
Abstract
The tumor microenvironment (TME) is a complex biological structure surrounding tumor cells and includes blood vessels, immune cells, fibroblasts, adipocytes, and extracellular matrix (ECM) [1, 2]. These heterogeneous surrounding structures provide nutrients, metabolites, and signaling molecules to provide a cancer-friendly environment. The metabolic interplay between immune cells and cancer cells in the TME is a key feature not only for understanding tumor biology but also for discovering cancer cells’ vulnerability. As cancer immunotherapy to treat cancer patients and the use of metabolomics technologies become more and more common [3], the importance of the interplay between cancer cells and immune cells in the TME is emerging with respect to not only cell-to-cell interactions but also metabolic pathways. This interaction between immune cells and cancer cells is a complex and dynamic process in which immune cells act as a determinant factor of cancer cells’ fate and vice versa. In this chapter, we provide an overview of the metabolic interplay between immune cells and cancer cells and discuss the therapeutic opportunities as a result of this interplay in order to define targets for cancer treatment. It is important to understand and identify therapeutic targets that interrupt this cancerpromoting relationship between cancer cells and the surrounding immune cells, allowing for maximum efficacy of immune checkpoint inhibitors as well as other genetic and cellular therapies.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Key Points-
Tumor cells produce numerous substances to create an immunosuppressive tumor microenvironment.
-
The tumor microenvironment physically constitutes a barrier against T-cell infiltration.
-
Activated T cells reprogram OXPHOS and FAO to glycolysis and glutaminolysis.
-
Tumors escape immunity via T-cell dysfunction or hyporesponsiveness by upregulation of several inhibitory receptors.
-
Increased glucose uptake by cancer cells restricts T-cell function by decreasing mTOR activity.
-
Treatments using immune checkpoint inhibitors increase extracellular glucose levels to improve T-cell infiltrating lymphocytes’ function.
1 Introduction
The tumor microenvironment (TME) is a complex biological structure surrounding tumor cells and includes blood vessels, immune cells, fibroblasts, adipocytes, and extracellular matrix (ECM) [1, 2]. These heterogeneous surrounding structures provide nutrients, metabolites, and signaling molecules to provide a cancer-friendly environment. The metabolic interplay between immune cells and cancer cells in the TME is a key feature not only for understanding tumor biology but also for discovering cancer cells’ vulnerability. As cancer immunotherapy to treat cancer patients and the use of metabolomics technologies become more and more common [3], the importance of the interplay between cancer cells and immune cells in the TME is emerging with respect to not only cell-to-cell interactions but also metabolic pathways. This interaction between immune cells and cancer cells is a complex and dynamic process in which immune cells act as a determinant factor of cancer cells’ fate and vice versa. In this chapter, we provide an overview of the metabolic interplay between immune cells and cancer cells and discuss the therapeutic opportunities as a result of this interplay in order to define targets for cancer treatment. It is important to understand and identify therapeutic targets that interrupt this cancer-promoting relationship between cancer cells and the surrounding immune cells, allowing for maximum efficacy of immune checkpoint inhibitors as well as other genetic and cellular therapies.
2 Tumor Immunity and the Various Roles of Immune Cells
The immune system’s antitumor activity is mainly carried out by tumor antigen-specific cytotoxic T lymphocytes (CTL), T effector (Teff) cells, antibody-producing B cells, as well as antigen-presenting dendritic cells (DC), which lead to adaptive immunity by directly recognizing and eliminating cancer cells. Natural killer (NK) cells, macrophages, and NK-T cells also play crucial roles in suppressing tumor progression via a nonspecific immune response. Even though this defense system is well developed, the tumor often has the ability to develop an immunosuppressive microenvironment favorable to its progression. Specifically, myeloid-derived suppressor cells (MDSC), regulatory T (Treg) cells, and tumor-associated macrophages (TAMs) are well-known players. These tumor-friendly immune cells suppress the settlement of tumor-infiltrating lymphocytes (TILs) by expressing essential amino acid (EAA)-degrading enzymes including arginase 1 (Arg1) and indoleamine-2,3-dioxygenase (IDO) [4,5,6,7], and inhibitors targeting Arg1 and IDO are being investigated in ongoing clinical trials [8, 9]. The TME is an environment composed of multifaceted components with tumor-friendly or antitumoral characteristics where there is strong competition for metabolites and nutrients. Studies have shown that T-cell-mediated adaptive response is a promising therapeutic strategy to strengthen the antitumor activity of the immune system [10,11,12].
2.1 Metabolic Competition and Tumor Immunity
With strong evidence showing how T lymphocytes infiltrate into the tumor niche and how checkpoint inhibitors or chimeric antigen receptor (CAR) T cells inhibit the growth of cancer cells, a new era of immunotherapy has just begun with successful clinical development [13]. However, cancer cells can escape immune recognition via “immunoediting,” allowing cancer cell clones without detectable cancer antigens to dominate and escape from the pressure of immune checkpoint inhibitors [14].
Rapidly growing tumor cells require nutrients, oxygen, and essential metabolites to proliferate and, at the same time, create an immunosuppressive microenvironment. How immune cells and cancer cells share or compete in these harsh environmental conditions and how the TME alters immunometabolism are important questions to address. Specifically, it needs to be addressed how cancer cells and neighboring immune cells compete to take up nutrients and metabolites, which consequently influences signaling cascades, metabolic activities, and tumor progression.
