
Abstract
Common wheat is a hexaploid species crop that is widely recognized as an important staple food crop. The establishment of a gold standard reference genome sequences of the well-studied CHINESE SPRING, and its progenitors (including Triticum turgidum ssp. dicoccoides accession Zavitan, Triticum durum accession Svevo, Triticum urartu, Aegilops tauschii), in the last 5 years has dramatically promoted our understanding of wheat genome diversity and evolution through the resequencing of collections of wheat and its progenitors. In this chapter, we review progress in the analysis and interpretation of genome‑based studies of wheat focusing on geographic genome differentiation, interspecies gene flow, haplotype blocks, and gene diversity in breeding. We also consider approaches for efficiently discovering and integrating the genes and genome variations, hidden in Genebank collections, into wheat breeding programs.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
6.1 Wheat Origin and Spread in the World
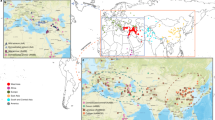
Common wheat (Triticum aestivum L.) provides approximately 20% of the total calories for human intake globally. The origins of the common hexaploid wheat species were through natural crosses between cultivated emmer (Triticum turgidum, AABB) and Aegilops tauschii (DD) and is considered to be the first domesticated crop in the “hilly flanks of the Fertile Crescent” in southwestern Asia between 10,000 and 7000 BC (Feldman and Levy 2012). Key advances for the domestication process included the absence of head brittleness and free-threshing grains. The dispersal of wheat selections prior to the 5th millennium BC was extensive as several Triticum taxa spread from the Fertile Crescent westwards across central Europe and along the northern coastal line of the Mediterranean (Fig. 6.1). To the East, wheat is documented in archaeological records to be present in Turkmenistan and Pakistan before 5000 BC. It was introduced into west China in 2000 BC and into central and east China in approximately 1500 BC (Liu et al. 2016, 2019a, b), based on archaeological discoveries.

Map showing hypothesized dispersals of domesticated Triticum and Hordeum taxa (i.e. wheat and barley) originated in southwestern Asia across the Old World dating between 5000 and 1500 cal BC. Black circles: sites older than 5000 BC; gray circles: sites dated between 5000 and 2500 BC; white circles: sites dated between 2500 and 1500 BC; solid line: parsimonious inference from botanical evidence from dated archaeological context (the density of which varies greatly across Eurasia). Map is originally presented in Liu et al. (2019a, b), modified with permission
Colonization of wheat in the very new and distinct environments eventually replaced native crops as the staple crop and resulted in field-level selections of traits with very strong geographic characters to meet the local cultivation and consumption of variant human populations. These genetic changes were basically retained in the genome variation between cultivars, especially the landraces. Establishment of the so-called gold standard wheat genome sequence, taken together with the assemblies of the reference genomes of its progenitor species as well as other hexaploid varieties (Avni et al. 2017; Luo et al. 2017; Zhao et al. 2017; Ling et al. 2018; IWGSC 2018; Zhu and Luo 2021), has provided the basis for high-density SNP-chips and resequencing analyses. Advances such as the production of SNP-chips with 90, 285, 660 K SNPs have provided the means for the wide use to elucidate genome diversity. Germplasm exchange and the development of genomics now provide a new opportunity to re-evaluate and reconsider the evolution and dispersal process of wheat from this new point of view. These works also pave the way for associating the allelic variation with phenotypes for physical mapping of variation in the genome (Varshney et al. 2021).
6.2 Global Distribution of Wheat Genome Diversity and the Leading Role of 3B in Geographic Differentiation
The genotyping of 632 world wheat landraces using the 285 K SNP array-markers on chromosome 3B, allowed Paux et al. (2008) to define the very strong geographic differentiation (Fig. 6.2). In a follow-up diversity analysis of Chinese wheat landraces using a 660 K SNP array, we found they could be basically classified into two sub-groups, the north-China sub-group and middle-south China sub-group (Wang et al. 2021). Among the 21 chromosomes, 3B and 7A were particularly prominent in being associated with the stratified domestication in China, based on the standard Fst values for SNP allele frequencies that differentiate populations in two groups, namely Triticum aestivum-L1 and T. aestivum-L2 (Fig. 6.3).

Haplotypes in landraces on 3B and their global distribution (provided by Dr. Etienne Paux, INRAE). The red, pink, blue, and green dots refer to different haplotypes (see Paux et al. 2008) and the clustering of the different colored haplotypes across the landscape from Europe to China is evident

Very strong geographic and genetic differentiation happened in Chinese wheat landraces, forming two subsets, L1 (blue) and L2 (red). The 3B and 7A lead the differentiation among the 21 chromosomes. a Quite distinct distribution of collections in L1 and L2. b The FST value between L1and L2 along the 21 chromosomes, which was estimated based on the 660 K SNP markers using CS R 1.0 as reference. c and d. The triangles/arrows indicate FST values between L1 (blue) and L2 (red), L1 and modern cultivars (M, green), and L2 and M on 3B and 7A. The red lettering along the arrows focuses on the crucial genomic regions (3B: 280–375 Mb) and (7A: 211.7–272.9 Mb). The data along the dashed circles were FST values between subsets in whole genome of wheat (adapted from Wang et al. 2021)
When the differentiation of populations T. aestivum-L1 and T. aestivum-L2 were narrowed down to the analysis of the crucial regions of 280–375 Mb on 3B and 211.7–272.9 Mb on 7A in the CS reference 1.0 (IWGSC 2018), the Fst reached 0.84 and 0.66, respectively (Fig. 6.3a; quantified in Fig. 6.3b), and were associated with grain size and length in multi-environment BLUP phenotype data (Wang et al. 2021). Accessions in T. aestivum-L1 were mainly distributed in northwestern China, whereas those in T. aestivum-L2 were mainly from central to eastern China (Fig. 6.4). The most distinct agronomic trait was grain size (TKW), i.e., the T. aestivum-L2 accessions usually had smaller grain size than the T. aestivum-L1 accessions, which was achieved by reduction in grain length (Wang et al. 2021).

