
Abstract
In this chapter we present a MATLAB-based computational pipeline for the quantification of monolayer migration assays. Wound healing assay (or scratch assay) is a commonly used in vitro assay to assess collective cell migration. Our pipeline outputs traditional and spatiotemporal readouts that quantify the group migration properties and was previously used for a screen that included thousands of time-lapse sequences. You will learn how to execute the pipeline, the principles behind the design and implementation choices we made, pitfalls, tips, and tricks in using it.
This Chapter has been reviewed by Simon F. Nrrelykke, ETH Zurich, Switzerland.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


In this chapter we present a MATLAB-based computational pipeline for the quantification of monolayer migration assays. Wound healing assay (or scratch assay) is a commonly used in vitro assay to assess collective cell migration. Our pipeline outputs traditional and spatiotemporal readouts that quantify the group migration properties and was previously used for a screen that included thousands of time-lapse sequences. You will learn how to execute the pipeline, the principles behind the design and implementation choices we made, pitfalls, tips, and tricks in using it.
8.1 Introduction
In vitro monolayer migration assays are a simple model for studying collective cell migration, a fundamental cellular function with vast implications in health and disease. Quantification of monolayer migration is required for the investigation of the molecular and cellular mechanisms that govern collective cell migration, and is the bottleneck in many projects. Data analysis and automated quantification become absolutely essential especially due to recent advances in automated imaging-based data acquisition through high content imaging platforms, making manual annotation impractical. Wound healing (or “scratch”) assay is the most common assay to quantify collective cell migration in vitro (Liang et al., 2007), and is performed by monitoring the “healing” of a scratch in a growing confluent monolayer of cells by still or time-lapse microscopy (Jonkman et al., 2014). The basic initial step of almost all monolayer migration analyses pipelines is the segmentation of each image to cellular and non-cellular regions. This segmentation can be then used to quantify the area covered by the monolayer in two snapshots (endpoint readout) or to calculate the rate of healing through time using live imaging (temporal readout). This type of analysis does not require fluorescent labeling and is usually performed using label-free imaging modalities such as phase contrast or differential interference contrast (DIC) microscopy. Accordingly, several open computational tools were designed to segment cellular and non-cellular image regions in label-free images with the purpose to quantify the wound healing progression (Geback et al., 2009; Zaritsky et al., 2017b; Masuzzo et al., 2016; Deforet et al., 2012; Milde et al., 2012), including several FIJI plugins (Caldas et al., 2015; Suarez-Arnedo et al., 2020) and a CellProfiler pipeline (Carpenter et al., 2006). Tracking the overall growth of confluent cell monolayers is not always sufficient to discriminate different modes of monolayer migration and to fully understand the collective dynamics. Indeed, live imaging can provide important and useful information beyond the healing rate: persistent migration (Ng et al., 2012), orientation (Milde et al., 2012; Ng et al., 2012), directional migration (Deforet et al., 2012; Milde et al., 2012; Ng et al., 2012), strain rate (Lee et al., 2013), monolayer front dynamics (Zaritsky et al., 2015b) and other measures for local or global coordinated migration (Deforet et al., 2012; Milde et al., 2012; Ng et al., 2012; Slater et al., 2013; Zaritsky et al., 2014; Zhou et al., 2019). These measures can be used to characterize and discriminate between effects of different treatments or experimental conditions (e.g. Simpson et al., 2008; Vitorino and Meyer, 2008). Importantly, valuable discriminative information can be extracted from cells within the bulk during monolayer migration (Deforet et al., 2012; Zaritsky et al., 2017b; Zhou et al., 2019), but these spatiotemporal measures are inherently less intuitive and are harder to process, analyze and interpret. In wound healing experiments with simple geometrical patterns of the monolayer front, spatiotemporal averaging of many cells based on their location relative to the monolayer’s front can generate qualitative visualization and quantitative measures that can help in interpreting observed phenotypes (Zaritsky et al., 2012). In this chapter we present a computational pipeline for automated visualization and quantification that was previously robustly applied to thousands of monolayer migration experiments (Zaritsky et al., 2017b). The pipeline was implemented in MATLAB and provides the traditional “wound healing” measures as well as more advanced spatiotemporal representations that can be used for visualization as well as for quantitative analysis. The chapter contains detailed information including practical usage instructions, parameter tuning, algorithms, troubleshooting and output interpretation.
8.2 Dataset
Accessing sample data: sample data from Zaritsky et al. (2013) is available in a Zenodo dataset (Zabary and Zaritsky, 2020). The dataset is in a compressed (ZIP) file within the following folders:
-
TimeLapseSamples—six .tif image stacks of representative time-lapse experiments for single expanding monolayers and two monolayers expanding toward each other in different cell systems and experimental conditions (Fig. 8.1). The specific cell system and imaging parameters are as follows:
-
The SingleExpandingMonolayer folder:
-
EXP_16HBE14o_1E_SAMPLE.tif—16HBE14o cells, imaged with pixel size of 1.267 \({\mu }m\) and time resolution of 5 min per frame.
-
EXP_DA3_PHA_1E_SAMPLE.tif—DA3 cells treated with PHA, imaged with pixel size of 1.24 \({\mu }m\) and time resolution of 14.5 min per frame.
-
EXP_MDCK_HGFSF_1E_SAMPLE.tif—MDCK cells treated with HGF/SF, imaged with pixel size of 0.879 \({\mu }m\) and time resolution of 15.7 min per frame.
-
-
The TwoExpandingFronts folder:
-
DA3_PHA_2E_SAMPLE.tif—DA3 cells, imaged with pixel size of 1.24 \({\mu }m\) and time resolution of 14.5 min per frame.
-
MDCK_HGFSF_2E_SAMPLE.tif—MDCK cells treated with HGF/SF, imaged with pixel size of 0.879 \({\mu }m\) and time resolution of 15.7 min per frame.
-
MDCK_ctrl_2E_SAMPLE.tif—MDCK cells, imaged with pixel size of 0.879 \({\mu }m\) and time resolution of 15.7 min per frame.
-
-
-
MultipleExperimentKymographs—kymographs from multiple time-lapse experiments for post-processing analysis.