Cancer-associated adipocytes: It is well known that adipocytes play an important role in communicating with cancer cells by excreting inflammatory factors, adipokines, and free fatty acids, which help cancer growth. In addition, immune cell functions are heavily regulated by lipids, lipoproteins, and cholesterol within the TME. For example, elevated levels of oxidized lipoproteins as a result of incorporation via scavenger receptors and formation of lipid droplets can compromise the ability of dendritic cells (DCs) to activate T cells by presenting tumor antigens [15, 16]. Also, it is well known that cancer cells instruct neighboring adipocytes to increase lipolysis [17].
Cancer-associated fibroblasts [18, 19]: It is reported that tryptophan catabolism by CAFs causes the starvation of immune cells and results in the production of kynurenine, an immunosuppressive metabolite [20]. Cancer cells also produce hydrogen peroxide (H2O2), which can lead to oxidative stress in CAFs. Oxidative stress is associated with impaired mitochondrial function, which results in upregulated glucose uptake and elevated reactive oxygen species (ROS) levels [21]. In addition, glucose is also a key metabolite for the antitumor activities of effector T (Teff) cells and M1 macrophages because aerobic glycolysis is necessary for their activation [22, 23].
Altered amino acid levels: Amino acids in the TME are not only a resource competed for by cancer and immune cells but also another metabolic checkpoint regulating antitumor immunity. For example, glutamine is a precursor for the tricarboxylic acid (TCA) cycle [24] and lipid synthesis in hypoxic cancer cells [25] and Teff cells [26]. As such, glutaminolysis, a series of biochemical reactions that start with the conversion of glutamine carbon to glutamate and aspartate, is essential for cancer cells by providing nutrients and metabolites through anaplerotic reactions, and leads to tumor cells’ and TILs’ competition for glutamine, the pathway’s starting material [27,28,29]. Moreover, it is known that glutamine activates the mammalian target of rapamycin (mTOR) signaling cascades in T cells and macrophages and is important for protein O-GlcNAcylation and synthesis of S-2-hydroxyglutarate (S-2GH), a regulator of effector T (Teff) cell function [30, 31]. Consequently, it was found that there is an upregulation of the major glutamine transporter alanine, serine, cysteine transporter 2 (ASCT2), also known as SLC1A5, for several types of cancer [32].
The proliferation of immune cells relies on growth factors for efficient nutrient utilization. For example, interleukin-2 (IL-2) promotes increased expression of glucose transporters (GLUT) and thus enhances glycolysis in activated T cells [33,34,35,36]. There is a question about how metabolites activate signaling pathways to induce changes in immune cell functions. A classic example is the binding of metabolites and energy substrates to G protein-coupled receptors (GPCRs) [37]. For example, succinate leads to increased chemotaxis and activation of dendritic cells after toll-like receptor (TLR) agonist treatment by binding to the succinate receptor GPR91 [38]. On the other hand, adenosine, by binding to A2B and A2A adenosine receptors, leads to increased interleukin 4 (IL-4)-induced M2 macrophage activation [39]. Moreover, it is recently reported that there is a significantly reduced arginine level in the TME as a result of inducible nitric oxide synthase (iNOS) and arginase expressed by myeloid-derived suppressor cells, indicating that rapid dynamic changes of amino acids can happen in the TME [40].
2.2 Antitumor T-Cell Metabolisms in the TME
As T cells play a critical role in antigen-specific adaptive immunity against the tumor, it is fundamentally important to understand T-cell biology. T lymphocytes respond to the presence of antigens and evolve rapidly. This response first requires T-cell growth; then their drastic expansion, differentiation, and death; and lastly, the formation and preservation of the memory of the immune response.
T lymphocyte proliferation in the TME requires a switch in its metabolism. While naïve T cells utilize fatty acid β-oxidation, activated T cells mainly use glycolysis, pentose phosphate pathway, and glutaminolysis [41, 42]. Additionally, it is reported that distinct transcriptional programs and signaling pathways are involved in this metabolic shift, including the transcription factor c-Myc [43, 44], estrogen-related receptor alpha (ERRα) [41, 45, 46], phosphatidylinositol-3-OH kinase (PI(3)K), and GLUT1-dependent Akt pathways [45]. This significant metabolic reprogramming of activated T cells is required for their proliferation and expansion. Consistently, it is also reported that activated T cells switch from oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) to glycolysis and glutaminolysis, which are characteristics of Teff cells, induced by antigenic stimulation through the T-cell receptor (TCR) and engagement of CD28 with a ligand on antigen-presenting cells (APC) [26]. Although glycolysis produces less ATP than OXPHOS, it is very efficient at producing biosynthetic precursors [47], which can further support the rapid proliferation and pro-inflammatory functions of Teff. Moreover, it is consistent with the findings that depletion of GLUT1 impaired T-cell proliferation and functions [36], while elevated expression of GLUT1 increased Teff cell functions [48]. In addition, demands for nutrients, such as glucose, glutamine, and other amino acids, lead to upregulations of transporters in T cells, including GLUT1 [36, 45, 49], glutamine transporters and sodium-coupled neutral amino acid transporters 1 and 2 (SNAT1 and SNAT2) [50], and monocarboxylate lactate transporters MCT1 and MCT4 to export lactate produced via aerobic glycolysis [51].
