
Abstract
Genomic prediction and selection models (GS) were deployed as part of DataBio project infrastructure and solutions. The work addressed end-user requirements, i.e., the need for cost-effectiveness of the implemented technologies, simplified breeding schemes, and shortening the time to cultivar development by selecting for genetic merit. Our solutions applied genomic modelling in order to sustainably improve productivity and profits. GS models were implemented in sorghum crop for several breeding scenarios. We fitted the best linear unbiased predictions data using Bayesian ridge regression, genomic best linear unbiased predictions, Bayesian least absolute shrinkage and selection operator, and BayesB algorithms. The performance of the models was evaluated using Monte Carlo cross-validation with 70% and 30%, respectively, as training and validation sets. Our results show that genomic models perform comparably with traditional methods under single environments. Under multiple environments, predicting non-field evaluated lines benefits from borrowing information from lines that were evaluated in other environments. Accounting for environmental noise and other factors, also this model gave comparable accuracy with traditional methods, but higher compared to the single environment model. The GS accuracy was comparable in genomic selection index, aboveground dry biomass yield and plant height, while it was lower for the dry mass fraction of the fresh weight. The genomic selection model performances obtained in our pilots are high enough to sustain sorghum breeding for several traits including antioxidants production and allow important genetic gains per unit of time and cost.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction, Motivation and Goals
Genomic selection (GS), fitting the big data generated from several sources such as phenomics, genomics, and Internet of Things (IoT), provides the enabling technologies to support crop breeding companies and research and development institutions. Genomic selection models were deployed as part of DataBio project infrastructure and solutions tailored to the end user requirements. Specific challenges, which GS addresses in agriculture, are mostly represented by the need for cost-effectiveness of the implemented technologies, simplified breeding schemes, and shortening the time to cultivar development selecting for genetic merit estimated through genomic modelling in order to sustainably improve productivity and profits. One of the interesting features of genomic selection is the possibility to customize the solutions to fit the farmer’s requirements such as putting major emphasis on a single characteristic or several plant characteristics aggregated in selection index. Genomic selection allows therefore to close the gap between agricultural business planning and the responsible and sustainable maximization of the profit deriving mainly from increased crop productivity and efficiency of resource use, and reduced uncertainty of management decisions.
Another key feature of genomic selection is its ability to decouple selection from phenotyping—the assessment of expressed plant characteristics as influenced by genetic make-up and changes in the environment—in the process of crop improvement (Fig. 16.1). Genomic selection is implemented in coherent steps starting from genotyping (determining the individual’s genetic constitution through Deoxyribonucleic acid sequencing) and phenotyping the training population, and then proceeding with calibrating the phenotypes against the genomic information, whole-genome genotyping the selection candidates, using calibration equation to predict plant characteristics, operating selection upon genetic merit (genomic estimated breeding values, GEBVs), and implementing repetitive cycles of crossing and selection based on GS-generated information.

Overall genomic prediction and selection operational steps. Refer to text for further description
The diagram below (Fig. 16.2) represents the generic pipeline for data flow of genomic selection and prediction: from data collection to data processing and decision-making, and its mapping to the steps of the top-level pipeline that is in compliance with the Reference Architecture for Big Data Application Providers [1].

Generic pipeline for data flow in genomic selection and prediction
One of the most compelling merits of the genomic selection technology is the possibility to integrate Marker Assisted Selection for yield into practical breeding programmes, particularly in the areas of population genetics and quantitative genetics. This has been a puzzle to breeders, geneticists and other scientists for the last 30 years of Quantitative Traits Loci (QTL, a chromosomal region that correlates with variation of a plant characteristic) breeding. Genomic selection represents the gold standard approach to expedite cultivar development, and for estimating breeding values upon which superior cultivars are identified and selected. Genomic selection allows superior response to selection, and hence superior breeding progress, due to its intrinsic attributes that expedite breeding works by shortening generation intervals through genomic prediction and selection-driven intercrosses. The genomic selection technology is therefore expected to significantly improve genetic gain by unit of time and cost, allowing farmers to grow a better variety sooner relative to conventional approaches, and hence make more income.