Experimental setup. Our pipeline supports experiments of single expanding monolayers (left) or two monolayers expanding toward each other (right). Left: a single expanding monolayer of Human bronchial epithelial cell monolayer (16HBE14o line) live imaged with phase contrast micrsocopy. Right: the traditional wound healing (or scratch) assay, two monolayers of Madin-Darby Canine Kidney (MDCK) cells live imaged with differential interference contrast (DIC) microscopy. For spatiotemrpoal quantification we recommend the single expanding monolayer setting (see text). Images from Zaritsky et al. (2015a)

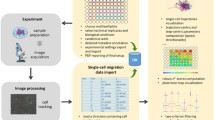
Pipeline overview: The entire data and analysis flow, discussed in this chapter. See text for full details
8.2.1 Experimental Considerations
The pipeline supports two experimental settings: (1) A single expanding monolayer and (2) the standard “wound healing” or “scratch” assay that includes two monolayers advancing toward one another. Examples are shown in Fig. 8.1. We recommend performing experiments with a single expanding monolayer for two reasons. First, the segmentation algorithm implemented in this pipeline performs less accurately once the two advancing monolayers are very close. Second, our spatiotemporal analysis is based on having sufficient bulk of cells behind the monolayer advancing front (for more spatial information) together with sufficient free space for the monolayer to expand (for more temporal information), both are easier to meet with single expanding monolayer experiments. This is especially true given that most studies do not use the information following the monolayers collision. For simplicity, we will use the term “wound healing” for both settings. Note that the pipeline supports monolayers expanding in the horizontal axis (x-axis). Thus, a \(90^{\circ }\) rotation preprocessing step (e.g. with FIJI) is required before analyzing vertically expanding monolayers.
8.3 Tools
The custom analysis pipeline was implemented in MATLAB. You must have a MATLAB license in order to use the pipeline. Note that many academic institutions have campus-wide MATLAB licenses, contact the IT in your institution for details. The pipeline was tested with MATLAB version 2019b on Mac/Windows/Linux operating systems, it will not function properly on earlier MATLAB versions. The pipeline produces multiple types of outputs, ranging from the standard “healing rate” to advanced spatiotemporal visualizations and quantifications (see Fig 8.2). Note, that the raw experimental data consists of live label-free imaging, and thus the pipeline is based on Particle Image Velocimetry (PIV) (Santiago et al., 1998), rather than single cell tracking. In the coming sections of this chapter we made efforts to provide detailed explanations for users inexperienced in MATLAB programming, as well as a modular and documented implementation to enable flexibility and customization for experienced users.
8.3.1 Setting up the MATLAB Environment and Executing the Analysis Pipeline
All the analysis pipeline code will be placed in the working directory.
All the raw data and the outputs will be placed in the data directory.
-
1.
Accessing the code: the complete code is available in a GitHub repository https://github.com/assafZaritskyLab/SpatiotemproalQuantificationMonolayerCellMigrationPipeline.git The repository includes MATLAB source code and a sample dataset.
-
2.
To analyse a single time-lapse, use the script quantifyMonolayerMigrationMain.m; for convenience we shall use the abbreviation main.m. The script requests as input a label-free image stack (the supported formats are .tiff/.zvi/.lsm), and a minimal set of parameters (the physical pixel size in \({\mu }m\), the time resolution in minutes, one/two expanding monolayers, the cell line’s approximate maximal speed and two more parameters for the time interval of the analysis). The script executes the analysis with the default parameters, and takes as default the 1st channel of a multi-channel data.
-
3.
To analyse a set of experiments use the script quantifyMonolayerMigrationBulkMain.m. For convenience we will use the abbreviation mainBulk.m. The script is a batch-processing version of main.m for multiple image stacks. It requests as input a path to a directory containing multiple label-free image stacks (supporting the same formats) and the same set of mandatory parameters. The script executes the analysis for each label-free image stack using the default parameters, followed by “meta analysis”, extracting information from the complete dataset (for advanced users, detailed information can be found later).
8.4 Workflow
In the following sub-sections we will provide a detailed description of the analysis pipeline. After reading it you will be able to understand the input/output of each step, tune parameters and troubleshoot the execution on your data.
8.4.1 Pipeline Overview
The pipeline receives as input the raw image data in one of the following formats: tiff stack, zvi (Zeiss Vision Image) or lsm (Zeiss tiff based proprietary format). Each data file is a single time-lapse experiment. The overview of the pipeline is presented in Fig. 8.2.
The pipeline includes four conceptual steps, which are reflected in four central MATLAB functions, each depending on the previous one and thus must be executed sequentially (see lines 88–104 in mainBulk.m script). The first two steps are performed at the single time-lapse level (see lines 74–79 main.m script). The rest of the pipeline is for the analysis of multiple experiments, enabling the comparison between different experiments and conditions, and might be challenging for novice users (see mainBulk.m script).
-
Part 1:
Segmenting each image to cellular (foreground) and background regions, and calculating the velocity fields. The output of this stage includes quantification of the wound healing over time, visualizations of the foreground/background segmentation, visualization of the velocity fields, and more detailed visualization of outputs for advanced debugging purposes (Fig. 8.2, 2nd row). The functions that produce each of these outputs are invoked by the function StepsScripts/calcSpatiotemporalRaw.m lines 25–30.
-
Part 2:
Calculating kymographs that capture the experiment’s spatiotemporal dynamics. The output of this stage includes visualization of the kymographs (Fig. 8.2, 3rd row) and is generated by the function StepsScripts/KymographsByMeasure.m.
-
Part 3:
Extracting spatiotemporal feature vectors from each kymograph (Fig. 8.2, 4th row) using the StepsScripts/kymographToFeaturesVec.m function on each single experiment.
-
Part 4:
Calculating the principal components of these features across experiments (Fig. 8.2, 5th row). This is performed with the StepsScripts/PCAOnAllExperimentsMeasurements.m function, which uses MATLAB’s built-in pca (2020) function.
Parameter Initialization
There are seven parameters that must be explicitly specified by the user: (1) the physical pixel size (in \({\mu }m\)), (2) the time resolution (in minutes), (3) the advancing monolayer estimated maximal speed (in \({\mu }m * {h^{-1}}\)), (4) the number of expanding monolayers (one or two), (5–6) the time interval: initial and final frame for the analysis, (7) the patch size (in \({\mu }m\), explained later). Other parameters can be set manually withing the code, for example, reuse enables or disables the use of past calculations (see line 3 in main.m). Default parameters are set in utils/initParamsDirs.m (invoked in main.m, line 73).
Table 8.1 summarizes the parameters that can be adjusted by the user via the GUI. The exact purpose of each parameter and the effect of altering parameters will be discussed later.
Output Directory Structure
The pipeline outputs will be automatically generated and placed in the data directory.
-
1.
The outputs of each time lapse sequence will be located in a directory named according to the raw data file name that contains the following sub-directories: (1) “images/” a separate raw image file for each frame in the time lapse sequence, (2) “VF/” debug outputs and results for the velocity fields analysis, and (3) “ROI/” debug outputs and segmentation results.
-
2.
The outputs that relate to a full time lapse experiment are located in designated directories that are generated in the data directory. For example, plots of the wound healing rate over time for all experiments will be located in a designated directory to allow straightforward comparison between different experiments. These directories are: (1) “segmentation/” videos with visualization of the segmentation results, (2) “monolayerMigrationMeasures/” plots and data relating to the wound healing readouts, (3) “kymographs/”, spatiotemporal quantification and visualization of the experiment, (4) “kymographFeatures/”, quantitative features extracted from the kymographs, and (5) “PCA_Results/”, dimensionality reduction results.
From here, each of the four steps of the analysis are explained in detail.
8.4.2 Part 1: Estimation of Velocity Fields, Semantic Segmentation, and Calculation of Wound Healing Measurements
This part starts with the raw image data, calculating the velocity fields followed by segmentation of the foreground cellular regions in each image and includes a correction for microscope re-positioning error. This step is implemented by the function StepsScripts/calcSpatiotemporalRaw.m. The output of this stage includes quantification of the wound healing rate over time, and visualizations of the foreground/background segmentation and velocity fields. This part also provides detailed visualization of the output in every frame for troubleshooting and debugging.
Estimating Velocity Fields
We start by estimating the velocity fields for each frame in the time-lapse sequence. Velocity fields were computed using custom cross-correlation-based particle image velocimetry (PIV), utilizing non-overlapping image patches. This is illustrated in Fig. 8.3a, and implemented in utils/whscripts/whLocalMotionEstimation.m function (below).
The frame-to-frame displacement of each patch was defined based on the maximal cross-correlation of a given patch with the subsequent image in the time-lapse image sequence.