This metabolic shift from OXPHOS and FAO to glycolysis and glutaminolysis during T-cell activation is mediated by several crucial regulators. It is reported that TCR directly induces PI3K/Akt/mTORC1 and MYC pathways, which not only activate effector T cells but are also crucial for their proliferation and biological functions [26]. Indeed, an activated mTOR pathway promotes glycolysis by upregulating c-MYC and hypoxia-inducible factor 1α (HIF1α) [41, 45, 48, 49, 52, 53]. MYC then induces the transcriptional factor AP4, which further upregulates glycolytic enzyme gene expressions [54]. Moreover, increased HIF1α expression and activity upregulate pyruvate dehydrogenase kinase (PDK1) and lactate dehydrogenase A (LDHA), leading to increased aerobic glycolysis and decreased OXPHOS [55, 56], thus switching pyruvate away from the TCA cycle to lactate production. HIF1α also promotes glycolysis by upregulating GLUT1 and MCT4 expression, in addition to glycolytic enzymes and regulators [57].
After fulfilling their duties, activated T cells undergo apoptosis during a time period called the contraction phase [58], while Treg cells and memory T (Tmem) cells, by using lipid oxidation for energy production, remain in peripheral tissues or secondary lymphoid organs without undergoing apoptosis [48, 57, 59, 60].
2.3 Cancer Cells’ Impacts on T-Cell Metabolism in the TME
It is well known that T-cell dysfunction, or hyporesponsiveness, can result in tumors escaping immunity. This dysfunction or hyporesponsiveness is due to exhaustion and senescence of T cells [61]. For instance, tumor cells are shown to express indoleamine 2,3-dioxygenase (IDO), an enzyme that results in decreased tryptophan levels and inhibition of T-cell proliferation [62, 63]. Lactate produced by tumor cells can also lead to reduced T-cell function by blocking their lactate export [64]. Intracellular lactate accumulation impairs their aerobic glycolysis and thus limits their function [65].
Moreover, increased glucose uptake and consumption by cancer cells [66] impair T-cell function by decreasing their mTOR activity, glycolysis, and INF-γ production. These negative consequences on T cells help promote tumor progression, which is also facilitated by decreased cytokine production due to the lack of glucose in the microenvironment. It is also supported by the fact that many types of tumors have high glycolysis rates [67, 68]. Moreover, lack of glucose impairs IFN-γ production of T cells and pro-inflammatory signals in macrophages [36, 65, 69]. In addition, increased glycolysis rate in tumor cells as a result of the overexpression of the glycolytic enzyme hexokinase 2 (HK2) reduced glucose uptake and IFN-γ production in TILs, which led to a more tumor-friendly microenvironment [69, 70].
2.4 Cancer Cell-Induced Metabolically Harsh Environment Impairs T-Cell Function
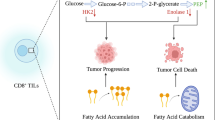
As the tumor grows larger, (1) oxygen supply becomes limited, thus creating a hypoxic condition; (2) nutrients become deficient; and (3) the microenvironment becomes acidic. Recent findings suggest that these harsh metabolic states significantly disrupt T-cell function. Therefore, the influence of cancer cell metabolism on the TME may directly control the metabolic pathways in surrounding T cells [71]. The tumor microenvironment physically constitutes a barrier against T-cell infiltration, as it is a compact structure with tight interactions among cancer cells, fibroblasts, immune cells, and ECM. Indeed, tumor cells generate numerous substances to create an immunosuppressive microenvironment. For example, hypoxic cancer cells release prostaglandin E2 (PGE2) and adenosine, which can result in T lymphocyte proliferation inhibition by activating G protein-coupled receptors (GPCR) and protein kinase A [72]. Among the GPCRs, chemokine (C-X-C motif) receptor 3 (CXCR3) and chemokine (C-C motif) receptor 5 (CCR5) are often expressed in active lymphocytes that have infiltrated the tumors in melanoma, breast, and colorectal cancers [73] (Fig. 1).

Potential immunometabolism-targeting strategy in the TME. APC antigen-presenting cells, CAR T chimeric antigen receptor T cell, CAF cancer-associated fibroblast, ECM extracellular matrix
In order to reach tumor cells and to enhance the efficacy of immunotherapy, T lymphocytes have to degrade the ECM and heparan sulfate proteoglycans (HSPGs) [74]. It is reported that chimeric antigen receptor (CAR) T cells need to release heparanase (HPSE) to successfully degrade HSPGs, which then allows T cells to gain access to the solid tumor [75] (Table 1 and Fig. 1).
