
Abstract
We propose a novel method which we call the Probabilistic Infection Model (PIM). Instead of stochastically assigning exactly one state to each agent at a time, PIM tracks the likelihood of each agent being in a particular state. Thus, a particular agent can exist in multiple disease states concurrently. Our model gives an improved resolution of transitions between states, and allows for a more comprehensive view of outbreak dynamics at the individual level. Moreover, by using a probabilistic approach, our model gives a representative understanding of the overall trajectories of simulated outbreaks without the need for numerous (order of hundreds) of repeated Monte Carlo simulations.
We simulate our model over a contact network constructed using registration data of university students. We model three diseases; measles and two strains of influenza. We compare the results obtained by PIM with those obtained by simulating stochastic SEIR models over the same the contact network. The results demonstrate that the PIM can successfully replicate the averaged results from numerous simulations of a stochastic model in a single deterministic simulation.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Two popular approaches for modeling infectious diseases are the simulation of disease spread through stochastic agent-based modelling; and the use of deterministic meta-population models [1, 10]. Stochastic agent-based models represent specific individuals or groups of individuals as agents. Each agent’s actions are governed by a set of rules which may themselves be functions of each agent’s characteristics, or of each agent’s environment. Interactions between pairs of agents which emerge as each one follows these rules establishes a contact network through which infectious disease can spread.
Many meta-population models use a system of differential equations to approximate the rate of change of the number of individuals in each disease state (e.g., susceptible, infected, etc.). The mathematical description of the SEIR (Susceptible, Exposed, Infected, and Recovered) frameworkFootnote 1 is given in Fig. 1. S, E, I, and R represent the number of individuals in Susceptible, Exposed, Infected, and Recovered states respectively. The total population is then given by \(N=S+E+I+R\). Parameter \(\beta \) is the proportion of contacts between members of S and members of I that lead to disease transmission. Parameter \(\sigma \) is the rate at which the exposed become infected. Parameter \(\gamma \) is the recovery rate at which the infected transition to the recovered state.

A pictoral representation of the SEIR model, along with the modeling equations.
Meta-population disease models are computationally efficient due to their deterministic nature. Further, closed form approximations of significant epidemiological parameters such as the basic reproduction number \(R_0\) (i.e. the expected number of secondary cases caused by a single infectious individual in a completely susceptible population) can be derived analytically using meta-population models. However, these models assume a homogeneous mixing rate within a homogeneous population. Thus, they do not take into account the diversity of a population which could lead certain individuals to have more contacts than others.
The stochastic agent-based approach incorporates population heterogeneity which could lead to variations in the numbers of contacts corresponding to each individual. Modeling interactions between pairs of individuals allows for flexibility in dictating specific patterns of behavior for individual agents. These models use stochastic processes to decide which contacts (i.e., edges) represented in the network lead to state transitions of agents (i.e., vertices) from Susceptible to Exposed at each simulated time step. However, due to the reliance on stochastic processes, a single run of an outbreak simulation using these models is not representative of an expected outcome. Thus, these models often require hundreds of repeated trials per unique set of parameters in order to properly estimate trends in the data. The computation required for this repetition of trials limits the scope of the analysis that can feasibly be done using these models. Analysis using stochastic models is complicated further by the fact that it is difficult to derive closed-form expressions for important quantities such as the basic reproduction number \(R_0\) without direct experimentation.
Our work is motivated by the advantages and drawbacks of these two popular epidemiological models. We introduce the Probabilistic Infection Model (PIM), which combines the heterogeneity of the stochastic models with the computational efficiency and deterministic nature of the meta-population models. The key idea of PIM is to calculate for each vertex in a contact network, the probabilities of the four SEIR states associated with that vertex. To compute the probability function, we leverage the research conducted in escape probabilities by Thomas and Weber [16]. The probabilities for each state and each vertex are compounded over windows of time corresponding to the latent and infectious periods of the given disease. This allows for probabilistic values of different states over time at the individual level and also provides the expected values of the sizes of the SEIR sub-populations corresponding to each state. As an added advantage, our proposed PIM model allows us to compute an expression for \(R_0 (v_0)\), which yields the value of \(R_0\) for specific single infective individuals in an otherwise susceptible contact network.