Haplotype network based on SNPs on 3B in cultivated emmer (CE), domesticated emmer (DE), Northeast landrace group 1 (L1), Southeast landrace group 2 (L2), modern Chinese cultivar (MC), and wild emmer (WE). Circle size is proportional to the number of accessions for a given haplotype. The short lines between two nodes indicate the number of mutations
Haplotype analysis in genotyped collections including wild emmer, domesticated emmer, common wheat landraces, and Chinese modern cultivars based on the 660 K SNP genotyped data clearly revealed wild emmer (WE) was the donor for the hap-block in L1 (see Haps 1, 2, 4, and 12 in Fig. 6.4). This is consistent with the suggested intercross and genome introgression between common wheat and wild emmer (He et al. 2019; Cheng et al. 2019).
GWAS based on the multi-year agronomic trait phenotypes revealed strong association of the crucial region on 3B (280–375 Mb) with spike length (− log10 (p) ≥ 5.0). In Chinese landraces, the northwest haplotype-group (L1) usually has longer spike and larger grains than the southeast haplotype-group (L2). Breeding selection in the seven decades from 1950 to 2020 favored the L1-haplotypes from the wild emmer (Fig. 6.4; Hao et al. 2020; Wang et al. 2021). Therefore, we estimate that this genomic region might also relate to NUE or WUE of cultivars. The great increase of L1-haplotypes (including Haps 1, 2, 4, and 12) in the modern Chinese cultivars (MC) at this genome location also correlated with the cooking style from full grain in history to wheat flour products today, because small grain was favored in full grain cooking, but larger grain was favored in flour-product consumption because of higher yield (Liu et al. 2014, 2016). Based on the analysis in the 10 + pangenome, we found large structure variation (SV) existing in this region across the 3B centromere (Fig. 6.5).

GWAS based on 660 K SNP array with multi-year phenotyping data of landraces (NP) and biparental population (BPP) indicated that high genome differentiation at 280–375 Mb was associated with spike length on 3B chromosome as indicated by the scores for Fst exceeding the significance cutoff, across the 280–375 Mb region
6.3 Frequent Gene Flow Between Species and Its Effects on Diversity
In Israel, wild emmer wheat with intermediate phenotypes grew at the boundaries of cultivated areas. These wild plants may have originated from hybridization of wild emmer with T. turgidum cultivars. They are indicative of gene flow between wild and domesticated populations (Matsuoka 2011). Dvorak et al. (2006) provided initial molecular evidence for existence of introgressions from wild emmer (Triticum dicoccoides) into common wheat, which was indirectly supported by the fact that wild emmer usually existed as an accompanying weed of durum and common wheat in wheat origin/domestication regions. Hexaploid and tetraploid wheats were also cultivated as a mixture in the field in these regions (Matsuoka 2011). The overall consequence was that the mixed cropping provided opportunities for gene flow between species through natural hybridization.
The identity score (IS) is widely used to reveal the parent’s genetic contribution to their derived cultivars in breeding. The IS is defined with reference to similar nucleotide sequences present in two, or more than two, individuals through replication of the same ancestral copy of respective sequences. Our IS graph file analysis based on resequencing analysis in common wheat (landraces and modern cultivars), wild emmer, domesticated emmer and durum wheat, revealed frequent genomic introgressions between common wheat and wild emmer, as well as cultivated emmer, where CS 1.0 was used as reference (light blue, IWGSC 2018). Of course, more frequent introgressions between the tetraploid species were detected as expected, because they shared the common genome AABB (Fig. 6.6a). Independent research by two other groups also revealed the wild existence of introgressions from wild emmer into common wheat (Fig. 6.6b) (Cheng et al. 2019; He et al. 2019). In addition, global wheat diversity research was also strongly promoted by the establishment golden standard reference genomes of common wheat and T. dicoccoides and Triticum durum (IWGSC 2018; Avni et al. 2017; Maccaferri et al. 2019; Pont et al. 2019; Sansaloni et al. 2020), all of which were sequentially perfected with the integration of more assembles based on 3rd generation sequence reads (Zhu et al. 2021).

Frequent genome introgressions between species in Triticum genus revealed by 1-IBD within the A sub-genome chromosomes, where CS 1.0 was used as reference and expressed in light blue. a Graph based on (1-IBD) indicated frequent introgressions from wild emmer to domesticated emmer and common wheat. It also revealed reverse introgression from common wheat to domesticated emmer and wild emmer. b Genomic introgressions detected in global common wheat on the 14 chromosomes of A and B sub-genomes from four wild emmer populations into common wheat (adapted from Cheng et al. 2019)
Alien introgression usually reduces the recombination frequency and leading to strong LD in natural or breeding population, which results in decline of diversity in the respective genome regions. However, the SNP density was usually increased because suppression of recombination has retained the intact of the introgression fragments, which retain regions rich SNPs in their comparison with CS reference genome (Fig. 6.7). We found that the evenness of recombination rate along the D sub-genome chromosomes is much better than either A- or B- sub-genome chromosomes. This might be caused by the introgressions from wild emmer, which mainly existed within the A- and B-sub-genomes of common wheat (Fig. 6.8). Sufficient genome differentiation should happen between the hexaploid and tetraploid AB genome, which prevents occurrence of recombination between the “introgressions” and original homology fragments. The great difference on recombination rate across the centromeres between 7A and 7B in Chinese modern cultivars directly supported our hypothesis of introgression suppression to recombination, because there is a large introgression detected on 7A across centromeric region (Fig. 6.8, ca 230–430 Mb, Cheng et al. 2019; Hao et al. 2020).