3 Targeting the Metabolism of Immune Cells for Cancer Treatment
Accumulating evidence from the past decade indicates that metabolic reprogramming greatly affects T cells. Indeed, when T cells recognize antigens, they are activated to proliferate and produce effector molecules to eliminate the foreign antigens. During this course of the immune response, immune cells respond to changes in the metabolic microenvironment, which serves as a “metabolic checkpoint” responsible for connecting the metabolic states with signaling pathways in immune cells, which further determines their immune functions [47]. Accordingly, metabolic reprogramming of cells, such as a switch from OXPHOS and FAO to glycolysis and glutaminolysis in naïve and memory T cells, helps provide energy and other building block materials to generate new biomass. The manipulation of metabolic enzyme expressions helps T cells adapt in the tumor-suppressive microenvironment and restore their functions. Specifically, overexpression of phosphoenolpyruvate carboxykinase 1 (PCK1) results in a high level of the glycolytic metabolite phosphoenolpyruvate (PEP). High PEP level then enhances T-cell effector functions through T-cell receptor-mediated Ca2+-dependent nuclear factor of activated T-cell (NFAT) signaling. PCK1-overexpressing T cells inhibited melanoma tumor growth in vivo [70]. Another example is the oxygen-sensing prolyl-hydroxylase (PHD) proteins, which, as oxygen sensors in T cells, support cancer metastasis to the lung. Indeed, targeting T-cell-intrinsic PHD proteins resulted in increased antitumor immunity [112]. Also, as TILs usually have impaired mitochondrial function after infiltrating the tumors, reactivation of PPAR-gamma coactivator 1α (PGC1α) by suppressing Akt signaling in these T cells can induce mitochondrial biogenesis. Thus, increasing the expression of PGC1α in these T cells also activates their functions [113]. These approaches may improve antitumor immunity for adoptive T-cell therapy, which is a personalized therapy for cancer via T-cell manipulation [70, 112, 113].
3.1 The Metabolism of the Immune Checkpoint Blockades
When T cells infiltrate the TME, they gradually lose several abilities, including responsiveness to T-cell receptor (TCR) stimuli and production of antitumor cytokines, in a phenomenon referred to as T-cell exhaustion or hyporesponsiveness. This is the result of the upregulation of several inhibitory receptors such as PD-1, LAG3, TIGIT, and CTLA-4 that make T cells less sensitive to tumor antigens [114]. In particular, the PD-1:PD-L1 axis and CTLA-4 are critical immune checkpoints for T cells, and targeting these receptors breaks down the cross talk between cancer cells and exhausted T cells. This result is supported by numerous clinical successes of immune checkpoint inhibitors, including ipilimumab, nivolumab, pembrolizumab, atezolizumab, avelumab, durvalumab, and cemiplimab-rwlc [115].
Interestingly, glucose deprivation caused by rapid uptake by actively growing cancer cells and glucose competition between cancer cells and other cells in the TME further upregulates PD-1 expression [114, 116]. In fact, PD-1 activation leads to suppressed T-cell receptor (TCR), PI3K, and mTOR signaling in T cells and reduced glycolysis, which may lead to increased accumulation of regulatory CD4 (Treg) cells in the TME [117,118,119]. If PD-L1 on the surface of tumor cells binds to PD-1 on T cells, referred to as the engagement of PD-1, T-cell proliferation, cytokine production, and cytolytic function are inhibited, which promotes cancer cell proliferation [120]. It has also been shown that the degree of PD-L1 expression correlates with glycolysis rates, as well as the expression levels of glycolytic enzymes [116]. Moreover, α-PD-L1 antibody treatment increases extracellular glucose levels in vivo, which results in improved TIL function and subsequently reduced tumor growth. Indeed, intrinsic PD-1 expression promotes mTOR signaling and tumor growth [121], while blockade of PD-1 signaling activates glycolysis and anabolic pathways in exhausted T cells via mTORC-1 [69, 122]. Thus, this metabolic shift provides the rationale for the clinical development of combination therapy with immune checkpoint blockade and mTOR inhibitors. Indeed, multiple clinical trials are under investigation with those drug combinations in patients with TNBC and renal cell carcinoma (NCT03805399, NCT04203901). Collectively, these results imply that the most promising therapy should target the co-inhibitory receptor-to-ligand interactions and re-sensitize exhausted T cells in the TME.
3.2 The Metabolism of Chimeric Antigen Receptor (CAR) T Cells
Recent clinical progress with genetically engineered chimeric antigen receptor (CAR) T cells for cancer therapy opens up a new era of cell/gene therapy. However, its success is limited thus far to acute lymphoblastic leukemia (ALL) and lymphoma, whereas it shows less promising results for solid tumor treatment [72]. It is widely accepted that the major cause of the limited efficacy of CAR T cells is the poor accessibility of T cells to the TME and the low-nutrient, hypoxic environment that provides suboptimal conditions for T-cell proliferation and cytokine production [123]. Thus, CAR T-cell infiltration into the tumor is a critical step to enhance their antitumor efficacy in solid tumors (Fig. 1).