The pilot trials for this work were run by a collaborative effort between Council for Agricultural Research and Economics, Italy (CREA) and Centre for Research and Technology Hellas, Greece (CERTH). Genomic data (SNPs) produced in tomato was enough to run genomic models, but the size of tomato population phenotyped was too low (less than 40) and it was not therefore possible to run genomic models in tomatoes as genomic models require a big size of the training population. We therefore report herein the results obtained from CREA’s sorghum pilot experiments where a sufficiently bigger population (380) had been genotyped and phenotyped, to improve yields of biomass and health-promoting compounds used to manufacture specialty foods.
In the GS approach, different assumptions of the distribution of marker effects were accommodated in order to account for different models of genetic variation including, but not limited to: (1) the infinitesimal model, (2) finite loci model, (3) algorithms extending Fisher’s infinitesimal model of genetic variation to account for non-additive genetic effects. Many problems were modelled including the performance of new and unphenotyped lines, untested environments, single trait, multi-traits, single environment, and multi-environment. Models were fed several data types: open-field phenotypic data, biochemical data, phenomic and genomic data and other data sources (environmental indoor/outdoor, farm data/log/profile) collected to describe the crop management and production environment. Next, the GS equations were used to predict the breeding values of genotyped but unphenotyped candidates and the outcome was encouraging as detailed below.
2 Pilot Set-Up
The first stage of the sorghum pilot trials started in 2018 in several locations in Emilia Romagna Region, Northern Italy. In this year, the CREA’s platform for genomic prediction and selection was specified to accommodate the requirements of the breeding programmes, particularly the upcoming genomic and phenomic/phenotypic data from sorghum field experiments. In the second stage of the trials in 2019, a second temporal replication of sorghum pilot trials was established in the same region of Emilia Romagna but in locations different from 2018 as dictated by the rules of crop rotations. Sorghum lines were genotyped using a genotyping-by-sequencing (GBS) strategy on Illumina next-generation sequencing platform.
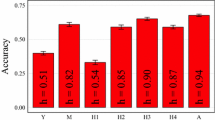
Genotypic variability is an important precondition for genomic selection and prediction. To evaluate the genotypic variability for the evaluated traits, the Bayesian regression model was implemented in R using the probabilistic programming language Stan, implementing Hamiltonian Monte Carlo and its extension, the no-u-turn sampler (NUTS). Our choice for these algorithms was motivated by their faster convergence relative to other commonly used Markov chain Monte Carlo algorithms, like the Metropolis Hastings and Gibbs sampler. The default rules were applied to choose hyperparameters. For each trait, the models were fitted using four chains, each with 50,000 iterations of which the first 10,000 were warmup (burn-in) to calibrate the sampler, leading to a total of 160,000 posterior samples upon which our analyses were based. Genotypic variability was measured using the mean (estimate) and the standard deviation (estimate error) of the posterior distribution as well as two-sided 95% credible intervals (l–95% CI and u–95% CI) based on quantiles. Variance components and trait broad-sense heritability (repeatability) were estimated by fitting the appropriate linear mixed model equation.
3 Technology Used
3.1 Phenomics
In this work, we measured a set of phenotypes from sorghum plants (physical and biochemical traits) that were produced over the course of development and in response to environmental stimuli. The biochemical analysis was carried out both with colorimetric and chromatographic methods. Total polyphenol content was measured with the Folin-Ciocalteu method, total antioxidant activity was assessed with DPPH (2,2-diphenyl-1-picrylhydrazyl) radical assay, and total flavonoid content was measured with AlCl3 method. The phenotypic characterization of sorghum lines was carried out according to international standard operating procedures following International Board for Plant Genetic Resources (IBPGR) and International Union for the Protection of New Varieties of Plants (UPOV) as described in previous works [2, 3].
