuss two possible routes for SN2 reactions, involving front-side and back-side approach, but note that the back-side approach ‘invariably’ occurs (Roberts & Caserio, 1965, 297). The front-side approach is (in some weak sense) geometrically possible, but is rendered physically impossible by the structure of the species being attacked. One might also say that, although the experimental evidence seems to rule out front-side attack for SN2 reactions, until that information was acquired it might have been considered epistemically possible. This suggests a role for eliminative reasoning about reaction mechanisms, something that Roald Hoffman (1995, Chapter 29), illustrates beautifully with a discussion of three possible mechanisms for the photolysis of ethane to ethene (known traditionally as ethylene), and how H. Okabe and J. R. McNesby used isotopic labelling to eliminate two of them. In photolysis, light energy (written hν) causes ethane (C2H6 or H3C—CH3) to eliminate hydrogen (H2), leaving ethene (a molecule with a carbon-carbon double bond; see Fig. 7.4).

Photolysis of ethane: the reaction. (From Hoffmann, 1995, 145)
The question is how this occurs, with three candidate pathways (see Fig. 7.5). Mechanism 1 involves two hydrogen atoms on neighbouring carbon atoms leaving by a ‘concerted reaction’. Mechanism 2 involves two steps: elimination of H2 from a single carbon atom followed by rearrangement of the fragment to give ethene. In mechanism 3, a light photon breaks a C-H bond, leaving two free radicals (C2H5• and H•, the dots representing unpaired electrons). This kicks off a chain reaction when the free radicals collide with other molecules, generating further free radicals. Mechanism 3 is eliminated because in a mixture of C2H6 and C2D6 (deuterated ethane), it would produce significant amounts of HD (isotopically mixed hydrogen molecules), whereas little is detected. That leaves mechanisms 1 and 2, of which mechanism 1 is eliminated because it would produce significant amounts of HD from isotopically mixed ethane (H3C—CD3), whereas little is detected.

Photolysis of ethane: three possible mechanisms. (From Hoffmann, 1995, 146)
That leaves mechanism 2: does the elimination of the other candidate mechanisms afford it any positive evidential support? Chemists often say that mechanisms cannot be proven (see for instance Sykes, 1981, 43; Carpenter, 1984, Chapter 1). Hoffmann notes the Popperian response but notes that scientists ‘want to do something positive’ (1995, 149). I think Hoffmann is right here, and the right framework for that is eliminative induction rather than Popperian refutation. For philosophers of science, the interest is whether structure theory (and knowledge of other mechanisms) can do any epistemic heavy lifting here, by limiting the number of conceivable pathways. Eliminative inductions, notoriously, are only as secure as the initial disjunctions of possibilities that constitute their major premises. A well-attested structure for the reagent will constrain how it might possibly transform into the product. The epistemic justification for the attribution of a structure is surely the source of any justification for the starting disjunction.
7.4 Mechanisms and Reduction
This section draws on published research arising from the Durham Emergence Project. The first part draws on arguments from Hendry (2017a, b); the second part draws on collaborative work with Robert Schoonmaker, which I have presented in Hendry (2019, 2022). I am most grateful to the John Templeton Foundation for funding the project (Grant ID 40485), and also to members of the project for many helpful conversations.
Many philosophers would expect the view I am defending of mechanisms in organic chemistry to support a robustly reductionist view of chemical substances. Firstly, it is committed to microstructural essentialism about chemical substances, which supports theoretical identity claims of the form ‘gold is the element with atomic number 79’ and ‘water is H2O’. Secondly, I have defended the claim that chemical reaction mechanisms involve transfers of conserved quantities, primarily charge and mass, in the form of electrons and nuclei. Isn’t it obvious that if water is H2O, then everything that water does is done by entities at the molecular level, namely H2O molecules? And isn’t the path to reductionism even clearer once we realise that the mechanisms by which water exerts its powers – how it does what it does – are properly described in terms of processes involving transfers of physical quantities governed by physical laws?