The lack of therapeutic effects of CAR T cells in solid tumors is due, in part, to the immunosuppressive TME, which acts as a critical barrier. As such, new strategies to increase CAR T cells’ accessibility to TME in solid tumors have been proposed. For example, stabilization of HIF1α under hypoxic conditions regulates cellular metabolism, which is a critical feature in the hypoxic TME. A recent study found that targeting an oxygen-sensitive subdomain of HIF1α enhances the CAR-T activity in solid tumors [124]. Another example of new strategies for CAR T therapy is the targeting of heparanase (HPSE). Stroma and tumor cells in the TME are linked together through the ECM which contains a considerable amount of heparan sulfate proteoglycan (HSPG) [75]. To explore whether HSPG can be targeted in solid tumors, Caruana et al. generated HPSE-expressing CAR T cells that showed ECM degradation ability in solid tumors, which resulted in increased infiltration and antitumor activity [75]. This approach may imply the therapeutic benefits of the use of CAR T immunotherapy coupled with HPSE degradation to access tumor niches.
Another approach of engineering CAR T cells to target solid tumors is the development of the nuclear factor of activated T cells, which is referred to as T cells redirected for antigen-unrestricted cytokine-initiated killing (TRUCKs). For instance, engineered CAR T cells with several cytokines, including interleukin-7, -12, -15, and -18, are being explored for TRUCKs [125, 126]. The underlying physiological functions of these interleukins in CAR T-cell therapies are summarized in the reference [126]:
-
Interleukin-2: proliferation of T-cell differentiation of Teff, development of Treg in thymus
-
Interleukin-4: differentiation of Th2 and Th9 cells, survial of B-cells and T-cells
-
Interleukin-7: development of T-cell in thymus, survival of and homeostasis in memory and naïve T cells
-
Interleukin-9: mast cell proliferation, increased antitumor immunity
-
Interleukin-15: development of CD8+ T-cell memory, survival of and homeostasis in CD8+ T-cells
-
Interleukin-21: suppression of Treg, survial and proliferation of CD4+ Th17 cells
Among them, engineered CAR T cells with IL-2, IL-7, IL-15, and IL-21 NFATs are being investigated in clinical trials [126] (Table 1 and Fig. 1).
In addition, it is also known that cytokines can be manipulated to control the metabolism of stem memory T cells (TSCM) and central memory (Tcm) T cells. Of note, T-cell activation by interleukin families, including IL-15 and IL-17, leads to an increased TSCM-like phenotype as well as increased interferon-gamma (IFNγ), tumor necrosis factor alpha (TNFα), and IL-2 production [127]. Moreover, it has been reported that IL-15 activates fatty acid oxidation (FAO) and mitochondrial spare respiratory capacity (SRC) as an alternate way for energy production in T cells [60]. Taken together, IL-15 may provide therapeutic benefits in the form of T memory cell differentiation and mitochondrial metabolism [60]. In addition to mitochondrial metabolism, manipulation of ion and pH levels in the tumor microenvironment, such as decreasing the concentration of potassium, can also enhance T-cell antitumor activity [128]. As such, these metabolism-targeting approaches will provide the rationales for future clinical developments and therapeutic use of CAR T-cell immunotherapy for cancer patients.
4 Conclusion
The immunosuppressive microenvironments in solid tumors are physically and functionally hostile for immune cells, including immune checkpoint inhibitors and CAR T cells. The reasons for less promising efficacy of immunotherapies vary and include the immune cells’ poor accessibility to tumor cells in the TME due to physical and metabolic barriers, including a lack of nutrients and acidosis. In order to improve the therapeutic efficacy of immunotherapies, the tightly controlled microenvironment has to be modified by targeting the metabolic vulnerability of cancer cells. This includes either targeting metabolic enzymes to regulate the metabolism of cancer cells or disrupting the tumor-friendly microenvironment. As metabolism is fundamental for biological and cellular functions, targeting the tumor microenvironment itself or modifying T-cell metabolism is a promising strategy to improve current treatment efficacy.
Abbreviations
- ASCT2:
-
Alanine, serine, cysteine transporter 2
- CAFs:
-
Cancer-associated fibroblasts
- CAR:
-
Chimeric antigen receptor
- CTL:
-
Cytotoxic T lymphocytes
- ECM:
-
Extracellular matrix
- ERRα:
-
Estrogen-related receptor alpha
- FAO:
-
Fatty acid oxidation
- GLUT:
-
Glucose transporter
- GPCRs:
-
G protein-coupled receptors
- HIF1α:
-
Hypoxia-inducible factor 1 α
- HPSE:
-
Heparanase
- HSPG:
-
Heparan sulfate proteoglycans
- IFNγ:
-
Interferon-gamma
- IL:
-
Interleukin
- LDHA:
-
Lactate dehydrogenase A
- MCT:
-
Monocarboxylate lactate transporters
- MDSC:
-
Myeloid-derived suppressor cells
- mTOR:
-
Mammalian target of rapamycin
- NK cells:
-
Natural killer cells
- OXPHOS:
-
Oxidative phosphorylation
- PCK1:
-
Phosphoenolpyruvate carboxykinase 1
- PDK1:
-
Pyruvate dehydrogenase kinase
- PEP:
-
Phosphoenolpyruvate
- PGC1α:
-
PPAR-gamma coactivator 1α
- PHD:
-
Prolyl-hydroxylase
- PI3K:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- SNAT:
-
Sodium-coupled neutral amino acid transporter
- TAM:
-
Tumor-associated macrophages
- Tcm:
-
Central memory T cells
- TCR:
-
T-cell receptor
- Teff:
-
Effector T cells
- TILs:
-
T-cell infiltrating lymphocytes
- TLR:
-
Toll-like receptor
- TNFα:
-
Tumor necrosis factor alpha
- Treg:
-
Regulatory T cells
- TRUCKs:
-
T cells redirected for antigen-unrestricted cytokine-initiated killing
- Tscm:
-
Stem memory T cells
References
Antonio, M. J., Zhang, C., & Le, A. (2021). Different tumor microenvironments lead to different metabolic phenotypes. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_10
Nabi, K., & Le, A. (2021). The intratumoral heterogeneity of cancer metabolism. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_11
Hoang, G., Udupa, S., & Le, A. (2019). Application of metabolomics technologies toward cancer prognosis and therapy. International Review of Cell and Molecular Biology, 347, 191–223.