We applied our model to a contact network created from class enrollment data from the University of North Texas. We conducted our experiments with three sets of disease parameters and compared the results with those produced by the stochastic models. Our results demonstrate that the PIM simulations are similar to those produced by averaging trials from Monte Carlo models. This similarity is most notable when simulating diseases that are highly infectious.
2 The Probabilistic Infection Model
In this section we describe our proposed Probabilistic Infection Model. In Table 1, we provide a list of the terms that we use in our computations, along with their definitions. In the standard stochastic model, for a given contact event, a vertex selects a single neighbor in the network to simulate a contact. Due to the stochasticity of the model, the simulation must be run multiple times to estimate how population sizes for each SEIR state change over simulated time.
In our probabilistic infection model, contact events occur between adjacent vertices. Thus, all neighbors of a specific vertex have a probability to make a contact. For any given contact event, we set the contact probability per pair of vertices to be proportional to the weight of their corresponding edge. The probability that vertex v will be contacted by vertex u as a result of a single contact expended by u is \(\varPsi (u,v)=\frac{w(u,v)}{\sum \limits _{x \in N(v)} w(u,x)}\); w(u, v) is the weight of the edge (u, v) and N(v) is the set of neighbors of vertex v. Note that this function is not commutative. The probability of a contact from vertex u to vertex v, will differ from the probability of a contact from vertex v to vertex u, depending on each vertex’s number of neighbors and weights of the adjacent edges.
Each time v is contacted by an infectious individual u, there is a transmission probability T(u, v). The probability that vertex v is infected by u on day t as a result of a single contact made by u is then given by
i.e. the product of the probability of contact between u and v, the probability the u is infected on day t, and the transmission probability between u and v.
Lemma 1
Given that a vertex v is in the exposed state, i.e. \(E_x(v)>0\) and \(I_x(v)=0\) on day x, v will have \(I_t(v) > 0\), i.e. be in an infectious state on day t for some \(t >x\), only if it was contacted by an infectious vertex within the critical infection window of \(t- (\gamma _v +\sigma _v )+1\) and \(t-\sigma _v\).
Proof
We note that since each partial infection received by v has a latent period \(\sigma _v\), the infection probability of v, for a day r prior to day t, will remain unchanged for \(t-\sigma _v +1 \le r \le t\). Moreover, because the infectious period is \(\gamma _v\), any infections that arose from interactions made by v on or before day \(t-(\gamma _v + \sigma _v\)) would have expired by day t. Thus, taking these together, the time between \(t- (\gamma _v +\sigma _v )+1\) and \(t-\sigma _v\) is the critical infection window where an infectious contact will take v to an infectious state on day t.
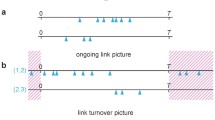
Figure 2 depicts how this critical window affects the state of the vertex. For ease of explanation, we consider the probabilities in this example to be 0 or 1, which can occur if there is only one successful infectious contact. Consider the vertex v to be in an exposed state \((E_x(v)=1)\) . In Case 1, if an infectious contact occurs within the critical infection window, then v will be in an infected state \((I_t(v)=1)\) on day t. If, Case 2, the infectious contact occurs after the critical infection window then v will remain in exposed state \((E_t(v)=1)\) on day t. If, Case 3, the infectious contact occurs before the critical infection window then v will be in recovered state \((R_t(v)=1)\) on day t.

A pictoral representation of the duration of infections with respect to the critical infection window
2.1 Computing the Probability for Each State
We now derive the expressions for computing the probability of each state for a given vertex v and a day t. We assume at the beginning of the simulation, i.e. at day 0, all vertices are either completely (with 100% probability) in the susceptible state or in the infected state.
Let \(\varOmega _t(u)\) denote the number of contacts that u makes on day t. The probability of v not being infected due to one contact made by u on day t is \(1-\delta _{t}(u,v)\). Taking all neighbors of v, the probability that v is not infected by any of the neighbors is \(\prod \limits _{u \in N(v)}(1-\delta _{t}(u,v))^{\varOmega (u)}\), where we make the approximation that each event where vertex v is not infected by some contact is independent.
