
Abstract
Nephronophthisis comprises a clinically and genetically heterogeneous group of autosomal recessive inherited tubulointerstitial diseases. It represents the most frequent genetic cause of end-stage kidney disease in children and young adults. Disease-causing mutations in 25 different genes have been identified to date. Almost all gene products localize either to primary cilia or the ciliary base which leads to the classification of nephronophthisis as a ciliopathy. Nephronophthisis is often accompanied by anomalies in other organs. Several well described complex clinical syndromes can feature the renal picture of nephronophthisis, including Senior-Løken syndrome, Joubert syndrome, COACH syndrome, Jeune syndrome, Meckel-Gruber syndrome and others.
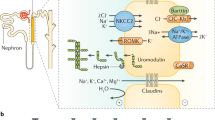
Autosomal dominant tubulointerstitial kidney disease (ADTKD) is a rare genetic disorder characterized by a slowly progressive tubulo-interstitial nephropathy leading to end-stage kidney disease in late adulthood. Although there is major clinical and histological overlap with nephronophthisis, autosomal dominant inheritance as well as progressive renal failure later in life are two main differences. So far five genes have been identified in which mutations lead to ADTKD: UMOD, REN, MUC1, HNF1B, and SEC61A1.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


References
Fanconi G, Hanhart E, von Albertini A, Uhlinger E, Dolivo G, Prader A. Die familiäre juvenile Nephronophthise. Helv Paediatr Acta. 1951;6:1–49.
Smith C, Graham J. Congenital medullary cysts of kidneys with severe refractory anemia. Am J Dis Child. 1945;69:369–77.
Titieni A, König J. Nephronophthise und assoziierte Ziliopathien. Medgen. 2018;30:461–8.
König J, Kranz B, König S, Schlingmann KP, Titieni A, Tönshoff B, Habbig S, Pape L, Häffner K, Hansen M, Büscher A, Bald M, Billing H, Schild R, Walden U, Hampel T, Staude H, Riedl M, Gretz N, Lablans M, Bergmann C, Hildebrandt F, Omran H, Konrad M. Phenotypic spectrum of children with nephronophthisis and related ciliopathies. Clin J Am Soc Nephrol. 2017;12(12):1974–83.
Srivastava S, Molinari E, Raman S, Sayer JA. Many genes-one disease? Genetics of nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr. 2017;5:287.
Waldherr R, Lennert T, Weber HP, Fodisch HJ, Scharer K. The nephronophthisis complex. A clinicopathologic study in children. Virchows Arch [Pathol Anat]. 1982;394:235–54.
Zollinger HU, Mihatsch MJ, Edefonti A, Gaboardi F, Imbasciati E, Lennert T. Nephronophthisis (medullary cystic disease of the kidney). A study using electron microscopy, immunofluorescence, and a review of the morphological findings. Helv Paediatr Acta. 1980;35:509–30.
Blowey DL, Querfeld U, Geary D, Warady BA, Alon U. Ultrasound findings in juvenile nephronophthisis. Pediatr Nephrol. 1996;10:22–4.
Luo F, Tao YH. Nephronophthisis: a review of genotype-phenotype correlation. Nephrology (Carlton). 2018;23(10):904–11.
Omran H. NPHP proteins: gatekeepers of the ciliary compartment. J Cell Biol. 2010;190(5):715–7.
Simms RJ, Eley L, Sayer JA. Nephronophthisis. Eur J Hum Genet. 2009;17(4):406–16.
Hildebrandt F, Strahm B, Nothwang HG, Gretz N, Schnieders B, Singh-Sawhney I, Kutt R, Vollmer M, Brandis M. Molecular genetic identification of families with juvenile nephronophthisis type 1: rate of progression to renal failure. APN Study Group. Arbeitsgemeinschaft fuer Paediatrische Nephrologie. Kidney Int. 1997b;51:261–9.
Konrad M, Saunier S, Heidet L, Silbermann F, Benessy F, Calado J, Le Paslier D, Broyer M, Gubler MC, Antignac C. Large homozygous deletions of the 2q13 region are a major cause of juvenile nephronophthisis. Hum Mol Genet. 1996;5:367–71.
Hildebrandt F, Rensing C, Betz R, Sommer U, Birnbaum S, Imm A, Omran H, Leipoldt M, Otto E. Arbeitsgemeinschaft fuer Paediatrische Nephrologie (APN) Study Group: establishing an algorithm for molecular genetic diagnostics in 127 families with juvenile nephronophthisis. Kidney Int. 2001;59(2):434–45.