To analyse total phenols, tannins, flavonoids and antioxidant capacity (TAC), a 10 g sample from each genotype was ground using a Cyclotec Udy Mill (sieve: 0.5 mm), the moisture in the sample was determined after they were oven-dried overnight at 105 °C, and antioxidants and TAC were analysed in duplicate using 100 mg of each sample. For the phenolic compounds, the absorbance of samples was measured at 750 nm and expressed as gallic acid equivalents (gGAEkg−1 dry mass basis). For condensed tannins and total flavonoids assays, the absorbances were measured at 500 nm and 510 nm, respectively, and expressed as μg CE (catechin equivalents) g−1 dry mass basis. The TAC was determined using the 2,20-azino-bis/3-ethylbenzthiazo-line-6-sulphonic acid (ABTS) assay and expressed as mmol TE (Trolox equivalents) kg−1 dry basis. Internet of things (IoT) technology was implemented to collect and characterize soil, plant, and environmental properties.
3.2 DNA Isolation, Next-Generation Sequencing/Genotyping, and Bioinformatics
In sorghums, DNA was isolated from plantlets using the GeneJET Plant Genomic DNA Purification Kit. The methylation sensitive restriction enzyme ApeKI was used for library preparation, and genotyping-by-sequencing (GBS) was carried out on an Illumina HiSeq X Ten platform. The final working matrix consisting of 61,976 high-quality SNPs was used in this work for genomic selection and prediction analytics.
3.3 Genomic Predictive and Selection Analytics
To evaluate the performance of GS models, the Monte Carlo (repeated hold-out) cross-validation approach [4, 5] was applied using 70% and 30%, respectively, as training and validation sets. In a standard hold-out cross-validation, the data is randomly divided into two subsets: a training and a test (validation) set. The test set represents new, unseen data to the model. To obtain a more robust performance estimate that was less variant to how the data was split into training and test sets, the hold-out method was repeated 50 times with different random seeds and the average performance was computed over these 50 repetitions. The repeated hold-out procedure provides a better estimate of how well our model may perform on a random test set, compared to the standard hold-out validation method [5]. In addition, it provides information about the model’s stability as to how the model, produced by a learning algorithm, changes with different training set splits. In the Monte Carlo method, models were implemented fitting best linear unbiased predictions (BLUP) data using Bayesian ridge regression (BRR), genomic best linear unbiased predictions (GBLUP), Bayesian least absolute shrinkage and selection operator (LASSO), and BayesB algorithms accounting for all spatial and temporal replications of the trials (Table 16.1).
In the case of multi-environment scenario, different cross-validation experiments (Table 16.2) were evaluated using GBLUP. Cross-validation CV1 reflected prediction of sorghum lines that have not been evaluated in any of the target environments, while cross-validation CV2 reflected prediction of lines that have been evaluated in some, but not all, target environments. The rationale being that prediction of non-field evaluated lines benefits from borrowing information from lines that were evaluated in other environments. This is critical in cutting costs for varietal adaptability trials of large numbers of lines in several target environments. The model was run on a single environment basis, across environments, marker-by-environment interaction, and using the reaction norm model.
Our findings show that genomic models perform comparably under single environments (Table 16.1, Fig. 16.3). On the other hand, under multiple environments, CV2 was superior to CV1. Under CV2 settings, single-environment model performed poorly. Accounting for environmental noise, marker information x environment or implementing the reaction norm model performed comparably and produced superior results relative to single environment model (Table 16.2).