Susceptible State: The probability that the vertex is in a susceptible state is the probability that v is not infected by any of the neighbors since day 0 to current day t. Thus;
Exposed State: Any susceptible vertex that was infected \(\sigma _v\) (the incubation period) days earlier will be exposed. Thus the probability of the exposed state is the probability of being in the susceptible state on day \(max(0, t - \sigma _ v)\) minus the current probability of the susceptible state on day t.
Infectious State: Any susceptible vertex that was infected \(\sigma _v\) + \(\gamma _v\) (the incubation period + infectious period) days earlier will be in an infectious state. The probability of the exposed state is the probability of being in the susceptible state on day \(max(0, t-\sigma _v)\) minus the current probability of the exposed state on day t.
Recovered State: Any susceptible vertex that was infected before the critical infection window \(t-(\sigma _v + \gamma _v)\) will have recovered by day t. The probability of the recovered state is 1 minus the probability that the vertex was still susceptible \(\gamma _v + \sigma _v\) days prior.
The total number of individuals ever infected at the end of an outbreak can be computed by several methods. One method is to take the expected number of recovered individuals by summing over \(R_L(v)\) for all v, where L is the last day of the simulation. Another way to approximate this quantity is to integrate the expected number of infected individuals \(\sum _{v \in V(G)} I_t(v)\) over time and divide the result by the disease’s infectious period to account for over-counting. Since time is counted in discrete steps, this integral can be reduced to a sum.
Thus, given an outbreak of length L in days;
This is satisfied in standard Monte Carlo models as well as in our PIM model.
Moreover, using PIM, we can calculate the value of the basic reproduction rate, \(R_0\), for a specific single infective \(v_0\) in a contact network where all other vertices are susceptible, as follows:
Here the \(\delta \) factor is replaced by just the product of transmission and contact probabilities, as \(I_n(v_0) =1\) for \(0 \le n < \gamma _{v_0}\).
2.2 Infection Redundancy Correction
One critical issue in using the PIM model is the effect of infection redundancy. This problem is illustrated in Fig. 3. Consider on day t, vertex v is exposed to the infection \(\delta _{t}(u,v)\) through contact with vertex u. Once v reaches an infected state on day \(t+\sigma _v\), it will expose vertex u to the infection \(\delta _{t+\sigma _v}(v,u)\). However, note that some of the infections contributing to the value of \(I_{t+\sigma _v}(v)\) have originated from u. This will result in u compounding its own probability of being infected, by incurring these redundant infections.

An illustration of the infection redundancy problem.
In order to correct this effect, we modify the infection from vertex u to vertex v by correcting each \(\delta _{t}(u,v)\) to only factor in u’s probability of being infectious as a result of contacts from vertices other than v. This ensures that infections originating from u will not be returned to u by any of u’s direct neighbors. Making this correction will improve the accuracy provided by PIM at the expense of computation time.
To calculate this, consider
and
Then X represents the probability that u was not infected in the critical infectious window by any of its neighbors (using the same logic as calculating for \(S_t(v)\) earlier). Y represents the probability that u was not infected in the critical infectious window by vertex v. Since the values are given as products, the ratio of \(\frac{X}{Y}\) approximates the probability that u was not infected in the critical infectious window by any of its neighbors and also discards the effect of infections from v. The probability that u is infected as a result of contacts with vertices other than v is then given by \(1-\frac{X}{Y}\). We thus modify the probability that v is infected by u on day t as a result of a single contact made by u to obtain
where the factor representing the probability that u was infectious on day t has been modified to prevent infection redundancy. We note that this is an approximate correction, as it is still possible for an infection to return to its source after passing through multiple vertices. Since an infection moving down a path of vertices gets exponentially smaller in magnitude as the length of the path increases, it is expected that the effect would be increasingly negligible for higher order corrections.
3 Experimental Results
In this section we present our experimental results of comparing the simulation of PIM with the stochastic Monte-Carlo simulations.