A wild emmer introgression (~ 172–448 Mb) and their effects on SNP density, recombination ratio (ρ), and genome diversity (π) in comparison with the neighbor region without introgression (468–530 Mb) on chromosome 4A. CL: Chinese landrace, IMC: introduced modern cultivars

Recombination ratio (ρ) along the three chromosomes of homologous group 7 in Chinese wheat landraces (blue) and modern cultivars (red). There is more recombination disequilibrium on 7A and 7B than on 7D chromosome
6.4 LD and Haplotype Blocks in Wheat Evolution and Breeding
Linkage disequilibrium (LD) is a common phenomenon in the population genetics analysis of crops. For a long time, it was believed that strong positive selection for a gene usually led to strong LD around the loci because of hitchhiking effect. LD was usually affected by population diversity, selection pressure at the crucial loci, as well as recombination rate. We found that chromosomes in A and B sub-genomes have larger and stronger LD blocks in wheat (Hao et al. 2020). This might be caused by two factors (1) more QTLs controlling agronomic traits on the A and B sub-genomes (Peng et al. 2003, 2011). (2) Partial suppression of recombination along A and B sub-genome chromosomes caused by the introgressions from wild T. turgidum species (Figs. 6.6, 6.7 and 6.8).
6.4.1 Haplotype Block Size Along a Chromosome in Wheat
On each of the 21 chromosomes, five chromosomal regions were defined by the IWGSC, based on the overall recombination pattern observed in wheat (IWGSC 2018). There are fewer but very large (> 10 kb, Fig. 6.9a example for chromosome 2B) haplotype blocks at regions across the centromeres, and smaller haplotype blocks at the R1 and R2 regions (Fig. 6.9). The identification of the R1–R3 blocks of chromosome regions in the wheat chromosomes is based on the recombination rate characteristics, gene density, and tissue-specific vs household expression variation across each of the 21 wheat chromosomes. The R1 and R3 designate the distal ends of the short and long chromosomal arms, respectively; R2a and R2b designate the interstitial regions of the short and long arms and the C region and identify the pericentromeric regions (IWGSC 2018).

The box plots in Fig. 6.9 provide a statistical assessment of the R1, R2a, R2b, and R3 designations across the wheat genome based on recombination frequency and indicated that the difference between C and the terminal blocks was significant. Consistent with this significant difference in recombination frequency, Jordan et al. (2020) found that DNS scores assessing DNA accessibility to Micrococcal Nuclease (MNase), and thus the more open or compacted state of the chromatin, were significantly higher (= more open chromatin) for the genome space in the R1 and R3 regions.
6.5 Large Haplotype Blocks and Their Role in Breeding
Identification of haplotype blocks and big PAVs and tracking their variants in evolution and breeding are notable aspects in self-pollination crops in the current genomics era. The investigation of gene-network contributions to the well-studied thousand grain weight (TGW) phenotype that contributes to yield in wheat, for example, provided an unexpected influence of structural variation for the presence/absence of the 5AS chromosome arm (Taagen et al. 2021). A combination of transcriptome data and high-resolution marker maps for the TGW QTL initially thought to be on 5AL, in fact indicated that the QTL resulted from linkage to the presence/absence of the 5AS arm. On a larger scale, the resequencing of 145 land marker cultivars in China, it was found that there were more long-range haplotypes on A and B sub-genomes rather than on D sub-genome in common wheat (Jordan et al. 2015; Hao et al. 2020). The first reason was that the gene flow occurred from the wild tetraploid T. dicoccoides during early co-cultivation of tetraploid and hexaploid wheat, where wild emmer was also present as a weed in wheat fields. This was therefore expected to result in a substantial increase in polymorphism on the A and B sub-genomes relative to the D sub-genome in modern bread wheat. This also partially, negatively, affected the homologous recombination occurrence within A and B sub-genomes at crucial genomic regions because of differentiation on the intergenic repeats among wild emmer, cultivated emmer, and common wheat, leading to a very uneven distribution of recombination ratio and SNPs along chromosomes (Fig. 6.8). The second reason is asymmetric distribution of agronomic traits among the three sub-genomes. There are more QTLs or genes controlling domestication and yield traits mapped on the A sub-genome than either on B or D sub-genomes, leading to stronger selection on the A sub-genomes (Peng et al. 2011; Jordan et al. 2015).
Haplotype-based breeding (HBB) can now be proposed following the genome resequencing of larger number of cultivars, because it represents a promising breeding approach for dealing with and identifying, superior haplotypes and their deployment in breeding programs (Varshney et al. 2021). We propose that for self-pollinated crops with a long breeding history, it will be possible to take advantage of hap-block identification to select ideal parent materials to achieve new high-performing cultivars via HBB (Figs. 6.9 and 6.10).

Diversity features at key haplotype blocks considering cultivars subsets released pre- and post-development of the two hallmark Chinese cultivars AIMENGNIU (AMN) and XIAOYAN 6 (XY6) as well as within Chinese landraces (CL) and modern Chinese cultivars (MCC). SNP similarity between AMN and XY6, population diversity for each sub-set (pre-AMN vs. post-AMN, pre-XY6 vs. post-XY6, MCC vs. CL) on chromosomes 6A (a), 1A (b). Adapted from Hao et al. (2020)
We dissected diversity features along chromosomes 6A (Fig. 6.10a) and 1A (Fig. 6.10b) in cultivar subsets released pre- and post-development of the two hallmark Chinese cultivars AIMENGNIU (AMN) and XIAOYAN 6 (XY6) based on their pedigrees. Fixation of big haplotype blocks from 224 to 442 Mb, on 6A in post-XY6 cultivars were detected but relatively higher diversity was retained in AMN-post cultivars. From 100 to 300 Mb, the haplotype block was fixed in post-AMN cultivars but not in post-XY6 on chromosome 1A. This indicated the haplotype block carried by XY6 on 6A and that carried by AMN on 1A provided sufficiently high-quality attributes for breeders to then retain them. An interesting but less pronounced trend was also found from 178 to 472 Mb on chromosome 2A, but with both XY6- and AMN-derived new cultivars, this genomic region maintained a higher diversity. This indicates haplotypes carried by either XY6 or AMN are not sufficiently high-quality enough for breeders to retain them. The very large sizes of the haplotype blocks also highlight the feasibility of HBB in wheat.