I think both inferences should be rejected. Microstructuralism about chemical substances is compatible with their being strongly emergent. Hilary Putnam once said that the extension of ‘water’ is ‘the set of all wholes consisting of H2O molecules’ (1975, 224). That is the first mistake, if a ‘whole’ is taken to be a mereological sum, or any other whole that can be formed without any interaction among the parts. ‘Water is H2O’ is true, but that doesn’t mean that H2O molecules are all there is to water. Water is formed from H2O molecules interacting: some of them self-ionise, producing protons (H+) and hydroxyl ions (OH−). Others form hydrogen-bonded oligomolecular structures. If Putnam had been right, then it would be safe to assume that water could not have any causal powers over and above those inherited from its constituent H2O molecules. Such a whole has no bulk properties, so there is no distinction to be made between its molecular and its bulk properties. In contrast steam, liquid water and ice (which has various structures) do have distinct properties produced by the distinct kinds of interactions between their parts. Wherever there is significant interaction between the H2O molecules, there is scope for that interaction to bring new powers into being. This is particularly clear if that interaction includes self-ionisation and the formation of oligomers. Now to mechanisms: as Eisenberg and Kauzmann put it, protons and hydroxyl ions both have ‘abnormal mobilities in both ice and water’ (1969, 226), which is possible because their excess charge can travel along the hydrogen bonded supramolecular structures without matter having to make the journey to carry them (see Fig. 7.6).

Mechanisms of proton and hydroxyl ion transfer in water. (From Eisenberg & Kauzmann, 1969, 227)
The mechanism by which that power is exercised requires some part of the molecular population to be charged. It also requires the supramolecular organisation. It therefore depends on a feature of a diverse population of molecular species. The reductionist will say at this point that the water can only acquire its causal powers from its parts, and interactions between them. Therefore, no novel causal powers have been introduced. The strong emergentist will ask why, when it is being decided whether they are novel, the powers acquired only when the molecules interact are already accounted for by the powers of H2O molecules. If the reductionists’ claim is that any power possessed by any molecular population produced by any interaction between H2O molecules is included, and we know that independently of any empirical information we might ever acquire about what water can do and how it does it, then it seems that we know a priori that there will be no novel causal powers, and therefore that reductionism is true. I take it that this would be for the reductionist to beg the question. This does not of course mean that the strong emergentist wins the argument by default: only that in making their case reductionists should deploy topic-specific scientific arguments. In the absence of such arguments, the reductionist and the strong emergentist conclude this discussion honours even. Anti-reductionists need not fear theoretical identities, and should even learn to love them.Footnote 10
Reductionists here point out that quantum mechanics provides an account of the structure of molecules. I agree, but a detailed examination of how molecular structures are in fact explained within quantum mechanics puts pressure on the idea that they can be said to be reduced to quantum mechanics. Quantum-mechanical explanations of structure depend on non-trivial assumptions about physical interactions between the electrons and nuclei within a molecule. Emergence provides a fruitful and flexible framework within which to think about these assumptions: it is well known that there are different kinds of emergence (e.g. strong vs. weak; ontological vs. epistemological), but for the purposes of this section it will be helpful to set aside the issue of which particular species of emergence is at stake. My strategy will be to identify the additional assumptions required to explain structure, and then argue that they fall under a widely accepted abstract characterization of emergence as dependent novelty.Footnote 11
Quantum mechanics is generally understood to describe many-body systems of electrons and nuclei in terms of the Schrödinger equation. The idea is that we will seek solutions to the Schrödinger equation that correspond to the possible stationary states of the molecular system. Electronic and nuclear motions are first separated, on account of the very different rates at which they move and respond to external interactions. This is the adiabatic approximation, which yields wavefunctions for two coupled systems (of electrons, and of nuclei), dynamically evolving in lockstep. Essentially the same assumption is widely referred to as the ‘Born-Oppenheimer’ approximation and justified solely in terms of the difference between the masses of electrons and nuclei.Footnote 12 That cannot be a sufficient justification, however, because the ratio of the nuclear and electronic masses is constant, but the adiabatic approximation breaks down in many systems. So what is the difference between adiabatic and non-adiabatic cases? Not the ratios of the nuclear and electronic masses, but rather the timescales over which electronic and nuclear wavefunctions are assumed to respond to each other. The justification for the adiabatic approximation should therefore be understood in terms of timescales too. Consider a single electron in a one-dimensional box: it is often observed that an adiabatic system (i.e. one in which the adiabatic approximation holds) is one in which the walls of the box move slowly enough for the electron’s wavefunction to respond smoothly and continuously to the change. If the walls of the box move too quickly, the system will jump to a different quantum state. Likewise, the adiabatic approximation allows us to think of the joint electronic and several nuclear wavefunctions as fixed parameters with respect to each other, each acting with respect to the other like the walls of the one-dimensional box on the electron’s wavefunction. The electrons see the nuclei as static, while the nuclei see the electrons as smeared-out charge distribution.