Constructing the Contact Network. Creating a reliable contact network presents a challenge in computational epidemiology [7]. This is because such as traditional methods of determining contacts such as surveys or sensor based tracking cannot scale. Surveys are also affected by recall bias, where part participants may not remember all of their contacts. As a solution to this problem, we observe that many of the daily routines of individuals are based on scheduled activities, such as going to meetings, going to appointments, attending classes etc. Thus if we have information about these scheduled activities we can create a reliable network of most of the frequently occurring contacts. Based on this assumption, we created a contact network of students based on the class-enrollment data for the Fall 2016 semester at the Discovery Park campus of the University of North Texas.
Our data contained information of 3700 students. Each student was assigned a randomly generated id to identify them uniquely, as well as to anonymize the data. The dataset contained the student ids and the classes in which each student was enrolled. Online classes and classes without regular meeting times were excluded. From this data, we constructed a graph where each student was a vertex, and two vertices (students) were connected by an edge if the corresponding students shared a class. The weight of an edge was the average duration of shared class time between the students.
3.1 Experiment Parameters
Experimentation was done with the parameters described in Table 1, and were run with the graph constructed from class-enrollment data. For each vertex v, 3 contacts were given per hour of average time spent in class over all weekdays by the student represented by v. Of the disease-specific parameters, the incubation and infection rates, measles parameters were adapted from [8, 15], whereas influenza parameters were adapted from [2, 4, 6]. Two sets of parameters were chosen for influenza that varied in length of incubation and infectious periods. We used the same values of \(\sigma \), \(\gamma \) and T for all vertices and edges.
In PIM simulations, a single vertex \(v_0\) was selected to be infected, with \(I_n(v_0)=1\) for \(0 \le n < \gamma \), and \(R_n(v_0)=1\) for \(n \ge \gamma \). The remaining vertices were initially completely susceptible. The probability values of the states of each vertex were obtained by computing the functions given in Eqs. 2–5 over the time period. The number of infected individuals at time t in days was determined by summing over \(I_t(v)\) for all \(v \in V(G)\). We terminated each simulation after day t if outbreak activity was sufficiently small, i.e. the total number of vertices with high probability of exposed and infected states was small. We quantitatively measured this using the following conditions:
The simulations were terminated if both these conditions were satisfied. In addition, simulations were not terminated before day 20. These bounds were selected to ensure that simulations do not end prematurely. Figure 4 shows the state of the vertices in the network as per the PIM model, on day 35. As can be seen, the measles epidemic spreads faster and takes longer time to recover (more red and less green nodes) than the influenza models.

States of the vertices in the contact network based on the PIM model on day 35. Yellow vertices are fully susceptible, whereas redder vertices have a higher probability of being infected at a given time. Green vertices have a probability of 95% or greater of being recovered. From left to right, the values are for Measles (left), Influenza 1 (middle) and Influenza 2 (right). (Color figure online)
In simulations using the stochastic model, the same graph, seed vertex of infection and parameters were used. 100 trials were run with a seeded random number generator for each of the three disease parameters. Contacts between vertices occurred randomly, with the probability of contact between vertices u and v for any given contact event proportional to w(u, v). Disease transmission occurred with probability T at the time of a successful contact between a susceptible and infectious individual.
3.2 Results
The results demonstrate that PIM produces results most similar to those produced by stochastic Monte Carlo models for diseases that are more highly infectious. As seen in Table 3, the Monte Carlo model and PIM produced similar values for the total number of infected individuals in an outbreak. Additionally, while the peak number of infected individuals and day of peak infection produced by PIM tended to be within one standard deviation of the mean values produced by the Monte Carlo trials, for all disease parameters, PIM outbreaks peaked slightly earlier and higher than the average Monte Carlo trial. This becomes more apparent when the parameters for less infectious diseases are used.
We believe that earlier peaks are observed partially due to an artifact of the stochastic method. In stochastic trials with low parameters, no outbreak of the disease is likely to be observed until multiple days have passed. Outbreak trials with peaks that are lower, occur later and show greater variance in the peak day of infection are observed as a result. This contrasts with PIM, which allows the seed of infection to partially contact multiple neighbors concurrently, possibly causing slightly earlier and higher peaks of infection. In addition, the approximation that events are independent may propel the initial spread of infection at a slightly greater rate, an effect that would be most noticeable for less infectious diseases.