Distribution (boxplot) of GS models validated accuracy in external sample (not used during model training) of 34 (30% of the total population) sorghum lines. FEN, FLA, TAC, TAN, respectively, polyphenols, flavonoids, total antioxidant capacity, and condensed tannins. Traits means are included within the boxplot. Trait means with same letter are not significantly different at the 5% level using the Tukey’s honestly significant difference (HSD) test. Refer to text for the description of the GS models. Reprinted from Habyarimana et al. [3] under a CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/), original copyright 2019 by the authors
When faced with the necessity to simultaneously improve more than one trait, a breeder can use three approaches: tandem selection, independent culling levels, and index selection [6]. In tandem selection, only one character is selected in each cycle; in independent culling levels, all genotypes with a phenotypic value below the culling threshold for at least one characteristic are discarded; the selection index aims at improving several traits simultaneously in such a way as to make the biggest possible improvement in overall genetic merit [7]. In this work, we implemented the optimum selection Index of Smith [2, 3, 8], the performance of which was demonstrated in previous studies [7, 9]. Our findings showed accuracy that was higher (acc = 0.52 – 0.59) and comparable in genomic selection index, aboveground dry biomass yield and plant height, while it was lower (acc = 0.36) for the dry mass fraction of the fresh weight (Fig. 16.4). In this work, the accuracy of the models was defined as the Pearson correlation coefficient (\(r\)) between observed (\(y\)) and predicted (\(\widehat{y}\), genomic estimated breeding values) phenotypic values as represented in the following formula:
where \(\overline{y}\) and \(\overline{{\widehat{y}}}\) are, respectively, the means of the observed and the predicted values.

Distribution (boxplot) of genomic selection index accuracy using single traits and all three traits of interest simultaneously in the entire panel. DMC, DMY, GSI, and PH, respectively, denote selection indices relative to dry mass fraction of fresh material, aboveground dry biomass yield, all the three traits simultaneously, and plant height. Means are indicated by open dots and are included within the boxplot. Means with same letter are not significantly different at the 5% level using the Tukey’s HSD (honestly significant difference) test. Refer to text for the description of the GS models. Reprinted from Habyarimana et al. [8] under a CC BY 4.0 license (http://creativecommons.org/licenses/by/4.0/), original copyright 2019 by the authors
4 Business Value and Impact
Genomic predictive and selection (GS) modelling was developed as response to the lengthier and costlier phenotypic selection. In business, time to market is important just as the production cost. In addition, specifically for plant breeding, the longer it takes to bring the new cultivar to the market, the shorter will that cultivar stay on the market, in virtue of the naturally occurring crop degeneration. Some of the most attractive GS attributes are enabling cutting time and cost to cultivar development with high selection accuracy. The high accuracy means that the plant lines selected will breed true to type, implying diminished risks in the breeding and production processes.
In this pilot, the GS technology showed meaningful and attractive results as reflected by the key performance indices (KPIs) presented in Table 16.3. The predictive performance obtained in this pilot was encouraging. Over the two-year trial, with data integration, the four genomic selection models implemented in this pilot performed comparably across traits and are considered suitable to sustain sorghum breeding for antioxidants production and allow important genetic gains per unit of time and cost. In comparison to conventional phenotypic breeding, the genomic predictive and selection modelling allows cutting costs five times and cutting four times the time of cultivar development (Table 16.3). The results produced in this pilot are expected to contribute to genomic selection implementation and genetic improvement of sorghum for several traits including grain antioxidants for different purposes including the manufacture of health-promoting and specialty foods in Europe in particular, and in the world in general. In addition, the NGS genotyping platforms were validated and were found to be usable for sequencing and genotyping (variants calling) services in other plant species and animal husbandry.
5 How to Guideline for Practice When and How to Use the Technology
The method for implementing genomic prediction and selection analytics was depicted in the above diagram (Fig. 16.1), while a reusable generic pipeline for data flow genomic selection and prediction was described in Fig. 16.2. Several scenarios can be modelled including a single trait, multiple traits as index selection, a single environment, and multi-environment. A generic technological flowchart is that, in the genomic predictive and selection modelling, phenotypic and marker data are scored in the training population and fitted into appropriate algorithm to produce individuals’ whole-genome marker effects. Most practically, the training set is the germplasm or a population that best samples the frequency of the genetic information (allele frequency) useful for the breeding programme. The marker effects are used in subsequent cycles of selection to compute the genomic estimated breeding values (GEBVs) that are used as predictors of breeding values in testing unphenotyped population. The genomic estimated breeding values are obtained as a product of the estimated marker effects in the training population and the coded marker values obtained in the testing population. To apply genomic selection, GEBVs are obtained in the selection candidates and then used to predict and rank the net genetic merit of the candidates for selection, and superior strains are selected in the process; GEBVs become the criteria for crossing block management and cultivar development. Genomic predictive and selection modelling is a gold standard for selecting for breeding values and is well poised to help breeders and seed industries to drastically cut breeding cost and time and bring new cultivar earlier on the market, thus generating higher incomes.