When modelling a molecular structure, the nuclei are assigned positions corresponding to the equilibrium positions of the known structure. Density-functional theory (DFT), which has revolutionised molecular quantum mechanics in the last few decades (see Kohn, 1999), then replaces the 3N-dimensional electronic wavefunction with a 3-dimensional electron density function: it can be shown that this can be done without approximation. According to the Hellmann-Feynman theorem, the overall force on a nucleus in the system is determined by the electron density, so effectively the nuclei are being pushed around by their interactions with the electrons.Footnote 13 As Richard Bader notes, this is quite intuitive:
Accepting the quantum mechanical expression for the distribution of electronic charge as given by ρ(r), the theorem is a statement of classical electrostatics and therein lies both its appeal and usefulness. (Bader, 1990, 315)
That a particular substance has a particular molecular structure is then explained by showing that that structure corresponds to a configuration in which the energy is a minimum (i.e. the forces on the nuclei are effectively zero). Two points are worth making, concerning scope and explanatory power. First consider scope: the Schrödinger equation for a molecule depends only on the electrons and nuclei present. Hence different isomers share their molecular Schrödinger equations, the starting point of the above explanation. Ethanol (CH3CH2OH) and dimethyl ether (CH3OCH3) share the same Schrödinger equation, as do enantiomers such as L- and D-tartaric acid (see Sutcliffe & Woolley, 2012). The starting point of the explanation – the molecular Schrödinger equation – does not respect the differences between isomers, and what results from localising the nuclei within the adiabatic approximation does respect these differences, so localising the nuclei in positions corresponding to the different isomers effectively inserts these differences. Reductionists may see this move as having a pragmatic justification: we cannot directly solve the molecular Schrödinger equations, and so must introduce approximations. But it is hard to see the Born-Oppenheimer approximation as a mere approximation if it changes the scope of the quantum-mechanical description, making it apply only to one isomer rather than to all of them.
A second point concerns explanatory power. Approximations are widely assumed to have no independent explanatory power because they are proxies for exact equations: anything that could be explained using an approximate or idealised model could, in principle, be explained using the exact equations. This would be a reasonable thing to conclude if every explanatorily relevant feature of the model could be grounded in the exact equations in some way. In my first paper on these topics (Hendry, 1998) I argued, drawing on the work of Brian Sutcliffe and Guy Woolley, that defences of Born-Oppenheimer models based on the proxy view must fail because they change symmetry properties, which are explanatorily relevant features with respect to which isomers may differ, and cannot therefore be grounded in the molecular Schrödinger equation alone. It seems hard to argue that the approximations have no independent explanatory power. The molecular structures could not, in principle, be explained without them. Hence the different structures, their different symmetry properties and the different causal powers they ground, are effectively introduced as unexplained explainers unless we regard the adiabatic separability of the nuclear and electronic motions and the localization of the nuclei as part of the explanation.
One might regard these conditions merely as initial or boundary conditions, and no more interesting than the ‘auxiliary assumptions’ that, according to received wisdom in the philosophy of science ever since Duhem, are required when we apply any theory. The story goes something like this: quantum mechanics (QM) implies the existence of molecular structure (MS) only in conjunction with statements describing the necessary boundary and initial conditions (BIC). Thus the conjunction of QM and BIC implies MS. This is all correct: adiabatic separability and nuclear localisation might plausibly be thought of as boundary conditions, while the choice of nuclear positions looks like an initial condition. However, this is no help at all if we want to see this explanation as a derivation from quantum mechanics, because meeting the adiabaticity and nuclear localisation conditions exactly is impossible for any genuine quantum system. The conjunction of QM and BIC is, in some important sense, incoherent.