6.6 Human Selection and Gene Diversity
Cloning the gene responsible of a trait or QTL and analyzing its natural variation to find valuable new alleles is one major task for scientists working in crop genetic resources. Fine mapping of QTL through advanced backcross QTL analysis was regarded as the best reliable method for a long time (Tanksley and Nelson 1996). However, QTL mapping-based cloning of genes in wheat is very time-consuming because of the complexity of genome and polyploid nature. The gold reference genome sequence promotes gene cloning via GWAS under the assistance of gene editing and transformation in wheat. The successful mapping of Rht24 through GWAS in large collections using the CHINESE SPRING genome as reference for 6A is a landmark indicator for gene mapping strategy that complements the QTL fine mapping in biparental recombination population to GWAS-fine mapping in natural population. Through anchoring the flanking markers on the RefSec v1.0, the candidate gene of Rht24 was narrowed down to 50 Mb region between 400 and 450 Mb on 6A chromosome, which was actively selected in breeding since 1990s (Würschum et al. 2017).
It is very hard to precisely map genes at pericentromeric region through recombination in biparental populations. But through GWAS, we can use long historic recombinations to carry out mapping and dissection of the crucial region. For example, we found a grain thickness-associated locus on the long arm of the chromosome 5A marked by the peak SNP chr5A_430246395 (− log10(p) = 4.17) because the region was overlapping with selection sweeps and contained the wheat homolog of the rice DEP1 gene (Fig. 6.11) that has been shown in rice to enhance grain yield by promoting nitrogen utilization efficiency (Huang et al. 2009; Xu et al. 2019). The LD block was ~ 1.3 Mb and contained 15 genes. A total of 33 SNPs were present in the region. Haplotype analysis of these SNPs showed that the grain thickness of accessions with haplotype 1 (Hap1) was significantly thicker (P < 0.001) than that of other accessions (Fig. 6.11), and these two sets of accessions also had significant increases in average thousand grain weight, but reduced plant height. The locus in fact shows pleiotropic effects on multiple agronomic traits and RNA-seq data showed that TaDEP1 expressed significantly different (P < 0.001) between accessions of thin-grain and thick-grain. We then used CRISPR/Cas9 editing to introduce deletion mutations into all three TaDEP1 homoeologs in cv. KENONG199. The edited plants displayed significant (P < 0.01) reductions in grain size, the edited mutants also showed short stem, more tillers, and compact spike (Fig. 6.11), confirming that TaDEP1 is a gene with pleiotropic effects and functionally essential for wheat grain size development (Li et al. 2022).

GWAS make it possible to precisely map and verify genes in the low recombination region using CS golden reference and historic recombinations under the assistance of genome re-sequence and gene editing. A grain thickness locus on chromosome 5A carrying the rice DENSE AND ERECT PANICLE ortholog TaDEP1. a GWAS signals at 430.24 Mb on 5A. b Three homologous of DEP1 and their mutated sites by CRISPR-Cas9 in KELONG 199. c Seed size difference between the triplet mutant (DEP1-MUT) and wild type (WT). d Statistics difference between the DEP1-MUT and WT on flowering time, grain length (GL), grain wide (GW), and grain thickness (GT). e and f phenotype difference on plant morphology and spike. Adapted from Li et al. (2022)
There are more PAV and other SV in common wheat than other crops. Therefore, if the agronomic target is located in the SV region, there are likely to be difficulties to map QTL precisely using biparent population, even in the natural population by GWAS using a single reference genome. A graph of the pangenome for functional genomics and HBB in wheat would be a major advance.
6.7 Yield Genes and Their Diversity
For yield genes, because of their conserved characters among cereals, much work was carried out based on the synteny and collinearity among cereal genomes, especially the good collinearity between wheat and rice. Three very interesting points were found. The first is dominance of the three homologous genes in the hexaploid species. The second is that most of the natural variations occurred within the promotor regions of the crucial genes among cultivars. The third is strong correlation of haplotypes with the water and fertility of the soil as well as sunlight and temperature resources in growing season. For example, the GS5 was recognized as one gene strongly influencing grain size in cereals (Li et al. 2011), and in wheat, it is preferentially expressing in young spikes and developing grains, and positively regulating grain size. Among the three homoelogous genes, GS5-3D has the dominant expression, GS5-3B is almost silenced with very low expression level, while the GS5-3A is seen to have medium expression levels (Fig. 6.12a). Only one SNP (T/G) was identified at 2334 bp downstream of the ATG start codon at the TaGS5-3A. Two alleles were detected on GS5-3A in world modern cultivars, with average 6–7 g difference on thousand grain weight. The global distribution of frequency of larger grain size allele TaGS5-3A-T exhibited very good correlation with water resources during wheat growing season (Fig. 6.12b). No diversity was detected at either 3B or 3D loci (Ma et al. 2016).

Dominance among the three GS5 homeology genes in wheat (a) and global distribution of alleles in modern cultivars (b). a Temporal and spatial expression of TaGS5 homoeologues. SL, seedling leaf; SR, seedling root; HR, root at the heading stage; HS, stem at the heading stage; FL, flag leaf; 1 YS, 3 YS, 5 YS, and 7 YS, young spikes of 1, 3, 5, and 7 cm in length; SP, spike at heading stage; various stages of grain development, including 1 DPA, 3 DPA, 5 DPA, 10 DPA, 15 DPA, 20 DPA, and 25 DPA. The expression of TaGS5-3A in the spike at heading stage was assumed to be 1. b Distributions of TaGS5-3A-T and TaGS5-3A-G alleles in wheat cultivars from different ecological regions. North America (I), CIMMYT (II), Europe (III); former USSR (IV); China (V); and Australia (VI)
6.8 Adaptation of Cultivars to Environments
Based on the whether or not a cold temperature vernalization is required to promote flowering, wheat cultivars are classified into winter and spring types. This vernalization requirement prevents temperate plants from flowering under freezing winter conditions. Wheat cultivars grown in different environments need diverse vernalization characteristics to ensure flowering and reproductive development at appropriate time to meet the need for higher yield and mature on time.