Snoek R, van Setten J, Keating BJ, et al. NPHP1 (Nephrocystin-1) gene deletions cause adult-onset ESRD. J Am Soc Nephrol. 2018;29:1772–9.
Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr Nephrol. 1989;3:50–5.
Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, Foreman JW, Goodship JA, Strachan T, Kispert A, Wolf MT, Gagnadoux MF, Nivet H, Antignac C, Walz G, Drummond IA, Benzing T, Hildebrandt F. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–20.
Senior B, Friedmann AI, Braudo JL. Juvenile familial nephropathy with tapetoretinal degeneration: a new oculo-renal dystrophy. Am J Opthalmol. 1961;52:625–33.
Leber T. Über Retinitis pigmentosa und angeborene Amaurose. Arch Ophthalmol. 1869;15:1–25.
Leber T. Über anormale Formen der Retinitis pigmentosa. Arch Ophthalmol. 1871;17:314–41.
François J. Leber’s congenital tapeto-retinal degeneration. Int Ophthalmol Clin. 1968;8:929–47.
Franceschetti A, Dieterle P. L’importance diagnostique de l’éléctro-rétinogramme dans le dégénérescences tapéto-rétinennes avec rétrécissement du champ visuel et héméralopie. Conf Neurol. 1954;14:184–6.
Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan SL, Muerb U, O’Toole JF, Helou J, Attanasio M, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282–8.
Adamiok-Ostrowska A, Piekiełko-Witkowska A. Ciliary genes on renal cystic diseases. Cells. 2020;9(4):907.
Grochowsky A, Gunay-Aygun M. Clinical characteristics of individual organ system disease in non-motile ciliopathies. Transl Sci Rare Dis. 2019;4(1–2):1–23.
Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten JC, Dede DE, Fennell E. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14:368–76.
Summers AC, Snow J, Wiggs E, Liu AG, Toro C, Poretti A, et al. Neuropsychological phenotypes of 76 individuals with Joubert syndrome evaluated at a single center. Am J Med Genet A. 2017;173(7):1796–812.
Poretti A, Snow J, Summers AC, Tekes A, Huisman T, Aygun N, et al. Joubert syndrome: neuroimaging findings in 110 patients in correlation with cognitive function and genetic cause. J Med Genet. 2017;54(8):521–9.
Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DWW, McDonald R, Eddy A, Chance PF, Glass IA. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82–91.
Strongin A, Heller T, Doherty D, Glass IA, Parisi MA, Bryant J, et al. Characteristics of liver disease in 100 individuals with joubert syndrome prospectively evaluated at a single center. J Pediatr Gastroenterol Nutr. 2018;66(3):428–35.
Parisi MA. The molecular genetics of Joubert syndrome and related ciliopathies: the challenges of genetic and phenotypic heterogeneity. Transl Sci Rare Dis. 2019;4(1–2):25–49.
Fleming LR, Doherty DA, Parisi MA, Glass IA, Bryant J, Fischer R, et al. Prospective evaluation of kidney disease in joubert syndrome. Clin J Am Soc Nephrol. 2017;12(12):1962–73.
Bergmann C. Educational paper: ciliopathies. Eur J Pediatr. 2012;171(9):1285–300.
Parisi MA, Doherty D, Eckert ML, Shaw DW, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, Bakhsh E, Indridason OS, Dobyns WB, Bennett CL, Chance PF, Glass IA. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43:334–9.
Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen AJ, Gahl WA, Gentile M, Gorden NT, Hikida A, Knutzen D, Ozyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet. 2010;47(1):8–21.
Dahmer-Heath M, Schriever V, Kollmann S, Schleithoff C, Titieni A, Cetiner M, Patzer L, Tönshoff B, Hansen M, Pennekamp P, Gerß J, Konrad M, König J. Systematic evaluation of olfaction in patients with hereditary cystic kidney diseases/renal ciliopathies. J Med Genet. 2020.
Hartill V, Szymanska K, Sharif SM, Wheway G, Johnson CA. Meckel-gruber syndrome: an update on diagnosis, clinical management, and research advances. Front Pediatr. 2017;5:244.
Salonen R. The meckel syndrome: clinicopathological findings in 67 patients. Am J Med Genet. 1984;18(4):671–89.
Sergi C, Adam S, Kahl P, Otto HF. Study of the malformation of ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly. Pediatr Dev Pathol. 2000;3(6):568–83.
Alexiev BA, Lin X, Sun CC, Brenner DS. Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Arch Pathol Lab Med. 2006;130(8):1236–8.