Munn, D. H., et al. (2002). Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science, 297(5588), 1867–1870.
Lee, G. K., et al. (2002). Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology, 107(4), 452–460.
Rodriguez, P. C., et al. (2004). Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Research, 64(16), 5839–5849.
Uyttenhove, C., et al. (2003). Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nature Medicine, 9(10), 1269–1274.
Adams, J. L., et al. (2015). Big opportunities for small molecules in immuno-oncology. Nature Reviews. Drug Discovery, 14(9), 603–622.
Mondanelli, G., et al. (2019). Immunoregulatory interplay between arginine and tryptophan metabolism in health and disease. Frontiers in Immunology, 10, 1565.
June, C. H. (2007). Adoptive T cell therapy for cancer in the clinic. The Journal of Clinical Investigation, 117(6), 1466–1476.
Leen, A. M., Rooney, C. M., & Foster, A. E. (2007). Improving T cell therapy for cancer. Annual Review of Immunology, 25, 243–265.
Kershaw, M. H., Westwood, J. A., & Darcy, P. K. (2013). Gene-engineered T cells for cancer therapy. Nature Reviews. Cancer, 13(8), 525–541.
Ribas, A. (2015). Adaptive immune resistance: How cancer protects from immune attack. Cancer Discovery, 5(9), 915–919.
Vesely, S., et al. (2013). Parameters derived from the postoperative decline in ultrasensitive PSA improve the prediction of radical prostatectomy outcome. World Journal of Urology, 31(2), 299–304.
Cubillos-Ruiz, J. R., et al. (2015). ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell, 161(7), 1527–1538.
Ramakrishnan, R., et al. (2014). Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. Journal of Immunology, 192(6), 2920–2931.
Nieman, K. M., et al. (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature Medicine, 17(11), 1498–1503.
Sazeides, C., & Le, A. (2021). Metabolic relationship between cancer-associated fibroblasts and cancer cells. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_14
Jung, J. G., & Le, A. (2021). Targeting metabolic cross talk between cancer cells and cancer associated fibroblasts. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_15
Hsu, Y. L., et al. (2016). Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget, 7(19), 27584–27598.
Arcucci, A., et al. (2016). Cancer: An oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. BioMed Research International, 2016, 4502846.
Buck, M. D., O’Sullivan, D., & Pearce, E. L. (2015). T cell metabolism drives immunity. The Journal of Experimental Medicine, 212(9), 1345–1360.
O’Neill, L. A., & Pearce, E. J. (2016). Immunometabolism governs dendritic cell and macrophage function. The Journal of Experimental Medicine, 213(1), 15–23.
Le, A., et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metabolism, 15(1), 110–121.
Pavlova, N. N., & Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell Metabolism, 23(1), 27–47.
Andrejeva, G., & Rathmell, J. C. (2017). Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metabolism, 26(1), 49–70.
Li, T., Copeland, C., & Le, A. (2021). Glutamine metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_2
Jin, L., Alesi, G. N., & Kang, S. (2016). Glutaminolysis as a target for cancer therapy. Oncogene, 35(28), 3619–3625.
Perez-Escuredo, J., et al. (2016). Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle, 15(1), 72–83.
Swamy, M., et al. (2016). Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nature Immunology, 17(6), 712–720.
Tyrakis, P. A., et al. (2016). S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature, 540(7632), 236–241.
Wang, Q., et al. (2015). Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. The Journal of Pathology, 236(3), 278–289.
Fox, C. J., Hammerman, P. S., & Thompson, C. B. (2005). Fuel feeds function: Energy metabolism and the T-cell response. Nature Reviews. Immunology, 5(11), 844–852.
Rathmell, J. C., et al. (2001). IL-7 enhances the survival and maintains the size of naive T cells. Journal of Immunology, 167(12), 6869–6876.
Wofford, J. A., et al. (2008). IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood, 111(4), 2101–2111.
Macintyre, A. N., et al. (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metabolism, 20(1), 61–72.
Blad, C. C., Tang, C., & Offermanns, S. (2012). G protein-coupled receptors for energy metabolites as new therapeutic targets. Nature Reviews. Drug Discovery, 11(8), 603–619.
Rubic, T., et al. (2008). Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nature Immunology, 9(11), 1261–1269.
Csoka, B., et al. (2012). Adenosine promotes alternative macrophage activation via A2A and A2B receptors. The FASEB Journal, 26(1), 376–386.
Kidani, Y., & Bensinger, S. J. (2012). Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunological Reviews, 249(1), 72–83.