I therefore think it is much less puzzling to describe the situation as follows: the mathematics provides only what one might call a dynamical consistency proof: the conditions that define a Born-Oppenheimer model could not hold exactly in any fully quantum-mechanical system, but the two kinds of system will evolve dynamically in approximately similar ways, for some given level of accuracy, and over the timescales relevant to the calculation. As we have seen, the molecular structure calculations described above assume dynamical conditions—the adiabatic separability of electronic and nuclear motions, and the localisation of the nuclei—which could not hold exactly in any quantum system. All that can be concluded therefore is that a quantum-mechanical system of electrons and nuclei will display approximately similar dynamics to the model. No derivation of the model dynamics from the exact equations has been provided, nor even a demonstration of their consistency under the conditions. All that the mathematics provides is that the relevant approximations introduced in the model can be neglected for a given level of accuracy over relevant timescales.
In joint work with Robert Schoonmaker (Hendry & Schoonmaker, Forthcoming), rather than treating adiabatic separability and nuclear localisation as approximations, we interpret them as substantive special assumptions about dynamical interactions within a quantum-mechanical system of electrons and nuclei. As already noted, these conditions radically transform the dynamical behaviour of quantum systems, and the scope of the equations that describe them. Adiabatic separability makes the overall energy of the electrons and nuclei a function of the nuclear configuration, so that the dependence of energy on nuclear positions can be mapped by a potential energy (PE) surface (or rather a hypersurface). This is not a global assumption because it depends on adiabatic separability, and a system of electrons and nuclei will not have a global PE surface. PE surfaces are not foliated, and near where they cross, the adiabatic separability of nuclear and electronic motions breaks down (see Lewars, 2011: Chapter 2). The effect of nuclear localisation is just as radical and interesting, for it suppresses the dynamical expression of quantum statistics. In general, any quantum system of electrons and nuclei must obey nuclear permutation symmetries: the overall wavefunction must be symmetric (for bosons) or antisymmetric (for fermions). These symmetries correspond to real physical processes however: a molecule exploring the space of its possible nuclear permutations involves the exchange of identical particles. Physical conditions that tend to slow down the exchange processes allow the particle permutation symmetries to be neglected over timescales that are relatively short compared to the exchange. In a quantum system with a classical molecular structure, the nuclei can typically be regarded as being localised by their interaction with the rest of the system. The dynamical effect is that which kind of statistics are assumed to apply to the nuclei – whether the overall wavefunction is symmetric (Bose-Einstein statistics), anti-symmetric (Fermi-Dirac statistics) or indeed asymmetric (classical statistics) with respect to permutation of the nuclei – makes a negligible difference to the evolution of the system. Interaction with the rest of the system effectively transforms the nuclei from quantum entities into classical objects.
It should be emphasised that neither of these conditions is necessary for bonding as such: chemists and condensed matter physicists study systems such as metals and superconductors in which there is bonding (since they form cohesive materials), but in which nuclear and electronic motions are not adiabatically separable, and in which the nuclei are not localised, in the sense that quantum statistics must be taken into account in describing their structure and behaviour. These conditions should be regarded as necessary only for the kind of structure that is describable in terms of the classical chemical structures developed in organic chemistry during the nineteenth century, and some later generalizations. Interestingly, although the expression of nuclear permutation symmetries is generally suppressed, there are molecules, such as protonated methane, in which interactions between one pair of protons means that the symmetries are expressed in the dynamical behaviour of the molecule (see Marx & Parrinello, 1995; Hendry & Schoonmaker, Forthcoming). The above conditions—adiabatic separability of nuclear and electronic motions, and nuclear localisation—are not, moreover, sufficient for the emergence of classical molecular structure. Some molecules, such as cyclobutadiene, tunnel between two different structures each of which is expressed in the molecule’s interaction with radiation: in IR spectra the molecule exhibits square symmetry, while higher-frequency x-ray diffraction catches it in the rectangular states between which it tunnels (see Schoonmaker et al., 2018).Footnote 14 It should be emphasised that tunnelling between different classical structures is the normal quantum-mechanical behaviour (consider P.W. Anderson (1972) on ammonia), but the dynamical behaviour of many molecules can be understood in terms of a single classical structure. Hence dynamical restriction to a single structure is a third necessary condition for the classical kind of structure that is exhibited by many organic molecules and was discovered by organic chemists in the 1860s.