In wheat, flowering time is controlled by both vernalization system and photoperiod reaction system together. For the vernalization, there are four genes, TaVrn1, TaVrn2, TaVrn3, and TaVrn4, that have been positionally cloned; TaVrn4 was identified as a duplication of TaVrn1 (Yan et al. 2003, 2004, 2006; Kippes et al. 2015). The expression level of TaVrn3 (FT) is the key element determining flowering or not flowering. However, expression of TaVrn3 is strongly, positively, regulated by TaVrn1 and TaVrn4 and PPD1, but negatively regulated by TaVrn2. Any function mutants in TaVrn1, TaVrn2, TaVrn4, and PPD1 influence expression of TaVrn3, and subsequently the flowering time. This provides wheat with an extensive range of variation to adapt particular combinations of variants to grow environments through combining different alleles at the four loci. Mutations at promoter region of VRN3 that result in a loss of binding site for VRN2 lead to complete loss of suppression of VRN3 by VRN2 and result in a full spring type wheat (Yan et al. 2003, 2004, 2006; Kippes et al. 2015). Furthermore, it was found TaVrn1 had significant epistatic effects on flowering time (Xie et al. 2021). Copy number variation (CNV) was also detected at VRN1 loci, which negatively influences expression level of itself (Diaz et al. 2012). In addition, TFs binding with cis at promoter regions of VRN1, VRN2, and VRN3 often affecting wheat heading and flowering time (Liu et al., JIBP 2019a). Furthermore, genes in the pathway of auxin were also involved in the regulation of leaf senescence and re-mobility of nutrients from leave and stems to grains in wheat (Li et al. 2023). A detailed summary of the vernalization system and photoperiod reaction networks in wheat is provided by Sehgal et al. (see Chap. 11 in this volume).
6.9 Disease-Resistant Genes and Their Diversity
In one life cycle, wheat is threatened by many diseases and pests. Some pathogens’ races, such as rusts, powdery mildew change very quickly from year to year. Therefore, wheat disease resistance breeding is a constant evolutionary arms race with their pathogens. Therefore, there must be enough diversity in R genes for this race. Fortunately, disease-resistant genes usually mapped at the high recombination regions (R1 and R3, Fig. 6.9) on chromosomes. Frequent recombinations often create new variation and PAV and CNV, which bring great opportunity to create new genes for resistance. Therefore, cloning R genes has been a high priority in wheat molecular biology in the past 10 years and is expected to continue to be a high priority.
Until now, there are three major types of disease resistance genes cloned in wheat (see also Chap. 10). Resistance genes with typical CC-NBS-LRR domains, such as powdery mildew resistance genes Pm1, Pm2, and Pm3, leaf rust resistance genes Lr1 and Lr13, stem rust resistance genes Sr33 and Sr35. 2) R genes containing Kinase-MCTP structure, such as the Yr36, has a START-Kinase structure. The Pm4 has a Kinase-MCTP structure. The Yr15 (WTK1), Sr60 (WTK2), and Pm24 (WTK3) have a tandem kinase structure. (3) Disease-resistant genes cording proteins with transmembrane transport functions, such as durable resistance genes Lr34/Yr18/Sr57/Pm38 and Lr67/Yr46/Sr55/Pm46. There are rare natural mutants in landraces carrying good-resistant genes, such as the famous Fhb1, Pm5e which encode an amino acid mutation in the NLR protein; the deletion of two amino acids in the powdery mildew resistance gene Pm24 (WTK3) confers broad-scope resistance to powdery mildew. Besides common wheat collections, the ancestral species of wheat usually contain abundant disease resistance genes, such as Pm60 from T. urartu, the Yr15 (WTK1), Yr36, and Pm41 from the T dicoccoides, the Lr21 and the powdery mildew resistance gene WTK4 derived from A. tauschii. In addition, distantly related wild species were also good resources to transfer resistance genes to wheat, such as the Pm21 from Haynaldia villosa, conferring durable and broad-spectrum resistance to wheat powdery mildew (Xing et al. 2018). The Fhb7 from Thinopyrum elongatum has good resistance for fusarium head blight spreading in wheat (Wang et al. 2020).
6.10 Prospects
The value of germplasm resources is in the genes hidden within them. The value of a gene is determined by its activity per se as well as the genetic background in which it is recovered. Only by transferring them from un-adapted germplasm into a good genetic background, assaying their value, and integrating them into breeding, we can truly activate them and realize their value for human life.
Genome segment introgression is a major source of genetic variation in wheat. Genomic regions of introgression have provided the hot spots for structural variation that contains many dispensable genes such as tolerant genes to bio-stress and abio-stress. Wheat pangenomes will enable genome-wide high-resolution admixture mapping across species and help figure out causal genetic mutations underlying specific traits (Lei et al. 2021). Furthermore, the pangenome-based research of hallmark cultivars will break through the limitation of having a single reference genome, for revealing the important contributions of chromosomal structural variations (translocation, inversion, duplication/deletion, PAV) in the formation of variety traits. Therefore, a pangenome within and across Triticum species will be of interest for wheat genomics in the next 5–10 years for interpreting and utilizing variation at the genome level for breeding and evolution (Khan et al. 2020).
Crucial founder genotypes should be sequenced by the third-generation technology and carefully annotated. Using the founder genotype genome sequences as reference, a set of genetics relative cultivars can be sequenced by cheaper second-generation sequence technology to reveal the haplotype blocks contributed by the founder genotypes in their genomes. The tracking markers could then be developed for haplotype-based breeding. Using the newest breeding founder genotypes as the recurrent parents crosses with core collections selectively backcrossing the hybrid for 2–3 generations can then establish AB-NAM populations. It is envisaged that through intercrossing between good lines from different AB-NAMs would be efficient strategy to realize the integration of breeding-beneficial alleles with desired haplotype blocks for create super-lines for breeding (Tanksley and Nelson 1996; Hao et al. 2020; Varshney et al. 2021). In addition, tetraploid wheats (see Chap. 8) would be an important and good gene resource for the improvement of common wheat. In view of the fact that the chromosome segments from wild tetraploid wheat have suppression effect on recombination, it is recommended that in the construction process of AB-NAM population, priority should be given to founder genotypes containing more introgressions from wild emmer to increase the recombination ratio, to create more variation, and efficiently to exclude the genetic drag from the wild species.