Sattar S, Gleeson JG. The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev Med Child Neurol. 2011;53(9):793–8.
Cogan DG. Heredity of congenital ocular motor apraxia. Trans Am Acad Ophthal Otolaryng. 1972;76:60–3.
Schröder S, Li Y, Yigit G, Altmüller J, Bader I, Bevot A, Biskup S, Dreha-Kulaczewski S, Christoph Korenke G, Kottke R, Mayr JA, Preisel M, Toelle SP, Wente-Schulz S, Wortmann SB, Hahn H, Boltshauser E, Uhmann A, Wollnik B, Brockmann K. Heterozygous truncating variants in SUFU cause congenital ocular motor apraxia. Genet Med. 2020;23(2):341–51.
Betz R, Rensing C, Otto E, Mincheva A, Zehnder D, Lichter P, Hildebrandt F. Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J Pediatr. 2000;136(6):828–31.
Di Rocco M, Picco P, Arslanian A, et al. Retinitis pigmentosa, hypopituitarism, nephronophthisis, and mild skeletal dysplasia (RHYNS): a new syndrome? Am J Med Genet. 1997;73:1–4.
Hedera P, Gorski JL. Retinitis pigmentosa, growth hormone deficiency, and acromelic skeletal dysplasia in two brothers: possible familial RHYNS syndrome. Am J Med Genet. 2001;101:142–5.
Brancati F, Camerota L, Colao E, Vega-Warner V, Zhao X, Zhang R, Bottillo I, Castori M, Caglioti A, Sangiuolo F, Novelli G, Perrotti N, Otto EA, Undiagnosed Disease Network Italy. Biallelic variants in the ciliary gene TMEM67 cause RHYNS syndrome. Eur J Hum Genet. 2018;26(9):1266–71.
Stephen J, Vilboux T, Mian L, et al. Mutations in KIAA0753 cause Joubert syndrome associated with growth hormone deficiency. Hum Genet. 2017;136:399–08.
Vilboux T, Malicdan MC, Roney JC, et al. CELSR2, encoding a planar cell polarity protein, is a putative gene in Joubert syndrome with cortical heterotopia, microophthalmia, and growth hormone deficiency. Am J Med Genet A. 2017;173:661–6.
Braun DA, Hildebrandt F. Ciliopathies Cold Spring Harb Perspect Biol. 2017;9(3):a028191.
Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, Sanchez JAO, Nevarez L, Nickerson DA, Bamshad M, University of Washington Center for Mendelian Genomics, Lachman RS, Krakow D, Cohn DH. Expanding the genetic architecture and phenotypic spectrum in skeletal ciliopathies. Hum Mutat. 2018;39(1):152–66.
Arts H, Knoers N. Cranioectodermal dysplasia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, Amemiya A, editors. GeneReviews®. Seattle, WA, University of Washington, Seattle; 2013. p. 1993–2020.
Wallmeier J, Nielsen KG, Kuehni CE, Lucas JS, Leigh MW, Zariwala MA, Omran H. Motile ciliopathies. Nat Rev Dis Primers. 2020;6(1):77.
Lemaire M, Parekh RS. A perspective on inherited kidney disease: lessons for practicing nephrologists. Clin J Am Soc Nephrol. 2017;12(12):1914–6.
Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016;89:275–84.
Joly D, Beroud C, Grünfeld JP. Rare inherited disorders with renal involvement-approach to the patient. Kidney Int. 2015;87:901–8.
König J, Titieni A, Konrad M. Network for Early Onset Cystic Kidney Diseases (NEOCYST)—a comprehensive multidisciplinary approach to hereditary cystic kidney diseases in childhood. Front Pediatr. 2018;6:24.
Eckardt KU, et al. Autosomal dominant tubulo-interstitial kidney disease: diagnosis, classification, and management—KDIGO consensus report. Kidney Int. 2015;88:676–83.
Goldman SH, Walker SR, Merigan TCJ, Gardner KDJ, Bull JM. Hereditary occurrence of cystic disease of the renal medulla. N Engl J Med. 1966;274:984–92.
Strauss MB, Sommers SC. Medullary cystic disease and familial juvenile nephronophthisis. N Engl J Med. 1967;277:863–4.
Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, Bleyer AJ. Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers. 2019;5(1):60.
Wolf MT, Mucha BE, Attanasio M, Zalewski I, Karle SM, Neumann HP, Rahman N, Bader B, Baldamus CA, Otto E, Witzgall R, Fuchshuber A, Hildebrandt F. Mutations of the Uromodulin gene in MCKD type 2 patients cluster in exon 4, which encodes three EGF-like domains. Kidney Int. 2003;64:1580–7.