In my view these considerations provide a good argument for regarding adiabatic separability, nuclear localization and dynamical restriction to a single structure as substantive conditions that form a necessary part of the explanation of this kind of structure. In what sense should structure be regarded as emergent, however? Emergence is often understood as dependent novelty: emergent properties are borne by systems that depend for their existence on something more fundamental (typically their parts), but also display properties or behaviour that is in some significant way novel with respect to the parts. This applies readily to the foregoing discussion. Molecular structures are ontologically dependent on electrons and nuclei: they cannot exist without them. The novelty consists in the distinct dynamical behaviour displayed by electrons and nuclei in the context of structured systems: adiabatic separability, nuclear localization and restriction to a single classical structure, which in each case is a suspension of the normal behaviour of a quantum system. The adiabatic separability and localization are also examples of transformational emergence (see Santos, 2015; Humphreys, 2016, Chapter 2), in which the behaviour of the parts of an emergent system is so different that it makes sense to say that they have been transformed into a new kind of entity. The radical transformation of nuclei from entities that obey quantum statistics into localized, semi-classical entities would seem to be a good example of transformational emergence.
Notes
- 1.
Everything I say in this section is intended to be consistent with relevant definitions agreed by the International Union of Pure and Applied Chemistry (IUPAC: see https://goldbook.iupac.org)
- 2.
I have no major objection to using levels, but in this paper I will opt for scales because (i) scales will do the job, and (ii) they are free of some distracting philosophical connotations associated with levels, which some metaphysicians fear give rise to important confusions in the context of debates about reduction and emergence.
- 3.
Here I am using ‘structure’ inclusively, to include elemental composition. Structure is sometimes contrasted with composition, but to know that a particular substance is composed of certain elements in certain proportions is to know something about its structure at the molecular scale.
- 4.
An additional argument is that if thermodynamic properties are what make a substance what it is, they should be metaphysically necessary. This is not, I believe, the perspective of chemistry, which allows thermodynamic properties such as boiling point to vary across nomologically different worlds (see Hendry, 2023; Hendry & Rowbottom, 2009).
- 5.
David Knight (1995, Chapter 13) calls chemistry a ‘service science’ on account of its analytical expertise, which I think nicely captures its status with respect to milk and wool.
- 6.
Potential energy (PE) surfaces aren’t quite fundamental. For the evolution of a physical system to be describable in terms of a PE surface, the energy of the system must be a function of just the nuclear coordinates. As we shall see in the final section of this paper, this is a substantive physical condition (adiabatic separability of electronic and nuclear motions), and one that quantum-mechanical systems can only approximate. In general, quantum-mechanical systems do not even approximate it.
- 7.
Note that this is only a tendency: if one of the reactants in another step is scarce enough in the local environment, then that step will become the slowest step.
- 8.
This explains the Walden inversion, known to chemists since the 1890s (see Brock, 1992, 544–5).
- 9.
‘Curly arrow’ is the official name among chemists. For a discussion of their interpretation see Suckling et al. (1978, Section 4.3.3), who argue that curly arrows mean formal transfer of electrons. I take it that that means that the net difference between the starting and end structures is equivalent to the charge transfer indicated by the arrow.
- 10.
For similar reasons I have never understood why ‘pain is c-fibres firing’ should have the significance for the reductionism debate that it has. The identification would only work from an explanatory point of view by appealing to significant levels of organisation above the level of the c-fibres.
- 11.
I am most grateful to Stewart Clark, Tom Lancaster and Robert Schoonmaker for conversations on these topics. This section draws on joint work with Schoonmaker, but the position I set out is my own interpretation of the various scientific facts, and I would not wish to implicate these interlocutors in any of my misunderstandings.
- 12.
See for instance Atkins (1986, 375).
- 13.