References
Avni R, Nave M, Barad O et al (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357:93–97. https://doi.org/10.1126/science.aan0032
Balfourier F, Bouchet S, Robert S, De Oliveira R, Rimbert H, Kitt J, Choulet F, International Wheat Genome Sequencing C, BreedWheat C, Paux E (2019) Worldwide phylogeography and history of wheat genetic diversity. Sci Adv 5:eaav0536. https://doi.org/10.1126/sciadv.aav0536
Cheng H, Liu J, Wen J, Nie X, Xu L, Chen N, Li Z, Wang Q, Zheng Z, Li M, Cui L, Liu Z, Bian J, Wang Z, Xu S, Yang Q, Appels R, Han D, Song W, Sun Q, Jiang Y (2019) Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. Genome Biol 20:136. https://doi.org/10.1186/s13059-019-1744-x
Díaz A, Zikhali M, Turner AS, Isaac P, Laurie DA (2012) Copy number variation affecting the Photoperiod-B1 and Vernalization-A1 genes is associated with altered flowering time in wheat (Triticum aestivum). PLoS ONE 7:e33234. https://doi.org/10.1371/journal.pone.0033234
Dvorak J, Akhunov ED, Akhunov AR, Deal KR, Luo MC (2006) Molecular characterization of a diagnostic DNA marker for domesticated tetraploid wheat provides evidence for gene flow from wild tetraploid wheat to hexaploid wheat. Mol Biol Evol 23:1386–1396. https://doi.org/10.1093/molbev/msl004
Feldman M, Levy AA (2012) Genome evolution due to allopolyploidization in wheat. Genetics 192:763–774. https://doi.org/10.1534/genetics.112.146316
Hao C, Jiao C, Hou J, Li T, Liu H, Wang Y, Zheng J, Liu H, Bi Z, Xu F, Zhao J, Ma L, Wang Y, Majeed U, Liu X, Appels R, Maccaferri M, Tuberosa R, Lu H, Zhang X (2020) Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Mol Plant 13:1733–1751. https://doi.org/10.1016/j.molp.2020.09.001
He F, Pasam R, Shi F, Kant S, Keeble-Gagnere G, Kay P, Forrest K, Fritz A, Hucl P, Wiebe K, Knox R, Cuthbert R, Pozniak C, Akhunova A, Morrell PL, Davies JP, Webb SR, Spangenberg G, Hayes B, Daetwyler H, Tibbits J, Hayden M, Akhunov E (2019) Exome sequencing highlights the role of wild-relative introgression in shaping the adaptive landscape of the wheat genome. Nat Genet 51:896–904. https://doi.org/10.1038/s41588-019-0382-2
Huang X, Qian Q, Liu Z, Sun H, He S, Luo D, Xia G, Chu C, Li J, Fu X (2009) Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet 41:494–497. https://doi.org/10.1038/ng.352
IWGSC (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191. https://doi.org/10.1126/science.aar7191
Jordan KW, Wang S, Lun Y, Gardiner LJ, MacLachlan R, Hucl P, Wiebe K, Wong D, Forrest KL, Consortium I, Sharpe AG, Sidebottom CH, Hall N, Toomajian C, Close T, Dubcovsky J, Akhunova A, Talbert L, Bansal UK, Bariana HS, Hayden MJ, Pozniak C, Jeddeloh JA, Hall A, Akhunov E (2015) A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol 16:48. https://doi.org/10.1186/s13059-015-0606-4
Jordan KW, He F, de Soto MF, Akhunova A, Akhunov E (2020) Differential chromatin accessibility landscape reveals structural and functional features of the allopolyploid wheat chromosomes. Genome Biol 21:176. https://doi.org/10.1186/s13059-020-02093-1
Khan AW, Garg V, Roorkiwal M, Golicz AA, Edwards D, Varshney RK (2020) Super-pangenome by integrating the wild side of a species for accelerated crop improvement. Trends Plant Sci 25:148–158. https://doi.org/10.1016/j.tplants.2019.10.012
Kippes N, Debernardi JM, Vasquez-Gross HA, Akpinar BA, Budak H, Kato K, Chao S, Akhunov E, Dubcovsky J (2015) Identification of the VERNALIZATION 4 gene reveals the origin of spring growth habit in ancient wheats from South Asia. Proc Natl Acad Sci USA 112:e5401-5410. https://doi.org/10.1073/pnas.1514883112
Lei L, Goltsman E, Goodstein D, Wu GA, Rokhsar DS, Vogel JP (2021) Plant pan-genomics comes of age. Annu Rev Plant Biol 72:411–435. https://doi.org/10.1146/annurev-arplant-080720-105454
Li Y, Fan C, Xing Y, Jiang Y, Luo L, Sun L, Shao D, Xu C, Li X, Xiao J, He Y, Zhang Q (2011) Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet 43:1266–1269. https://doi.org/10.1038/ng.977
Li AL, Hao CY, Wang ZY, Geng SF, Jia ML, Wang F, Han X, Kong XC, Yin LJ, Tao S, Deng ZY, Liao RY, Sun GL, Wang K, Ye XG, Jiao CZ, Lu HF, Zhou Y, Fu XD, Zhang XY, Mao L (2022) Wheat breeding history reveals synergistic selection of pleiotropic genomic sites for plant architecture and grain yield. Mol Plant. https://doi.org/10.1016/j.molp.2022.01.004
Ling HQ, Ma B, Shi X, Liu H, Dong L, Sun H, Cao Y, Gao Q, Zheng S, Li Y, Yu Y, Du H, Qi M, Li Y, Lu H, Yu H, Cui Y, Wang N, Chen C, Wu H, Zhao Y, Zhang J, Li Y, Zhou W, Zhang B, Hu W, van Eijk MJT, Tang J, Witsenboer HMA, Zhao S, Li Z, Zhang A, Wang D, Liang C (2018) Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557:424–428. https://doi.org/10.1038/s41586-018-0108-0
Li HF, Liu H, Hao CY, Li T, Wang XL, Yang YX, Zheng J, Zhang XY (2023) The auxin response factor TaARF15-A1 negatively regulates senescence in common wheat (Tritcum aestivum L.). Plant Physiol, 191:1254–1271