Figure 5 demonstrates that the attributes of the SEIR curves produced by PIM are similar to those of the average outbreak curves obtained from 100 stochastic trials. This similarity is most notable in simulations of highly infectious diseases, such as when using the parameters for measles; in Fig. 6 left, we show the simulation time series using measles parameters for all four states, showing that the PIM model closely follows the averaged curves of 100 trials of the stochastic model. In addition, we compare the infectious state probability curves of individual vertices produced by the PIM model: Fig. 6 right shows the \(I_t(v)\) curves produced by PIM for the seed infected node as well as for 100 vertices that were randomly sampled from the set of initially susceptible vertices for the measles simulation. Most vertices reached their peak probability of being infected around day 38, which is consistent with the peak day of infection given in Fig. 5.

A comparison between PIM and 100 simulations of the stochastic SEIR model with respect to the number of infectious individuals over the entire simulation. From left to right, the curves are for Measles (left), Influenza 1 (middle) and Influenza 2 (right). (Color figure online)

State of vertices in the measles simulation. Left: Comparison between the number of vertices in each state over time for PIM and the Monte Carlo (MC) method averaged over 100 trials. Right: Probability of infection of 100 randomly selected vertices of the network. The peak occurs around days 35–45.

The percent difference between the peak number of infected individuals is shown for simulations for measles produced by PIM with and without backflow correction for every possible initially infectious \(v_0\).
Influence of Correction Parameter. We now test by how much the correction due to redundant infection (as discussed in Sect. 2.2) affects the simulations. Figure 7 shows a comparison between simulations with PIM when correction the probability of vertex v infecting vertex u uses the modified version as in Eq. 7, and one where the original Eq. 1 is used. For each \(v_0\), the percent difference between the peak number of infected individuals produced by PIM with and without correction was less than \(0.2 \%\), suggesting that one-level-deep backflow correction is a sufficient approximation.
4 Related Research
Computational epidemics is an active area of research. Several software tools for simulating disease over a population have been developed including EpiSims [9] and DiSimS [5] that use high performance computing, and Broadwick [14] which uses a sequential, but modular framework that can be modified for various disease parameters. Our PIM method can also be implemented to be parallel, and thus can be executed on large networks.
The challenges of creating reliable contact networks are discussed in [7]. In 2008 [13], a cross-sectional survey on 7,290 participants conducted by different public health institutes or commercial companies was conducted to build a contact network. Another study [12], performed through the 2009 H1N1 flu pandemic on a population of 36 people based on communication using sensors. However, neither of these methods are scalable as compared to our method of utilizing scheduled data. Recent studies have also looked into the dynamic contact networks [3] and the effect of misinformation in developing contact networks [11].
5 Conclusion and Future Work
In this paper, we introduce a probabilistic infection model for simulating the spread of infectious diseases on contact networks. Our model encapsulates the advantages of both deterministic meta-population models as well as stochastic models on contact networks. We further propose a method of obtaining contact networks based on the scheduled activities of individuals in specific environments (e.g., businesses, schools, etc.), and simulate our model on a contact network built from a university’s class enrollment data. Comparisons of the results obtained from stochastic modelling and PIM on the contact network of university students demonstrate that our approach produces similar results to the stochastic model, but with significantly reduced computational overhead. Moreover, our model gives a tractable framework for probabilistic analysis of outbreak dynamics at the individual level.
As part of our future work, we will experiment with latent periods, infectious periods and transmission probabilities selected from distributions rather than as static values. In addition, we will pursue further studies of vaccine distribution and other individual-level outbreak intervention strategies by applying PIM’s approximations for individual SEIR state-probabilities.
Change history
15 June 2020
The original version of this chapter was revised. The first author’s first name contained a typo. It has been corrected to “William”.
Notes
- 1.
Births and deaths among the population are not considered in the model.