Bates JM, et al. Tamm-Horsfall protein knockout mice are more prone to urinary tract infection rapid communication. Kidney Int. 2004;65:791–7.
Gudbjartsson DF, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet. 2010;6:e1001039.
Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, Raffi H, Rampoldi L, Uchida S, Hille S, Dosche C, Kumar S, Castaneda-Bueno M, Gamba G, Bachmann S. Na+K+2Cl-Cotransporter (NKCC2) is regulated by Tamm-Horsfall-Protein (THP). American Society of Nephrology Annual Meeting, 2008; abstract 134.
Renigunta A, Renigunta V, Saritas T, Decher N, Mutig K, Waldegger S. Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function. J Biol Chem. 2011;286:2224–35.
Ayasreh N, et al. Autosomal dominant tubulointerstitial kidney disease: clinical presentation of patients with ADTKD-UMOD and ADTKD-MUC1. Am J Kidney Dis. 2018;72:411–8.
Kirby A, Gnirke A, Jaffe DB, Barešová V, Pochet N, Blumenstiel B, Aird D, Stevens C, Robinson JT, Cabili MN, Gat-Viks I, Kelliher E, Daza R, DeFelice M, Hůlková H, Sovová J, Vylet’al P, Antignac C, Guttman M, Handsaker RE, Perrin D, Steelman S, Sigurdsson S, Scheinman SJ, Sougnez C, Cibulskis K, Parkin M, Green T, Rossin E, Zody MC, Xavier RJ, Pollak MR, Alper SL, Lindblad-Toh K, Gabriel S, Hart PS, Regev A, Nusbaum C, Kmoch S, Bleyer AJ, Lander ES, Daly MJ. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. 2013;45:299–303.
Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B associated renal and extrarenal disease—an expanding clinical spectrum. Nat Rev Nephrol. 2015;11:102–12.
Vlahakos DV, et al. Renin-angiotensin system stimulates erythropoietin secretion in chronic hemodialysis patients. Clin Nephrol. 1995;43:53–9.
Zivná M, Hůlková H, Matignon M, Hodanová K, Vylet’al P, Kalbácová M, Baresová V, Sikora J, Blazková H, Zivný J, Ivánek R, Stránecký V, Sovová J, Claes K, Lerut E, Fryns JP, Hart PS, Hart TC, Adams JN, Pawtowski A, Clemessy M, Gasc JM, Gubler MC, Antignac C, Elleder M, Kapp K, Grimbert P, Bleyer AJ, Kmoch S. Dominant renin gene mutations associated with early onset hyperuricemia, anemia and chronic kidney failure. Am J Hum Genet. 2009;85:204–13.
Gribouval O, et al. Mutations in genes in the renin angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet. 2005;37:964–8.
Bolar NA, et al. Heterozygous loss-of-function SEC61A1 mutations cause autosomaldominant tubulo-interstitial and glomerulocystic kidney disease with anemia. Am J Hum Genet. 2016;99:174–87.
Schubert D, et al. Plasma cell deficiency in human subjects with heterozygous mutations in Sec61 translocon alpha 1 subunit (SEC61A1). J Allergy Clin Immunol. 2018;141:1427–38.
Lang S, et al. An update on Sec 61 channel functions, mechanisms, and related diseases. Front Physiol. 2017;8:887.
Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, Shirts B, Xu L, Zhu H, Barmada MM, Bleyer AJ. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39(12):882–92.
Scolari F, Caridi G, Rampoldi L, Tardanico R, Izzi C, Pirulli D, Amoroso A, Casari G, Ghiggeri GM. Uromodulin storage disease: clinical aspects and mechanisms. Am J Kidney Dis. 2004;44:987–99.
Bollée G, Dahan K, Flamant M, Morinière V, Pawtowski A, Heidet L, Lacombe D, Devuyst O, Pirson Y, Antignac C, Knebelmann B. Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin J Am Soc Nephrol. 2011;6:2429–38.
Scolari F, Puzzer D, Amoroso A, Caridi G, Ghiggeri GM, Maiorca R, Aridon P, De Fusco M, Ballabio A, Casari G. Identification of a new locus for medullary cystic disease, on chromosome 16p12. Am J Hum Genet. 1999;64:1655–60.
Rezende-Lima W, Parreira KS, Garcia-Gonzalez M, Riveira E, Banet JF, Lens XM. Homozygosity for uromodulin disorders: FJHN and MCKD-type 2. Kidney Int. 2004;66:558–63.