Liu X, Lightfoot E, O’Connell TC, Wang H, Li S, Zhou L, Hu Y, Motuzaite-Matuzeviciute G, Jones MK (2014) From necessity to choice: dietary revolutions in west China in the second millennium BC. World Archaeol 46:661–680. https://doi.org/10.1080/00438243.2014.953706
Liu X, Lister DL, Zhao Z, Staff RA, Jones PJ, Zhou L, Pokharia AK, Petrie CA, Pathak A, Lu H, Matuzeviciute GM, Bates J, Pilgram TK, Jones MK (2016) The virtues of small grain size: potential pathways to a distinguishing feature of Asian wheats. Quat Int 426:107–119. https://doi.org/10.1016/j.quaint.2016.02.059
Liu H, Li T, Wang YM, Zheng J, Li HF, Hao CY, Zhang XY (2019a) TaZIM-A1 negatively regulates flowering time in common wheat (Triticum aestivum L.). J Integr Plant Biol 61(3):359/376. https://doi.org/10.1111/jipb.12720
Liu X, Jones PJ, Motuzaite Matuzeviciute G, Hunt HV, Lister DL, An T, Przelomska N, Kneale CJ, Zhao Z, Jones MK (2019b) From ecological opportunism to multi-cropping: mapping food globalisation in prehistory. Quat Sci Rev 206:21–28. https://doi.org/10.1016/j.quascirev.2018.12.017
Luo MC, Gu YQ, Puiu D, Wang H, Twardziok SO, Deal KR, Huo N, Zhu T, Wang L, Wang Y, McGuire PE, Liu S, Long H, Ramasamy RK, Rodriguez JC, Van SL, Yuan L, Wang Z, Xia Z, Xiao L, Anderson OD, Ouyang S, Liang Y, Zimin AV, Pertea G, Qi P, Bennetzen JL, Dai X, Dawson MW, Muller HG, Kugler K, Rivarola-Duarte L, Spannagl M, Mayer KFX, Lu FH, Bevan MW, Leroy P, Li P, You FM, Sun Q, Liu Z, Lyons E, Wicker T, Salzberg SL, Devos KM, Dvorak J (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551:498–502. https://doi.org/10.1038/nature24486
Ma L, Li T, Hao C, Wang Y, Chen X, Zhang X (2016) TaGS5-3A, a grain size gene selected during wheat improvement for larger kernel and yield. Plant Biotechnol J 14:1269–1280. https://doi.org/10.1111/pbi.12492
Maccaferri M, Harris NS, Twardziok SO, Pasam RK, Gundlach H, Spannagl M, Ormanbekova D, Lux T, Prade VM, Milner SG, Himmelbach A, Mascher M, Bagnaresi P, Faccioli P, Cozzi P, Lauria M, Lazzari B, Stella A, Manconi A, Gnocchi M, Moscatelli M, Avni R, Deek J, Biyiklioglu S, Frascaroli E, Corneti S, Salvi S, Sonnante G, Desiderio F, Mare C, Crosatti C, Mica E, Ozkan H, Kilian B, De Vita P, Marone D, Joukhadar R, Mazzucotelli E, Nigro D, Gadaleta A, Chao S, Faris JD, Melo ATO, Pumphrey M, Pecchioni N, Milanesi L, Wiebe K, Ens J, MacLachlan RP, Clarke JM, Sharpe AG, Koh CS, Liang KYH, Taylor GJ, Knox R, Budak H, Mastrangelo AM, Xu SS, Stein N, Hale I, Distelfeld A, Hayden MJ, Tuberosa R, Walkowiak S, Mayer KFX, Ceriotti A, Pozniak CJ, Cattivelli L (2019) Durum wheat genome highlights past domestication signatures and future improvement targets. Nat Genet 51:885–895. https://doi.org/10.1038/s41588-019-0381-3
Matsuoka Y (2011) Evolution of polyploid triticum wheats under cultivation: the role of domestication, natural hybridization and allopolyploid speciation in their diversification. Plant Cell Physiol 52:750–764. https://doi.org/10.1093/pcp/pcr018
Paux E, Sourdille P, Salse J, Saintenac C, Choulet F, Leroy P, Korol A, Michalak M, Kianian S, Spielmeyer W, Lagudah E, Somers D, Kilian A, Alaux M, Vautrin S, Berges H, Eversole K, Appels R, Safar J, Simkova H, Dolezel J, Bernard M, Feuillet C (2008) A physical map of the 1-gigabase bread wheat chromosome 3B. Science 322:101–104. https://doi.org/10.1126/science.1161847
Peng J, Ronin Y, Fahima T, Roder MS, Li Y, Nevo E, Korol A (2003) Domestication quantitative trait loci in Triticum dicoccoides, the progenitor of wheat. Proc Natl Acad Sci USA 100:2489–2494. https://doi.org/10.1073/pnas.252763199
Peng JH, Sun D, Nevo E (2011) Domestication evolution, genetics and genomics in wheat. Mol Breed 28:281–301. https://doi.org/10.1007/s11032-011-9608-4
Pont C, Leroy T, Seidel M, Tondelli A, Duchemin W, Armisen D, Lang D, Bustos-Korts D, Goue N, Balfourier F, Molnar-Lang M, Lage J, Kilian B, Ozkan H, Waite D, Dyer S, Letellier T, Alaux M, Russell J, Keller B, van Eeuwijk F, Spannagl M, Mayer KFX, Waugh R, Stein N, Cattivelli L, Haberer G, Charmet G, Salse J (2019) Tracing the ancestry of modern bread wheats. Nat Genet 51:905–911. https://doi.org/10.1038/s41588-019-0393-z
Sansaloni C, Franco J, Santos B, Percival-Alwyn L, Singh S, Petroli C, Campos J, Dreher K, Payne T, Marshall D, Kilian B, Milne I, Raubach S, Shaw P, Stephen G, Carling J, Pierre CS, Burgueno J, Crosa J, Li H, Guzman C, Kehel Z, Amri A, Kilian A, Wenzl P, Uauy C, Banziger M, Caccamo M, Pixley K (2020) Diversity analysis of 80,000 wheat accessions reveals consequences and opportunities of selection footprints. Nat Commun 11:4572. https://doi.org/10.1038/s41467-020-18404-w
Taagen E, Tanaka J, Gul A, Sorrells ME (2021) Positional-based cloning ‘fail-safe’ approach is overpowered by wheat chromosome structure variation. Plant Genome 14:e20106