The quantum-mechanical electron-density distribution carries information about the nuclei too, so each of the nuclei is really being ‘pushed around’ by its interactions with the entire system.
- 14.
This is an expression of the scale-relativity of structure, for which I have argued elsewhere (see Hendry, 2023).
References
Anderson, P. W. (1972). More is different. Science, 177, 393–396.
Atkins, P. W. (1986). Physical chemistry (Third ed.). Oxford University Press.
Bader, R. F. W. (1990). Atoms in molecules: A quantum theory. Oxford University Press.
Brock, W. H. (1992). The Fontana history of chemistry. Fontana Press.
Carpenter, B. (1984). Determination of organic reaction mechanisms. Wiley.
Carroll, F. A. (1998). Perspectives on structure and mechanism in organic chemistry. Brooks/Cole.
Craver, C., & Tabery, J. (2016). Mechanisms in science. In E. N. Zalta (Ed.), The Stanford encyclopedia of philosophy (Fall 2016 ed.) http://plato.stanford.edu/archives/spr2016/entries/science-mechanisms/
Eisenberg, D., & Kauzmann, W. (1969). The structure and properties of water. Oxford University Press.
Goodwin, W. M. (2007). Scientific understanding after the Ingold revolution in organic chemistry. Philosophy of Science, 74, 386–408.
Goodwin, W. M. (2012). Mechanisms and chemical reaction. In R. F. Hendry, P. Needham, & A. I. Woody (Eds.), Handbook of the philosophy of science, volume 6: Philosophy of chemistry (pp. 309–327). North-Holland.
Gould, E. S. (1959). Mechanism and structure in organic chemistry. Holt, Rinehart and Wilson.
Havstad, J. C. (2018). Messy chemical kinds. British Journal for the Philosophy of Science, 69, 719–743.
Hendry, R. F. (1998). Models and approximations in quantum chemistry. In N. Shanks (Ed.), Idealization in contemporary physics: Poznan studies in the philosophy of the sciences and the humanities 63 (pp. 123–142). Rodopi.
Hendry, R. F. (2006). Elements, compounds and other chemical kinds. Philosophy of Science, 73, 864–875.
Hendry, R. F. (2010a). The elements and conceptual change. In H. Beebee & N. Sabbarton-Leary (Eds.), The semantics and metaphysics of natural kinds (pp. 137–158). Routledge.
Hendry, R. F. (2010b). Entropy and chemical substance. Philosophy of Science, 77, 921–932.
Hendry, R. F. (2017a). Prospects for strong emergence in chemistry. In M. Paolini Paoletti & F. Orilia (Eds.), Philosophical and scientific perspectives on downward causation (pp. 146–163). Routledge.
Hendry, R. F. (2017b). Mechanisms and reduction in organic chemistry. In M. Massimi, J. W. Romeijn, & G. Schurz (Eds.), EPSA15 selected papers: The 5th conference of the European Philosophy of Science Association in Düsseldorf (pp. 111–124). Springer.
Hendry, R. F. (2019). Emergence in chemistry: Substance and structure. In S. C. Gibb, R. F. Hendry, & T. Lancaster (Eds.), The Routledge handbook of emergence (pp. 339–351). Routledge.
Hendry, R. F. (2022). Quantum mechanics and molecular structure. In O. Lombardi et al. (Eds.), Philosophical perspectives on quantum chemistry (pp. 147–171). Springer.
Hendry, R. F. (2023). Structure, essence and existence in chemistry. Ratio, 36. In press, forthcoming.
Hendry, R. F. (Forthcoming). How (not) to argue for microstructural essentialism.
Hendry, R. F., & Rowbottom, D. P. (2009). Dispositional essentialism and the necessity of laws. Analysis, 69, 668–677.
Hendry, R. F., & Schoonmaker, R. (Forthcoming). The emergence of the chemical bond. Unpublished manuscript.
Hoffmann, R. (1995). The same and not the same. Columbia University Press.
Humphreys, P. (2016). Emergence: A philosophical account. Oxford University Press.
Knight, D. M. (1995). Ideas in chemistry: A history of the science. Athlone.
Kohn, W. (1999). Electronic structure of matter: Wave functions and density functionals. Reviews of Modern Physics, 71, 1253–1266.