6 Summary and Conclusions
Current empirical evidence for genomic selection efficiency in plant breeding is set to r = 0.5 as the baseline for genomic selection prediction accuracy in plant breeding. Also, recent research works demonstrated that genomic selection accuracy as low as 0.2 can allow substantial within-generation yield improvement [10]. Therefore, the genomic selection model performances obtained in our pilots are high enough to sustain sorghum breeding for several traits including antioxidants production and allow important genetic gains per unit of time and cost. In addition to the accuracy, the importance of the genomic selection strategy is also evaluated using other criteria such as the possibility that this technology offers to shorten the breeding cycle with significant economic returns due to intercrosses driven by genetic predictions, the quick delivery of novel superior cultivars onto the market. In the case of antioxidants, genomic selection offers the possibility to select for or against this trait early (e.g. at the seed or seedling stages) without waiting for seed setting or harvest. The genomic selection algorithms developed in this work can be directly used in sorghum breeding programmes and can be adapted to other plant species and animal husbandry. The genomic selection results presented herein and the experimental designs used in this pilot can be implemented in antioxidants and other traits genetic investigations and in breeding programmes to qualitatively and quantitatively improve plant characteristics and the antioxidant production for different purposes including the manufacture of health-promoting and specialty foods.
References
NIST Big Data Public Working Group Reference Architecture Subgroup (2015) NIST big data interoperability framework: Reference architecture, (Vol. 6). National Institute of Standards and Technology.
Habyarimana. E., Dall’Agata, M., De Franceschi, P., Baloch, F. S. (2019). Genome-wide association mapping of total antioxidant capacity, phenols, tannins, and flavonoids in a panel of Sorghum bicolor and S. bicolor × S. halepense populations using multi-locus models. PLoS ONE 14:e0225979. https://doi.org/10.1371/journal.pone.0225979
Habyarimana, E., Lopez-Cruz, M. (2019). Genomic selection for antioxidant production in a panel of sorghum bicolor and S. bicolor × S. halepense Lines. Genes 10:841. https://doi.org/10.3390/genes10110841
Scutari, M., Mackay, I., & Balding, D. (2016). Using genetic distance to infer the accuracy of genomic prediction. PLOS Genetics, 12, e1006288. https://doi.org/10.1371/journal.pgen.1006288.
Raschka, S. (2018). Model evaluation, model selection, and algorithm selection in machine learning. arXiv:181112808 [cs, stat].
Wricke, G., Weber, E. (1986). Quantitative genetics and selection in plant breeding, Reprint 2010 ed. edition. De Gruyter.
Bradshaw, J. E. (2017). Plant breeding: Past, present and future. Euphytica, 213, 60. https://doi.org/10.1007/s10681-016-1815-y.
Habyarimana, E., Lopez-Cruz, M., & Baloch, F. S. (2020). Genomic selection for optimum index with dry biomass yield, dry mass fraction of fresh material, and plant height in biomass sorghum. Genes, 11, 61. https://doi.org/10.3390/genes11010061.
Baker, R. J. (1986). Selection indices in plant breeding. CRC Press
Habyarimana, E. (2016). Genomic prediction for yield improvement and safeguarding genetic diversity in CIMMYT spring wheat (Triticum aestivum L.). Australian Journal of Crop Science, 10, 127–136.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Habyarimana, E., Michailidou, S. (2021). Genomic Prediction and Selection in Support of Sorghum Value Chains. In: Södergård, C., Mildorf, T., Habyarimana, E., Berre, A.J., Fernandes, J.A., Zinke-Wehlmann, C. (eds) Big Data in Bioeconomy. Springer, Cham. https://doi.org/10.1007/978-3-030-71069-9_16
Download citation
DOI: https://doi.org/10.1007/978-3-030-71069-9_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-71068-2
Online ISBN: 978-3-030-71069-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)