Stavrou C, Koptides M, Tombazos C, Psara E, Patsias C, Zouvani I, Kyriacou K, Hildebrandt F, Christofides T, Pierides A, Deltas CC. Autosomal-dominant medullary cystic kidney disease type 1: clinical and molecular findings in six large Cypriot families. Kidney Int. 2002;62:1385–94.
Wolf MT, Mucha BE, Hennies HC, Attanasio M, Panther F, Zalewski I, Karle SM, Otto EA, Deltas CC, Fuchshuber A, Hildebrandt F. Medullary cystic kidney disease type 1: mutational analysis in 37 genes based on haplotype sharing. Hum Genet. 2006;119:649–5.
Ulinski T, et al. Renal phenotypes related to hepatocyte nuclear factor1β (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17:497–503.
Decramer S, et al. Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol. 2007;18:923–33.
Gondra L, et al. Hyperechogenic kidneys and polyhydramnios associated with HNF1B gene mutation. Pediatr Nephrol. 2016;31:1705–8.
Shuster S, et al. Prenatal detection of isolated bilateral hyperechogenic kidneys: etiologies and outcomes. Prenat Diagn. 2019;39:693–700.
Mitchel MW, et al. 17q12 recurrent deletion syndrome. GeneReviews. https://www.ncbi.nlm.nih.gov/books/NBK401562/. Accessed 8 December 2016.
Verhave JC, Bech AP, Wetzels JF, Nijenhuis T. Hepatocyte nuclear factor 1ß-associated kidney disease: more than renal cyst and diabetes. J Am Soc Nephrol. 2016;27(2):345–53.
Faguer S, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011;80:768–76.
Oram RA, et al. Mutations in the hepatocyte nuclear factor1β (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am J Obstet Gynecol. 2010;203:364.e1–5.
MorenoDeLuca D, et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet. 2010;87:618–30.
Clissold RL, et al. Chromosome 17q12 microdeletions but not intragenic HNF1B mutations link developmental kidney disease and psychiatric disorder. Kidney Int. 2016;90:203–11.
Gimpel C, Avni FE, Bergmann C, Cetiner M, Habbig S, Haffner D, König J, Konrad M, Liebau MC, Pape L, Rellensmann G, Titieni A, von Kaisenberg C, Weber S, Winyard PJD, Schaefer F. Perinatal diagnosis, management and follow-up of cystic renal diseases: a clinical practice recommendation with systematic literature reviews. JAMA Pediatr. 2018;172(1):74–86.
Okorn C, Goertz A, Vester U, Beck BB, Bergmann C, Habbig S, König J, Konrad M, Müller D, Oh J, Ortiz-Brüchle N, Patzer L, Schild R, Seeman T, Staude H, Thumfart J, Tönshoff B, Walden U, Weber L, Zaniew M, Zappel H, Hoyer PF, Weber S. HNF1B nephropathy has a slow-progressive phenotype in childhood—with the exception of very early onset cases. Results of the German Multicenter HNF1B Childhood Registry. Pediatr Nephrol. 2019;34(6):1065–75.
Bleyer AJ, Hart TC, Shihabi ZAK, Robins V, Hoyer JR. Mutations in the uromodulin gene decrease urinary excretion of Tamm-Horsfall protein. Kidney Int. 2004;66:974–7.
Wenzel A, et al. Single molecule real time sequencing in ADTKD-MUC1 allows complete assembly of the VNTR and exact positioning of causative mutations. Sci Rep. 2018;8:4170.
Ross LF, Saal HM, David KL, Anderson RR. Technical report: ethical and policy issues in genetic testing and screening of children. Genet Med. 2013;15:234–45.
Liu X, et al. Effects of uric acid-lowering therapy on the progression of chronic kidney disease: a systematic review and meta-analysis. Ren Fail. 2018;40:289–97.
Hamada T, et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am J Hypertens. 2008;21:1157–62.
Acknowledgements
We would like to graciously acknowledge Andrea Titieni, Joachim Gerß and Carsten Bergmann for their contribution of figures and clinical pictures.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
König, J., Omran, H. (2023). Nephronophthisis and Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD). In: Schaefer, F., Greenbaum, L.A. (eds) Pediatric Kidney Disease. Springer, Cham. https://doi.org/10.1007/978-3-031-11665-0_11
Download citation
DOI: https://doi.org/10.1007/978-3-031-11665-0_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-11664-3
Online ISBN: 978-3-031-11665-0
eBook Packages: MedicineMedicine (R0)