References
Ajelli, M., et al.: Comparing large-scale computational approaches to epidemic modeling: agent-based versus structured metapopulation models. BMC Infectious Diseases
Balcan, D., et al.: Seasonal transmission potential and activity peaks of the new influenza a (h1n1): a monte carlo likelihood analysis based on human mobility. BMC Med. 7(1), 45 (2009)
Bansal, S., Read, J., Pourbohloul, B., Meyers, L.A.: The dynamic nature ofcontact networks in infectious disease epidemiology. J. Biol. Dyn. 4(5), 478–489 (2010).https://doi.org/10.1080/17513758.2010.503376, pMID: 22877143
Cori, A., Valleron, A.J., Carrat, F., Tomba, G.S., Thomas, G., Boëlle, P.Y.: Estimating influenza latency and infectious period durations using viralexcretion data. Epidemics 4(3), 132 (2012)
Deodhar, S., Bisset, K.R., Chen, J., Ma, Y., Marathe, M.V.: An interactive, web-based high performance modeling environment for computational epidemiology. ACM Trans. Manage. Inf. Syst. 5(2), 7 (2014). https://doi.org/10.1145/2629692
Drewniak, K., Helsing, J., Mikler, A.R.: A method for reducing the severity of epidemics by allocating vaccines according to centrality. In: ACM Conference on Bioinformatics, Computational Biology, and Health Informatics (2014)
Eames, K., Bansal, S., Frost, S., Riley, S.: Six challenges in measuring contact networks for use in modelling. Epidemics 10, 72 – 77 (2015). https://doi.org/10.1016/j.epidem.2014.08.006,http://www.sciencedirect.com/science/article/pii/S1755436514000413, challenges in Modelling Infectious Disease Dynamics
Enanoria, W.T., et al.: The effect of contact investigations and public health interventions in the control and prevention of measles transmission: A simulation study
Halloran, M.E., et al.: Modeling targeted layered containment of an influenza pandemic in the United States. Proc. Nat. Acad. Sci. 105(12), 4639–4644 (2008). https://doi.org/10.1073/pnas.0706849105,https://www.pnas.org/content/105/12/4639
Henson, S., Brauer, F., Castillo-Chavez, C.: Mathematical models in population biology and epidemiology. Am. Math. Monthly 110(3), 1 (2003)
Holme, P., Rocha, L.E.C.: Impact of misinformation in temporal network epidemiology. Netw. Sci. 7(1), 52–69 (2019). https://doi.org/10.1017/nws.2018.28
Jain, S., Benoit, S.R., Skarbinski, J., Bramley, A.M., Finelli, L.: For the 2009 pandemic influenza A (H1N1) virus hospitalizations investigation team: influenza-associated pneumonia among hospitalized patients with 2009 pandemic influenza A (H1N1) virus-United States, 2009. Clinical Infectious Diseases 54(9), 1221–1229 (2012). https://doi.org/10.1093/cid/cis197
Mossong, J., Hens, N., Jit, M., Beutels, P., Auranen, K., Mikolajczyk, R.T., Massari, M., Salmaso, S., Tomba, G.S., Wallinga, J., Heijne, J.C.M., Sadkowska-Todys, M., Rosińska, M., Edmunds, W.J.: Social contacts and mixing patterns relevant to the spread of infectious diseases. PLoS Med. 5, 1083–1087 (2008)
O’Hare, A., Lycett, S., Doherty, T., Monteiro Salvador, L., Kao, R.: Broadwick: a framework for computational epidemiology. BMC Bioinformatics 17, 65 (2016). https://doi.org/10.1186/s12859-016-0903-2
Ponciano, J.M., Capistrán, M.A.: First principles modeling of nonlinear incidence rates in seasonal epidemics. PLoS Computat. Biol. 7(2), e1001079 (2011)
Thomas, J.C., Weber, D.J.: Epidemiologic Methods for the Study of Infectious Diseases. Oxford University Press, Oxford (2001)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this paper

Cite this paper
Qian, W., Bhowmick, S., O’Neill, M., Ramisetty-Mikler, S., Mikler, A.R. (2020). A Probabilistic Infection Model for Efficient Trace-Prediction of Disease Outbreaks in Contact Networks. In: Krzhizhanovskaya, V., et al. Computational Science – ICCS 2020. ICCS 2020. Lecture Notes in Computer Science(), vol 12137. Springer, Cham. https://doi.org/10.1007/978-3-030-50371-0_50
Download citation
DOI: https://doi.org/10.1007/978-3-030-50371-0_50
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-50370-3
Online ISBN: 978-3-030-50371-0
eBook Packages: Computer ScienceComputer Science (R0)