The search radius was constrained by the searchRadiusInPixels parameter that was set based on the estimated maximal cell speed and the temporal resolution:


Part 1: velocity fields, segmentation and wound healing measurements. (a) Particle Image Velocimetry (PIV). Depiction of velocity estimation for a patch (blue square). The maximal correlation is calculated between the patch at time t and all potential translations in frame \(t+1\), and the corresponding velocity vector is recorded. (b) The segmentation problem is reduced to a narrow band that is defined based on the current contour (blue) and the maximal cell speed (green). (c) Wound healing plot, calculated using the segmentation masks
Processing time is dependent on the experiment data (size in pixels of each frame) and parameters (such as cell maximal speed and patch size). Processing of a single frame in the sample data with the experiment-specific default parameters may take up to 4 s on a standard laptop.
Segmenting the Cellular Foreground
For each frame in the time-lapse sequence, each patch is assigned as foreground or background, and this binary classification (segmentation) is used to calculate the contour of the migrating monolayer. The segmentation algorithm relies on two priors: (1) each frame contains one/two continuous “cellular foreground” segment/s and one continuous “background” segment, and (2) the contour advances monotonically over time toward the empty space. These assumptions allow us to compute the initial contour at time 0, as implemented in the custom function

which takes as input the image I, the patchSize, and lbpMapping (an internal structure calculated beforehand and required for the segmentation), and outputs the segmentation mask roiTexture.
Then, we use the segmentation at time t as a seed to expand the “cellular foreground” to time \(t+1\). The only patches to be resolved at time \(t+1\) are those labeled as “background” at time t and within a cell motion reach in respect to the monolayer contour (based on maxSpeed); this is illustrated in Fig. 8.3b. The function

computes ROI, a binary mask of the estimated cellular foreground at time \(t+1\). curRoi is calculated from thresholding the PIV cross-correlation scores followed by morphological operators.
Calculating the Wound Healing Over Time
The wound healing can be calculated as the expansion (in \({\mu }m\)) of the monolayer front over time:

ROI1 is the segmentation mask at the given time point, ROI0 is the segmentation mask at the previous time point. HealingUm(t) is the accumulated edge expansion at time t. This is illustrated in Fig. 8.3c.
The wound healing rate is calculated as the instantaneous or the average change in the wound healing over time (temporal derivative) in \({\mu }m*hour^{-1}\):

Part 1: Outputs
The execution of each part in the analysis pipeline is dependent on the successful execution of the previous steps. Thus, each part generates outputs (.mat format) to be used as input for the following step/s, as well as outputs for quantification, visualization and debugging (in multiple formats). Table 8.2 contains full description of the outputs of Part 1 of the pipeline.
Part 1: Parameter Sensitivity and Trade-Offs
The two parameters that have the most influence on the velocity fields calculation are the patch size and the cells’ maximal speed, as illustrated in Figs. 8.4 and 8.5. Patches that are too small do not contain sufficient image texture to statistically establish the optimal translation to the next time-frame, leading to spatial inconsistencies in the velocity fields. On the other hand, patches that are too large may include texture from multiple entities that move in different directions, leading to conflicting local motion patterns within the patch and impairing coherent motion estimation. An example is shown in Fig. 8.4b, with patch size equal to 30 \({\mu }m\). Other considerations include patch-size dependent (quadratic) velocity fields calculation time as patches decrease, and reduction in the resulting spatial resolution in a (quadratic) patch-size dependent manner. These inherent trade-offs are optimized by selecting a patch size smaller than the size of an average cell, and visually validating the coherency and resolution of the velocity fields outputs. A second validation that relies on the resulting kymographs will be discussed in the description of Part 2 of the pipeline. From our experience, a patch size of 15 \({\mu }m\) performs well for several cell lines and microscopy objectives.
The parameter related to the cell’s maximal speed is used to determine the search radius for the PIV calculations, where larger maximal speed requires a larger search radius. The actual value for this parameter is dependent on cell type and exact experimental setting, and should be determined based on the data. A too small search radius will lead to underestimation of the velocity fields magnitude (illustrated in Fig. 8.4b, for max speed equal to 10 \({\mu }m*hr^{-1}\)). A search radius beyond the one defined based on the true maximal cell speed will lead to more errors in the velocity field estimation, due to the quadratic increase in the number of possible translations. For example, detection of high motility patches in the background is presented in Fig. 8.4b, where the background speed for 200 \({\mu }m*hr^{-1}\) and for 90 \({\mu }m*hr^{-1}\) are to be compared, with patch size of 15 \({\mu }m\). In addition, the execution time is quadratic in the search radius size. Note that the pipeline is not very sensitive to the value of this parameter. Our recommendation is to assign this parameter using prior knowledge regarding the cell system and the experiment and validate visually using the resulting velocity fields (and/or kymographs, as we will discuss in Part 2).

Parameters trade-offs in PIV calculation. The maximal correlation (a) and the estimated speed (b), as a function of the parameters for patch size (x-axis) and maximal cell speed (y-axis). The middle frame (red boundaries) corresponds to the pipeline’s default parameters. The image frame used in this figure is shown at the lower-right corner

Patch size sensitivity and trade-offs. A cartoon illustrating how PIV is affected by the patch size. Subcellular (top row) versus supercellular (bottom row) patch size. The patch is marked in dark blue, and the search radius, which is calculated from the cell’s maximal speed and time resolution (black delimiter), is marked in orange. Columns represent (left-to-right) frame t, frame \(t+1\) and estimated velocities. The estimated displacement of the patch is marked in pink (second column). (Impaired displacement calculation for supercellular size patches containing texture from multiple cells is shown in the bottom-right, as red arrow)
The segmentation algorithm was optimized for robust high-content automated analysis. To achieve robust segmentation we relied on the assumption that the image includes one/two continuous monotonically expanding monolayers, thus dramatically reducing the number of patches to be resolved in each iteration (as shown in Fig. 8.3b, green). Active contours and graph cut algorithms in general produced accurate segmentation masks that were not limited by the patch size resolution (Zaritsky et al., 2011), however these approaches also led to large errors in segmentation of some image frames. We have observed that small segmentation inaccuracies have a minor effect on the wound healing measurements, as well as on the kymographs (Part 2), whereas large segmentation errors, even if only in one frame in a time lapse, may cause major artifacts in the calculated readotus (especially in the kymographs). Thus, we compromise for slightly reduced (overall) segmentation accuracy, to achieve enhanced robustness. This approach was found to be very effective in a large screen, allowing us to exclude less than 1% of the experiments due to major segmentation errors (Zaritsky et al., 2017a). Our recommendation is to perform a visual validation of the segmentation outcome. In case of identifying an erroneous frame, its corresponding segmentation mask (ROI) can be replaced with a previous or following frame without major effects (Table 8.3).
Part 1: Practical Usage of the Outputs
The outputs of Part 1 include the traditional wound healing readouts of the wound healing over time and the wound healing rate. These measurements can be compared across experiments and treatments. The visualizations can be used for setting and validating parameter values (as discussed above, in the the previous subsection). The visualization of the segmentation masks is important to verify that the segmentation follows the evolving contour, key for proper quantification of the wound healing readouts and spatiotemporal quantification (which we will discuss later in this Chapter)
Exercise 1
Several algorithms were proposed for monolayer segmentation. For example, Geback et al. (2009) used discrete curvelet transform, wheras Candès et al. (2006) and Zaritsky et al. (2011) used Support Vector Machine and Graph Cuts. These algorithms usually produce segmentation masks with higher accuracy, in comparison to the algorithm used here. Explain what was the reason for the algorithmic design choice in this pipeline.
8.4.3 Part 2: Kymographs
The spatiotemporal dynamics of a full time-lapse experiment can be quantified and visualized in kymographs. At each time point, the distance from the monolayer front was calculated using the segmentation masks, and the velocity fields were used to measure the cells migration properties at a given time and location with respect to the front. More specifically, in each frame, the cellular foreground is divided into bands of constant distances from the monolayer front, termed strips. Each bin in the kymographs records the cells’ mean speed or directionality in a specific strip at a particular time point. This is illustrated in Fig. 8.6. Speed is calculated as the magnitude of the corresponding velocity vector, while directionality is the absolute ratio between the mean velocity component perpendicular to the monolayer front and the velocity component parallel to the monolayer front:

DIST is an image where each pixel encodes its Euclidean distance from the monolayer front. inDist is a binary mask of all pixels within a specific strip in index d.

Kymograph construction. Speed (top right) and directionality (bottom right) kymographs provide a compact representation of the complete time-lapse sequence. Each bin (t,d) holds the average speed and directionality (accordingly) of all patches at time t and distance d from the monolayer front
Part 2: Outputs
The outputs of Part 2 of the pipleine are speed and directionality kymographs for visualization and further analysis; they will be passed on to Part 3. Table 8.4 contains their detailed description.
Part 2: Parameter Sensitivity and Trade-Offs
As the kymographs are calculated from the results of the previous analysis steps, potential errors in calculations in Part 1 will lead to inaccuracies in the kymographs. For example, if the search radius is set to a value smaller than the actual cell speed, the resulting vector field magnitudes and the kymograph values will be lower than the actual velocities, as illustrated in Fig. 8.7 (left). When the search radius is much higher than the maximal cell speed, the potential matching translations for each patch grow quadratically, leading to over-estimation of the velocities. Fig. 8.7 (right), illustrates this situation. Faulty segmentation can also lead to erroneous kymographs by altering the bands in relation to the monolayer front, as shown in Fig. 8.8.

Effect of the search radius on the kymographs. Top: Speed kymographs for different search radius values. The search radius is determined by the cell’s maximal speed parameter. Underestimation (left) and (minor) overestimation (right) of cell speed due to low and high maximal cell speed values, correspondingly. Bottom: Speed snapshots (at time \(=\) 250 min), corresponding to the magenta vertical band in the kymograph directly above

Effect of faulty segmentation on the kymograph. The top-left panel shows the speed kymographs altered by the defective segmentation. The colored (cyan, blue, green) kymographs’s columns represent the corresponding corrupted segmentation of individual frames (bottom). Importantly, the segmentation algorithm considers these faulty frames as the segmentation which includes a temporal continuity assumption (see Part 1). The top panel visualizes the deviation of the kymograph caused by the corrupted segmentations. The kymograph on the right is the subtraction of the kymograph (middle) from the defected kymograph (left)
Four parameters control the spatial and temporal ranges for which the kymograph is calculated for; this is illustrated in Fig. 8.9. The parameters include kymoMinDistMu and kymoMaxDistMu that define the spatial region in relation to the monolayer front, and kymoMinTimeFrameNum and kymoMaxTimeFrameNum that define the temporal range for kymograph calculation (Fig. 8.9). These parameters are used to calculate the internal parameter strips, an array of masks for each strip in the cellular foreground, allowing for fast retrieval of all velocity fields within each strip:

The purpose of setting these parameters is to enable focusing the spatiotemporal visualization and quantification to specific regions of interest in space and time. For example, if the research question relates exclusively to cells deep within the bulk then the spatial parameters can be set such that the range kymoMinDistMu to kymoMaxDistMu captures these cells of interest (Table 8.5).

Controlling the kymograph’s spatial and temporal range. Top left: A speed kymograph (using the pipeline’s default parameters, see Table 8.5). Top middle: The kymograph calculated with reduced temporal and spatial ranges (kymoMinDistMu = 60 \({\mu }m\), kymoMaxDistMu \(=\) 105 \({\mu }m\), kymoMinTimeMinutes \(=\) 40 min, kymoMinTimeMinutes \(=\) 130 min). Bottom: Snapshots from the time-lapse sequence, arrows point to the kymoMinTimeMinutes (cyan) and to the kymoMaxTimeMinutes (red). Top right: The spatial region defined by the kymoMinDistMu (orange) and the kymoMaxDistMu (purple) parameters
Part 2: Practical Usage of the Outputs
Kymographs can serve for visualization (e.g., Gan et al., 2016) and/or quantification, enabling comparison of the effect of different experimental conditions on spatiotemporal monolayer migration dynamics. For example, in Zaritsky et al. (2017b), we used the kymographs to visualize an overall motility impairment following inhibition of some proteins, and to reveal a rapid front-to-back motility synchronization, as a response to inhibition of a specific pathway, that could not be discovered without systematic spatiotemporal visualization and quantification. This is illustrated in Fig. 8.10. The kymographs can also be used as indication for a successful or defective analysis. For example, if most values in the directionality kymograph are close to 0, that might indicate that the monolayer advances vertically and should be rotated to advance horizontally (more examples follow in the section on parameters sensitivity and trade-offs). In Parts 3 and 4 we use the kymographs as high-dimensional quantitative readouts.