Wang, R., et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity, 35(6), 871–882.
Gerriets, V. A., & Rathmell, J. C. (2012). Metabolic pathways in T cell fate and function. Trends in Immunology, 33(4), 168–173.
Dang, C. V., Le, A., & Gao, P. (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical Cancer Research, 15(21), 6479–6483.
Le, A., & Dang, C. V. (2013). Studying Myc’s role in metabolism regulation. Methods in Molecular Biology, 1012, 213–219.
Frauwirth, K. A., et al. (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity, 16(6), 769–777.
Michalek, R. D., et al. (2011). Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proceedings of the National Academy of Sciences of the United States of America, 108(45), 18348–18353.
Wang, R., & Green, D. R. (2012). Metabolic checkpoints in activated T cells. Nature Immunology, 13(10), 907–915.
Michalek, R. D., et al. (2011). Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. Journal of Immunology, 186(6), 3299–3303.
Jacobs, S. R., et al. (2008). Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. Journal of Immunology, 180(7), 4476–4486.
Carr, E. L., et al. (2010). Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. Journal of Immunology, 185(2), 1037–1044.
Murray, C. M., et al. (2005). Monocarboxylate transporter MCT1 is a target for immunosuppression. Nature Chemical Biology, 1(7), 371–376.
Doedens, A. L., et al. (2013). Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nature Immunology, 14(11), 1173–1182.
Finlay, D. K., et al. (2012). PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. The Journal of Experimental Medicine, 209(13), 2441–2453.
Chou, C., et al. (2014). c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nature Immunology, 15(9), 884–893.
Kim, J. W., et al. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metabolism, 3(3), 177–185.
Papandreou, I., et al. (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metabolism, 3(3), 187–197.
Shi, L. Z., et al. (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. The Journal of Experimental Medicine, 208(7), 1367–1376.
Harty, J. T., & Badovinac, V. P. (2008). Shaping and reshaping CD8+ T-cell memory. Nature Reviews. Immunology, 8(2), 107–119.
Rosenblum, M. D., Way, S. S., & Abbas, A. K. (2016). Regulatory T cell memory. Nature Reviews. Immunology, 16(2), 90–101.
van der Windt, G. J., & Pearce, E. L. (2012). Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunological Reviews, 249(1), 27–42.
Crespo, J., et al. (2013). T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Current Opinion in Immunology, 25(2), 214–221.
Munn, D. H., & Mellor, A. L. (2013). Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends in Immunology, 34(3), 137–143.
Munn, D. H., et al. (1999). Inhibition of T cell proliferation by macrophage tryptophan catabolism. The Journal of Experimental Medicine, 189(9), 1363–1372.
Fischer, K., et al. (2007). Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood, 109(9), 3812–3819.
Cham, C. M., et al. (2008). Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. European Journal of Immunology, 38(9), 2438–2450.
Bose, S., Zhang, C., & Le, A. (2021). Glucose metabolism in cancer: The Warburg effect and beyond. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_1
Gatenby, R. A., & Gillies, R. J. (2004). Why do cancers have high aerobic glycolysis? Nature Reviews. Cancer, 4(11), 891–899.
Warburg, O. (1956). On the origin of cancer cells. Science, 123(3191), 309–314.
Chang, C. H., et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell, 162(6), 1229–1241.
Ho, P. C., et al. (2015). Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell, 162(6), 1217–1228.
Sugiura, A., & Rathmell, J. C. (2018). Metabolic barriers to T cell function in tumors. Journal of Immunology, 200(2), 400–407.
D’Aloia, M. M., et al. (2018). CAR-T cells: The long and winding road to solid tumors. Cell Death & Disease, 9(3), 282.
Mikucki, M. E., et al. (2015). Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nature Communications, 6, 7458.
Stewart, M. D., & Sanderson, R. D. (2014). Heparan sulfate in the nucleus and its control of cellular functions. Matrix Biology, 35, 56–59.
Caruana, I., et al. (2015). Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nature Medicine, 21(5), 524–529.
Tahmasebi, S., Elahi, R., & Esmaeilzadeh, A. (2019). Solid tumors challenges and new insights of CAR T cell engineering. Stem Cell Reviews and Reports, 15(5), 619–636.
Wang, L. C., et al. (2014). Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunology Research, 2(2), 154–166.
Nishio, N., & Dotti, G. (2015). Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology, 4(2), e988098.
Whilding, L. M., et al. (2019). CAR T-cells targeting the integrin alphavbeta6 and co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (Basel), 11, 5.
Hosen, N., et al. (2017). The activated conformation of integrin beta7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nature Medicine, 23(12), 1436–1443.
Posey, A. D., Jr., et al. (2016). Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity, 44(6), 1444–1454.
Zhou, R., et al. (2019). CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Frontiers in Immunology, 10, 1149.
Koneru, M., et al. (2015). IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology, 4(3), e994446.
Zhang, L., et al. (2013). Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Therapy, 20(5), 575–580.
Mohammed, S., et al. (2017). Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Molecular Therapy, 25(1), 249–258.
Adachi, K., et al. (2018). IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nature Biotechnology, 36(4), 346–351.