Tanksley SD, Nelson JC (1996) Advanced backcross QTL analysis: a method for the simultaneous discovery and transfer of valuable QTLs from un-adapted germplasm into elite breeding lines. Theor Appl Genet 92:191–203. https://doi.org/10.1007/Bf00223376
Varshney RK, Bohra A, Yu JM, Graner A, Zhang QF, Sorrells ME (2021) Design future crops: genomics-assisted breeding comes of age. Trends Plant Sci 26:631–649. https://doi.org/10.1016/j.tplants.2021.03.010
Wang H, Sun S, Ge W, Zhao L, Hou B, Wang K, Lyu Z, Chen L, Xu S, Guo J, Li M, Su P, Li X, Wang G, Bo C, Fang X, Zhuang W, Cheng X, Wu J, Dong L, Chen W, Li W, Xiao G, Zhao J, Hao Y, Xu Y, Gao Y, Liu W, Liu Y, Yin H, Li J, Li X, Zhao Y, Wang X, Ni F, Ma X, Li A, Xu SS, Bai G, Nevo E, Gao C, Ohm H, Kong L (2020) Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 368:eaba5435. https://doi.org/10.1126/science.aba5435
Wang Z, Hao C, Zhao J, Li C, Jiao C, Xi W, Hou J, Li T, Liu H, Zhang X (2021) Genomic footprints of wheat evolution in China reflected by a Wheat660K SNP array. Crop J 9:29–41. https://doi.org/10.1016/j.cj.2020.08.006
Würschum T, Langer SM, Longin CFH, Tucker MR, Leiser WL (2017) A modern Green Revolution gene for reduced height in wheat. Plant J 92:892–903. https://doi.org/10.1111/tpj.13726
Xu X, Wu K, Xu R, Wang S, Gao Z, Zhong Y, Li X, Liao H, Fu X (2019) Pyramiding of the dep1-1 and NAL1NJ6 alleles achieves sustainable improvements in nitrogen-use efficiency and grain yield in japonica rice breeding. J Genet Genomics 46:325–328. https://doi.org/10.1016/j.jgg.2019.02.009
Xie L, Zhang Y, Wang K, Luo X, Xu D, Tian X, Li L, Ye X, Xia X, Li W, Yan L, Cao S (2021) TaVrt2, an SVP-like gene, cooperates with TaVrn1 to regulate vernalization-induced flowering in wheat. New Phytol 231:834–848. https://doi.org/10.1111/nph.16339
Xing L, Hu P, Liu J, Witek K, Zhou S, Xu J, Zhou W, Gao L, Huang Z, Zhang R, Wang X, Chen P, Wang H, Jones JDG, Karafiatova M, Vrana J, Bartos J, Dolezel J, Tian Y, Wu Y, Cao A (2018) Pm21 from Haynaldia villosa encodes a CC-NBS-LRR protein conferring powdery mildew resistance in wheat. Mol Plant 11:874–878. https://doi.org/10.1016/j.molp.2018.02.013
Yan L, Loukoianov A, Tranquilli G, Helguera M, Fahima T, Dubcovsky J (2003) Positional cloning of the wheat vernalization gene VRN1. Proc Natl Acad Sci USA 100:6263–6268. https://doi.org/10.1073/pnas.0937399100
Yan L, Loukoianov A, Blechl A, Tranquilli G, Ramakrishna W, SanMiguel P, Bennetzen JL, Echenique V, Dubcovsky J (2004) The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303:1640–1644. https://doi.org/10.1126/science.1094305
Yan L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yasuda S, Dubcovsky J (2006) The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc Natl Acad Sci USA 103:19581–19586. https://doi.org/10.1073/pnas.0607142103
Zhao G, Zou C, Li K, Wang K, Li T, Gao L, Zhang X, Wang H, Yang Z, Liu X et al (2017) The Aegilops tauschii genome reveals multiple impacts of transposons. Nat Plant 3:946–955. https://doi.org/10.1038/s41477-017-0067-8
Zhu T, Wang L, Rimbert H, Rodriguez JC, Deal KR, De Oliveira R, Choulet F, Keeble-Gagnère G, Tibbits J, Rogers J, Eversole K, Appels R, Gu YQ, Mascher M, Dvorak J, Ming-Cheng Luo M-C (2021) Optical maps refine the bread wheat Triticum aestivum cv Chinese spring genome assembly. Plant J 107:303–314. https://doi.org/10.1111/tpj.15289
Acknowledgements
We appreciate Dr. Xinyi Liu, Washington University, St Louis, for his valuable help on the archaeological and transmission and permission for use Fig. 6.1. We also appreciate Dr. Zhiyong Liu, Institute of Genetics and Development Biology, CAS for his help on the disease-resistant gene diversity. Thanks should be given to Drs Paux, E and Balfourier, F, GDEC-INREA for providing Figs. 6.2 and 6.9 and fruitful discussion under umbrella of CAAS-INREA joint LAB. We also appreciate Mr Jiao CZ, Drs. Hao CY, Li T in Zhang group, ICS-CAAS, for their professional work in preparing most of the genome diversity figures. Zhang X was supported by the Key Research and Development Program of China (2016YFD01003) and CAAS Innovation Program. Appels R was supported by the University of Melbourne as an Honorary Professor.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2024 The Author(s)
About this chapter

Cite this chapter
Zhang, X., Appels, R. (2024). Genome Sequence-Based Features of Wheat Genetic Diversity. In: Appels, R., Eversole, K., Feuillet, C., Gallagher, D. (eds) The Wheat Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-031-38294-9_6
Download citation
DOI: https://doi.org/10.1007/978-3-031-38294-9_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-38292-5
Online ISBN: 978-3-031-38294-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)