Kymograph visualization enables new insight. Top: Speed kymographs for a control experiment (left), RAC1 inhibited cells (middle) and for RHOA inhibited cells (right). RAC1 inhibition leads to reduced motility in space and time. The steeper arrow indicates a faster front-to-back motility propagation for the RHOA depleted cells. Bottom: A snapshot of the control (left) and RHOA inhibited experiment (right) (time \(=\) 60 min, border color code matches the corresponding kymograph’s column) with overlaid velocity fields. The velocities of the RHOA treated cells at the front and the back of the monolayer are more synchronized than the control cells. Adapted from Zaritsky et al. (2017b)
Exercise 2
The velocity fields estimation is sensitive to the values assigned to the parameters patchSizeUm and maxSpeed. In this and the following exercises you will explore how alteration in these parameters affects the resulting kymographs.
Download the image sequences EXP_16HBE14o_1E_SAMPLE.tif and
EXP_MDCK_HGFSF_1E_SAMPLE.tif (see Sect. 8.2). Calculate and visually compare their speed and directionality kymographs under the following parameter configurations:
-
1.
patchSizeUm = 15 \({\mu }m\), maxSpeed = 90 \({\mu }m*h^{-1}\), being the default values of the parameters;
-
2.
patchSizeUm = 5 \({\mu }m\), maxSpeed = 30 \({\mu }m*h^{-1}\).
Discuss the obtained results.
Exercise 3
Visualize the kymographs computed in Exercise 2, utilizing the function utils/plotKymograph.m. Describe and explain the effect that the different parameters have on the resulting kymographs.
Exercise 4
The traditional analysis of wound healing experiments includes measurement of the wound healing rate, the change in the monolayer’s front evolution over time. Think of, and describe scenarios where, upon a perturbation, the wound healing rate remains unchanged, but other collective migration properties change.
Exercise 5
Visualizing the subtraction of two kymographs can provide valuable insights regarding the corresponding differences in their spatiotemporal dynamics. Write a code snippet to compute the subtraction of the two speed kymographs that you obtained in Exercise 2, for the case when the default parameters are used. Visualize the results.
8.4.4 Part 3: Feature Extraction
This part of the analysis pipeline compresses the kymograph to a feature vector as a more compact representation of the monolayer’s spatiotemporal dynamics. This is achieved by averaging the bins of a kymograph in space and time and starts by dividing the kymograph into a grid of (timePartition x spatialPartition) tiles:

The kymograph partition is defined by the indices iSpace (spatial partition) and iTime (temporal partition).

Reducing the representation of the spatiotemporal dynamics to a multi-dimensional feature vector. To obtain a compact representation of a speed kymograph, we average it across space and time to timePartition x spatialPartition features. Each feature (right) encodes the average speed (color) in the corresponding kymograph’s bins (left). The same process is applied to directionality kymographs
After that, each feature is computed as the mean value of all the kymograph bins that reside in the corresponding tiles (as illustrated in Fig. 8.11):

This creates a feature vector as a compressed representation of the spatiotemporal information encoded in the kymograph. In an example shown in Fig. 8.11, we use timePartition = 4 and spatialPartition = 3, inducing a 12-dimensional feature vector, where features 1–4 encode the acceleration of cells at the monolayer front, features 5–8 encode the acceleration of cells 50–100 \({\mu }\)m behind the monolayer front, feature 1, 5, and 9 encode the spatial variations in speed at the onset of the experiment, and features 3, 7, and 11 encode the spatial variation at later times. This representation was first described in Zaritsky et al. (2012).
Part 3: Outputs (See Table 8.6)
Part 3: Parameter Sensitivity and Trade-Offs
The two parameters used in Part 3 are timePartition (default = 4) and spatialPartition (default = 3), the number of temporal and spatial bins correspondingly (as illustrated in Fig. 8.11). Larger values of these parameters lead to more features, smaller values to a more compressed representation. For example, for timePatition = 2, each feature will encode the temporal information for half of the experiment’s duration, the same applies for the spatial component (Table 8.7).
Part 3: Practical Usage of the Outputs
Having a compact quantitative representation for a time-lapse experiment is key for systematic spatiotemporal statistical characterization and quantification of high-content and screening projects, where barely staring at kymographs and describing them is not sufficient (or feasible). Note that Part 3 is an intermediate step, and is usually followed by supervised (classification) or unsupervised (clustering, dimensionality reduction—see Part 4) machine learning that take high-dimensional feature vectors as input.
8.4.5 Part 4: Principal Component Analysis: PCA
Principal Components Analysis (PCA) is a dimensionality-reduction method transforming high-dimensional data sets to a set of individual linearly uncorrelated (orthogonal) dimensions (called Principal Components, or PCs), while preserving most of the variability in the data (Pearson, 1901). Such dimensionality reduction is performed in Part 4 of the pipeline, which receives a set of kymograph-extracted features (Part 3) from multiple experiments, normalizes the features and transforms them to a new representation of PCs, ranked by the variability that they explain:


Here, normalizedSingleMeasureFeatures is the normalized vector of either the speed or directionality kymograph features, and pca (2020) is a MATLAB built-in function.
The obtained PCs can be used to visualize, quantify, and sometimes interpret spatiotemporal alterations between different experimental conditions (Zaritsky et al., 2012, 2017b).
Part 4: Outputs (See Table 8.8)
Part 4: Practical Usage of the Outputs
Each PC can be used as a quantitative readout of the monolayer migration’s spatiotemporal dynamics. By focusing on the few first PCs that capture most of the variability in the data, one can visualize, cluster and interpret to distinguish between different experimental conditions / treatments, as illustrated in Fig. 8.12. To demonstrate the processing of multiple experiments, we provide a small dataset of previously computed kymographs (in the MultipleExperimentKymographs/ directory) and the code snippets bellow.
The function getLabelsAndPaths retrieves the experiments’ labels and paths to speed (or directionality) kymographs:


Depiction of the analysis of multiple experiments. Time-lapse sequences (left column) are the raw data used to calculate the speed and directionality kymographs (second column from the left), which are then used to calculate the monolayer’s spatiotemporal dynamics features vectors (third column), that are further compressed with PCA for visualization or quantification (right-most column)
Note that getLabelsAndPaths is a custom function that is dependent on a specific file-organization scheme. It was implemented such that each sub-directory within pathToKymographFolder contains a collection of kymograph of either the speed or directionality measurements, named according to the experiment’s name (see Sect. 8.4.1). For a different directory arrangement or labeling scheme, the getLabelsAndPaths function should be re-implemented accordingly.
Next, we set the necessary parameters (see Part 3 and the Parameters initialization section for details):

To extract the high-dimensional feature representation of each experiment, we use:

To calculate the PCs, we use:

To calculate the proportion of variance in the data attributed to each PC (here for PC1), we use:

Select k PCs, plot them with the corresponding labels:

8.4.6 Tips and Troubleshooting for Advanced Users
The pipeline was designed to be executed “as is”, to analyze monolayer migration experiments. However, advanced users may want to customize components or tweak the pipeline. For these users we recommend to go through the documented code in quantifyMonolayerMigrationMain.m, and quantifyMonolayerMigrationBulkMain.m. We list common issues in Table 8.9, and errors in Table 8.10, that may arise while customizing the code and we give some suggestions how to handle them.
Take-Home Message
Our pipeline provides an analysis suite for monolayer migration experiments. It is designed for both users inexperienced in programming, as well as those more experienced users who wish to customize it further. The pipeline can be applied to extract traditional “wound healing” measures and/or more advanced spatiotemporal visualizations and qualifications. Its robustness was verified through experiments on data from multiple labs and cell systems (Zaritsky et al., 2015a; Gan et al., 2016; Zaritsky et al., 2017b).
Solutions to the Exercises
Exercise 1
The segmentation implemented for this pipeline is optimized for robustness with the goal to enable high-content automated analysis. This was achieved by relying on the assumption that the monolayer advances over time and never goes “backwards” (Part 1). This assumption significantly reduces the number of image patches that must be segmented as foreground or background at each time frame, since it enables to focus only on patches close to the previous segmentation. Importantly, our kymograph-based quantification is not sensitive to small deviations in the segmentation (Part 2), but is very sensitive to large segmentation errors, even if they occur only in a few frames. Thus, although other segmentation algorithms in the majority of the cases produce more accurate segmentation, we preferred robustness at the cost of reduced segmentation accuracy.
Exercise 2
Run the following code snippet on each of the configurations, for both files EXP_ 16HBE14o_1E_SAMPLE.tif and EXP_MDCK_HGFSF_1E_SAMPLE.tif. The function stepsScripts/KymograhpsByMeasure computes and visualizes the kymographs. The visualization is rendered and saved using the custom function utils/plotKymograph, which is called from
KymographsByMeasure.

Exercise 3
When the maxSpeed parameter is set to a value below the true cells’ maximal speed (e.g., 30 \({\mu }\)m*h\(^{-1}\)), the estimated magnitude of the vector fields will be bounded by the underestimated search radius, leading to reduced velocities (as illustrated in Fig. 8.7, left). On the other hand, when the search radius is overestimated (by setting maxSpeed to a value higher than the true maximal speed) the potential cross-correlation matches grow quadratically leading to overestimation of the velocities magnitude (Fig. 8.7, right). Increasing patchSize leads to a trade-off between having more information for the cross-correlation analysis at the cost of lower resolution in the segmentation and velocity granularity. See Part 1 for a thorough discussion.
Exercise 4
For example, the wound healing rate may remain unchanged when a perturbation induces both increased directionality and impaired motility, canceling each other in the wound healing rate measurement. Another example is a perturbation that slightly reduces cells’ speed, while enhancing front-to-back inter-cellular communication, together leading to unchanged wound healing rate. The latter phenotypes were reported for RHOA-inhibited cells in Zaritsky et al. (2017b) and were first identified by visualizing the spatiotemporal dynamics using kymographs (Fig. 8.10).
Exercise 5
Lines #4-#10 in the code below enable a simple visualization of the kymographs. An alternative is to use the function utils/plotKymograph to visualize the kymograph.