Kragh, H. (2000). Conceptual changes in chemistry: The notion of a chemical element, ca. 1900–1925. Studies in History and Philosophy of Modern Physics, 31B, 435–450.
LaPorte, J. (2004). Natural kinds and conceptual change. Cambridge University Press.
Leigh, G. J., Favre, H. A., & Metanomski, W. V. (1998). Principles of chemical nomenclature: A guide to IUPAC recommendations. Blackwell Science.
Lewars, E. (2011). Computational chemistry: Introduction to the theory and applications of molecular and quantum mechanics (Second ed.). Springer.
Lewis, G. N. (1916). The atom and the molecule. Journal of the American Chemical Society, 38, 762–785.
Machamer, P., Darden, L., & Craver, C. (2000). Thinking about mechanisms. Philosophy of Science, 67, 1–25.
Marx, D., & Parrinello, M. (1995). Structural quantum effects and three-centre two-electron bonding in CH5+. Nature, 375, 216–218.
McLeish, T. (2019). Soft matter: An emergent interdisciplinary science of emergent entities. In S. C. Gibb, R. F. Hendry, & T. Lancaster (Eds.), The Routledge handbook of emergence (pp. 248–264). Routledge.
Needham, P. (2002). The discovery that water is H2O. International Studies in the Philosophy of Science, 16, 205–226.
Needham, P. (2008). Is water a mixture? Bridging the distinction between physical and chemical properties. Studies in History and Philosophy of Science, 39, 66–77.
Needham, P. (2011). Microessentialism: What is the argument? Noûs, 45, 1–21.
Popelier, P. (2000). Atoms in molecules: An introduction. Pearson.
Putnam, H. (1975). The meaning of “meaning”. In Mind language and reality (pp. 215–271). Cambridge University Press.
Roberts, J. D., & Caserio, M. C. (1965). Basic principles of organic chemistry. W.A. Benjamin.
Rocke, A. J. (2010). Image and reality: Kekulé, Kopp and the scientific imagination. Chicago University Press.
Salmon, W. (1984). Scientific explanation and the causal structure of the world. Princeton University Press.
Santos, G. (2015). Ontological emergence: How is that possible? Towards a new relational ontology. Foundations of Science, 20, 429–446.
Santos, G., Vallejos, G., & Vecchi, D. (2020). A relational constructionist account of protein macrostructure and function. Foundations of Chemistry, 22, 363–382.
Schoonmaker, R. T., Lancaster, T., & Clark, S. (2018). Quantum mechanical tunneling in the automerization of cyclobutadiene. Journal of Chemical Physics, 148, 104109.
Sidgwick, N. V. (1936). Structural chemistry. Journal of the Chemical Society, 149, 533–538.
Suckling, C. J., Suckling, K. E., & Suckling, C. W. (1978). Chemistry through models: Concepts and applications of modelling in chemical science, technology and industry. Cambridge University Press.
Sutcliffe, B. T., & Woolley, R. G. (2012). Atoms and molecules in classical chemistry and quantum mechanics. In R. F. Hendry, P. Needham, & A. I. Woody (Eds.), Handbook of the philosophy of science, volume 6: Philosophy of chemistry (pp. 387–426). North-Holland.
Sykes, P. (1981). A guidebook to mechanism in organic chemistry (Fifth ed.). Longmans.
van Brakel, J. (2000). Philosophy of chemistry. Leuven University Press.
van der Vet, P. (1979). The debate between F.A. Paneth, G. von Hevesy and K. Fajans on the concept of chemical identity. Janus, 92, 285–303.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2024 The Author(s)
About this chapter

Cite this chapter
Hendry, R.F. (2024). Mechanisms in Chemistry. In: Cordovil, J.L., Santos, G., Vecchi, D. (eds) New Mechanism. History, Philosophy and Theory of the Life Sciences, vol 35. Springer, Cham. https://doi.org/10.1007/978-3-031-46917-6_7
Download citation
DOI: https://doi.org/10.1007/978-3-031-46917-6_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-46916-9
Online ISBN: 978-3-031-46917-6
eBook Packages: Religion and PhilosophyPhilosophy and Religion (R0)