Arab, S., & Hadjati, J. (2019). Adenosine blockage in tumor microenvironment and improvement of cancer immunotherapy. Immune Network, 19(4), e23.
Beavis, P. A., et al. (2017). Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. The Journal of Clinical Investigation, 127(3), 929–941.
Srivastava, S., & Riddell, S. R. (2018). Chimeric antigen receptor T cell therapy: Challenges to bench-to-bedside efficacy. Journal of Immunology, 200(2), 459–468.
Arab, S., et al. (2017). Increased efficacy of a dendritic cell-based therapeutic cancer vaccine with adenosine receptor antagonist and CD73 inhibitor. Tumour Biology, 39(3), 1010428317695021.
Ligtenberg, M. A., et al. (2016). Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. Journal of Immunology, 196(2), 759–766.
Ninomiya, S., et al. (2015). Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood, 125(25), 3905–3916.
Newick, K., Moon, E., & Albelda, S. M. (2016). Chimeric antigen receptor T-cell therapy for solid tumors. Molecular Therapy Oncolytics, 3, 16006.
Scheffel, M. J., et al. (2016). Efficacy of adoptive T-cell therapy is improved by treatment with the antioxidant N-acetyl cysteine, which limits activation-induced T-cell death. Cancer Research, 76(20), 6006–6016.
Peggs, K. S., et al. (2009). Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. The Journal of Experimental Medicine, 206(8), 1717–1725.
Ren, J., et al. (2017). A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget, 8(10), 17002–17011.
John, L. B., et al. (2013). Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clinical Cancer Research, 19(20), 5636–5646.
Rupp, L. J., et al. (2017). CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports, 7(1), 737.
Liu, X., et al. (2016). A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Research, 76(6), 1578–1590.
Yoon, D. H., et al. (2018). Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): Combination or built-In CAR-T. International Journal of Molecular Sciences, 19, 2.
Fourcade, J., et al. (2012). CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Research, 72(4), 887–896.
Boice, M., et al. (2016). Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell, 167(2), 405–418. e13.
Johnston, R. J., Yu, X., & Grogan, J. L. (2015). The checkpoint inhibitor TIGIT limits antitumor and antiviral CD8(+) T cell responses. Oncoimmunology, 4(9), e1036214.
Kuhn, N. F., et al. (2019). CD40 ligand-modified chimeric antigen receptor T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell, 35(3), 473–488. e6.
Kershaw, M. H., et al. (2002). Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Human Gene Therapy, 13(16), 1971–1980.
Long, A. H., et al. (2016). Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunology Research, 4(10), 869–880.
Zhou, Q., et al. (2010). Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood, 116(14), 2484–2493.
Markley, J. C., & Sadelain, M. (2010). IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood, 115(17), 3508–3519.
Yao, X., et al. (2012). Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood, 119(24), 5688–5696.
Spear, P., et al. (2012). Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-gamma and GM-CSF. Journal of Immunology, 188(12), 6389–6398.
Chmielewski, M., & Abken, H. (2017). CAR T cells releasing IL-18 convert to T-Bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Reports, 21(11), 3205–3219.
Clever, D., et al. (2016). Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell, 166(5), 1117–1131. e14.
Scharping, N. E., et al. (2016). The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity, 45(2), 374–388.
Wherry, E. J., & Kurachi, M. (2015). Molecular and cellular insights into T cell exhaustion. Nature Reviews. Immunology, 15(8), 486–499.
Vaddepally, R. K., et al. (2020). Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel), 12, 3.
Chang, K. C., et al. (2013). Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Critical Care, 17(3), R85.
Bengsch, B., et al. (2016). Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity, 45(2), 358–373.
Parry, R. V., et al. (2005). CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and Cellular Biology, 25(21), 9543–9553.
Patsoukis, N., et al. (2015). PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nature Communications, 6, 6692.
Riley, J. L. (2009). PD-1 signaling in primary T cells. Immunological Reviews, 229(1), 114–125.
Kleffel, S., et al. (2015). Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell, 162(6), 1242–1256.
Staron, M. M., et al. (2014). The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity, 41(5), 802–814.
Ma, S., et al. (2019). Current progress in CAR-T cell therapy for solid tumors. International Journal of Biological Sciences, 15(12), 2548–2560.
Juillerat, A., et al. (2017). An oxygen sensitive self-decision making engineered CAR T-cell. Scientific Reports, 7, 39833.
Petersen, C. T., & Krenciute, G. (2019). Next generation CAR T cells for the immunotherapy of high-grade glioma. Frontiers in Oncology, 9, 69.
Dwyer, C. J., et al. (2019). Fueling cancer immunotherapy with common gamma chain cytokines. Frontiers in Immunology, 10, 263.
Gomez-Eerland, R., et al. (2014). Manufacture of gene-modified human T-cells with a memory stem/central memory phenotype. Human Gene Therapy Methods, 25(5), 277–287.
Eil, R., et al. (2016). Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature, 537(7621), 539–543.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Jung, J.G., Le, A. (2021). Metabolism of Immune Cells in the Tumor Microenvironment. In: Le, A. (eds) The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology, vol 1311. Springer, Cham. https://doi.org/10.1007/978-3-030-65768-0_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-65768-0_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65767-3
Online ISBN: 978-3-030-65768-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)