References
Caldas VE, Punter CM, Ghodke H, Robinson A, van Oijen AM (2015) iSBatch: a batch-processing platform for data analysis and exploration of live-cell single-molecule microscopy images and other hierarchical datasets. Mol Biosyst 11(10):2699–2708
Candès E, Demanet L, Donoho D, Ying L (2006) Fast discrete curvelet transforms. Multiscale Model Simul 5(3):861–899. https://doi.org/10.1137/05064182X
Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7(10):R100
Deforet M, Parrini MC, Petitjean L, Biondini M, Buguin A, Camonis J, Silberean P (2012) Automated velocity mapping of migrating cell populations (AVeMap). Nat Methods 9(11):1081–1083
Gan Z, Ding L, Burckhardt CJ, Lowery J, Zaritsky A, Sitterley K, Mota A, Costigliola N, Starker CG, Voytas DF, Tytell J, Goldman RD, Danuser G (2016) Vimentin intermediate filaments template microtubule networks to enhance persistence in cell polarity and directed migration. Cell Syst 3(3):252–263
Geback T, Schulz MM, Koumoutsakos P, Detmar M (2009) TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. BioTechniques 46(4):265–274
Jonkman JE, Cathcart JA, Xu F, Bartolini ME, Amon JE, Stevens KM, Colarusso P (2014) An introduction to the wound healing assay using live-cell microscopy. Cell Adh Migr 8(5):440–451
Lee RM, Kelley DH, Nordstrom KN, Ouellette NT, Losert W (2013) Quantifying stretching and rearrangement in epithelial sheet migration. New J Phys 15(2):025036
Liang CC, Park AY, Guan JL (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2(2):329–333
Masuzzo P, Van Troys M, Ampe C, Martens L (2016) Taking aim at moving targets in computational cell migration. Trends Cell Biol 26(2):88–110
Milde F, Franco D, Ferrari A, Kurtcuoglu V, Poulikakos D, Koumoutsakos P (2012) Cell Image Velocimetry (CIV): boosting the automated quantification of cell migration in wound healing assays. Integr Biol (Camb) 4(11):1437–1447
Ng MR, Besser A, Danuser G, Brugge JS (2012) Substrate stiffness regulates cadherin-dependent collective migration through myosin-II contractility. J Cell Biol 199(3):545–563
pca (2020) Xpca. https://www.mathworks.com/help/stats/pca.html
Pearson K (1901) LIII. On lines and planes of closest fit to systems of points in space. Lond Edinb Dublin Philos Mag J Sci 2(11):559–572. https://doi.org/10.1080/14786440109462720
Santiago JG, Wereley ST, Meinhart CD, Beebe D, Adrian RJ (1998) A particle image velocimetry system for microfluidics. Exp Fluids 25(4):316–319
Simpson KJ, Selfors LM, Bui J, Reynolds A, Leake D, Khvorova A, Brugge JS (2008) Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat Cell Biol 10(9):1027–1038
Slater B, Londono C, McGuigan AP (2013) An algorithm to quantify correlated collective cell migration behavior. BioTechniques 54(2):87–92
Suarez-Arnedo A, Figueroa FT, Clavijo C, Arbeláez P, Cruz JC, Muñoz-Camargo C (2020) An image j plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 15(7): e0232565. https://doi.org/10.1101/2020.04.20.050831
Vitorino P, Meyer T (2008) Modular control of endothelial sheet migration. Genes Dev 22(23):3268–3281
Zabary Y, Zaritsky A (2020) Quantifying monolayer cell migration sample dataset. https://doi.org/10.5281/zenodo.4308385
Zaritsky A, Natan S, Horev J, Hecht I, Wolf L, Ben-Jacob E, Tsarfaty I (2011) Cell motility dynamics: a novel segmentation algorithm to quantify multi-cellular bright field microscopy images. PLoS ONE 6(11):e27593
Zaritsky A, Natan S, Ben-Jacob E, Tsarfaty I (2012) Emergence of HGF/SF-induced coordinated cellular motility. PLoS ONE 7(9):e44671
Zaritsky A, Manor N, Wolf L, Ben-Jacob E, Tsarfaty I (2013) Benchmark for multi-cellular segmentation of bright field microscopy images. BMC Bioinf 14:319. https://doi.org/10.1186/1471-2105-14-319
Zaritsky A, Kaplan D, Hecht I, Natan S, Wolf L, Gov NS, Ben-Jacob E, Tsarfaty I (2014) Propagating waves of directionality and coordination orchestrate collective cell migration. PLoS Comput Biol 10(7):e1003747
Zaritsky A, Natan S, Kaplan D, Ben-Jacob E, Tsarfaty I (2015a) Live time-lapse dataset of in vitro wound healing experiments. Gigascience 4:8
Zaritsky A, Welf ES, Tseng YY, Angeles Rabadán M, Serra-Picamal X, Trepat X, Danuser G (2015b) Seeds of locally aligned motion and stress coordinate a collective cell migration. Biophys J 109(12):2492–2500
Zaritsky A, Obolski U, Gan Z, Reis CR, Kadlecova Z, Du Y, Schmid SL, Danuser G (2017a) Decoupling global biases and local interactions between cell biological variables. Elife 6:e22323
Zaritsky A, Tseng YY, Rabadán MA, Krishna S, Overholtzer M, Danuser G, Hall A (2017b) Diverse roles of guanine nucleotide exchange factors in regulating collective cell migration. J Cell Biol 216(6):1543–1556
Zhou FY, Ruiz-Puig C, Owen RP, White MJ, Rittscher J, Lu X (2019) Motion sensing superpixels (MOSES) is a systematic computational framework to quantify and discover cellular motion phenotypes. Elife 8:e40162
Acknowledgements
We thank Andres Nevarez for providing feedback and for testing the pipeline. We would like to thank Simon F. Nørrelykke and Kota Miura for providing insights that improved this chapter considerably.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Zabary, Y., Zaritsky, A. (2022). A MATLAB Pipeline for Spatiotemporal Quantification of Monolayer Cell Migration. In: Miura, K., Sladoje, N. (eds) Bioimage Data Analysis Workflows ‒ Advanced Components and Methods . Learning Materials in Biosciences. Springer, Cham. https://doi.org/10.1007/978-3-030-76394-7_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-76394-7_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-76393-0
Online ISBN: 978-3-030-76394-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)