
Abstract
This personal perspective focuses on small-molecule inhibitors of proteostasis networks in cancer—specifically the discovery and development of chemical probes and drugs acting on the molecular chaperones HSP90 and HSP70, and on the HSF1 stress pathway. Emphasis is on progress made and lessons learned and a future outlook is provided. Highly potent, selective HSP90 inhibitors have proved invaluable in exploring the role of this molecular chaperone family in biology and disease pathology. Clinical activity was observed, especially in non small cell lung cancer and HER2 positive breast cancer. Optimal use of HSP90 inhibitors in oncology will likely require development of creative combination strategies. HSP70 family members have proved technically harder to drug. However, recent progress has been made towards useful chemical tool compounds and these may signpost future clinical drug candidates. The HSF1 stress pathway is strongly validated as a target for cancer therapy. HSF1 itself is a ligandless transcription factor that is extremely challenging to drug directly. HSF1 pathway inhibitors have been identified mostly by phenotypic screening, including a series of bisamides from which a clinical candidate has been identified for treatment of ovarian cancer, multiple myeloma and potentially other cancers.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
It seems very fitting that a collection of articles on proteostasis networks in cancer – and one that is dedicated to the extraordinary life and work of Sue Lindquist – should conclude with a piece on experience with preclinical and clinical translation in this area. I feel privileged to be invited to provide a short article on my reflections on progress made, lessons learned and an outlook for the future in this field. This brief commentary is written very much as a personal perspective – based on more than two decades of involvement working on chemical biology and drug discovery around HSP90, HSP70 and HSF1 as molecular targets. I will mainly illustrate the progress and challenges based on the work carried out in my own laboratory – in association with many talented colleagues and collaborators. I will also refer to the work of others, but I apologise to the many excellent scientists in the field that it is in the nature of a short personal perspective that exhaustive referencing is not possible.
Sue Lindquist was an outstanding scientist and true pioneer in the field of proteostasis. Her curiosity and great aptitude for the basic science coupled with a strong desire to make a difference to human disease led her to work and collaborate across many research boundaries. Such an approach is often critically important to make real breakthroughs in fundamental research and is certainly essential to translate basic research into clinical benefit. In this and in her contributions to the development of the field and the careers of scientists who work in it, Sue was an inspiration to us all.
2 Therapeutic Targeting of HSP90
My lab started working on the Heat Shock Protein (HSP) and molecular chaperone HSP90 in the mid-to-late 1990’s as a serendipitous result of the convergence of the two main strands of our molecular pharmacology and drug discovery research at the time. The first strand was the design of drugs that would selectively kill cancer cells by virtue of their preferential bioreductive conversion to cytotoxic metabolites – either as a result of the hypoxic nature of solid tumours or/and the increased expression of enzymes that catalyse their bioreductive activation (Workman and Stratford 1993). The second strand was the design of drugs that exploit and counteract the oncogenic pathways involved in the maintenance and progression of cancer, now referred to as ‘oncogene addiction’ (Brunton and Workman 1993).
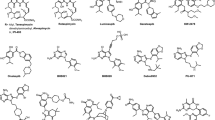
The coalescing of these two strands is illustrated by a study carried out in my lab to test two hypotheses to explain the molecular mode of action by which the natural product geldanamycin – a member of the benzoquinone ansamycin class of natural product antibiotic – exerts its nonclinical anticancer activity in human colorectal cancer cells. See Fig. 1 for the chemical structure of geldanamycin and other selected compounds and drugs referred to in this article. The two possibilities we tested were that the anticancer effect was mediated either through: (1) bioreductive activation of the quinone moiety catalysed by the flavoenzyme oxidoreductase NAD(P)H: quinone oxidoreductase (EC 1.6.99.2; known as NQO1 and previously as DT-diaphorase); or (2) depletion of the cellular c-SRC oncoprotein as a result of inhibition of the molecular chaperone HSP90. These two hypotheses did not appear from thin air. Exemplified by research on another natural product called mitomycin C, the bioreductive metabolic activation of quinone-group containing agents by NQO1 and other oxidoreductases to produce DNA-binding metabolites was well precedented as an approach to killing cancer cells through the work of the groups of Alan Sartorelli (Sartorelli 1988) and later David Ross (Ross et al. 2000) and others. Indeed, our own lab showed that cancer cell sensitivity to both mitomycin C and the synthetic indoloquinone EO9 is determined by the level of expression of NQO1 – which is often very high in some cancer cell lines that tend to be sensitive, or alternatively low or absent in others that tend to be resistant (Walton et al. 1992; Fitzsimmons et al. 1996). The alternative hypothesis that we tested was that the anticancer effect of geldanamycin and related agents such as herbimycin is a result of the inhibition of the activity of cellular SRC – a kinase that has been implicated in colorectal cancer initiation and progression (Cartwright et al. 1990; Garcia et al. 1991).

Chemical structures of selected examples of compounds and drugs referred to in this article
In our initial study we largely ruled out that geldanamycin kills human colorectal cancer cells through either bioreductive metabolism by NQO1 or inhibition of cellular c-SRC kinase activity (Brunton et al. 1998). Firstly, we showed that although NQO1 does reduce geldanamycin this could not explain its anticancer activity. And secondly, we observed no effect on cellular c-SRC at pharmacologically active concentrations. We did observe a decrease in c-SRC kinase activity in the colorectal cancer cells at very high concentrations and prolonged exposure to geldanamycin, but this is due to a reduction in overall c-SRC protein levels. Indeed, there had been previous reports of depletion of SRC and other tyrosine kinase oncoproteins in response to geldanamycin and herbimycin A. Furthermore, in what is now regarded as a seminal discovery in the field published by Luke Whitesell working in Len Neckers lab, it was shown that the major target to which geldanamycin is bound in the cell is a 90 kDa protein that was identified as the heat shock protein HSP90 – and moreover it was demonstrated that this led to the disruption of the known interaction between HSP90 and the oncogenic viral form of the tyrosine kinase v-SRC (Whitesell et al. 1994). These findings explained the previously demonstrated ability of geldanamycin to revert the oncogenic transformation of mammalian cells by viral SRC, rather than inhibiting SRC’s intrinsic tyrosine kinase activity, and pointed to HSP90 as a potential target in cancer by bringing about the depletion of oncogenic client proteins by proteosomal degradation. Despite this interesting and attractive mechanism of action, geldanamycin did not progress to clinical studies due to concerns about liver toxicity.
However, semi-synthetic derivatives of geldanamycin subsequently showed more promise as drugs. Thus we later returned, with more translational success this time, to study both the impact of bioreductive metabolism of the quinone moiety and also the depletion of oncogenic kinases and other ‘client’ proteins of the HSP90 molecular chaperone following exposure of cancer cells to the analogue of geldanamycin that was commonly (and still is) referred to as 17-AAG (17-allylamino-17-demethoxygeldanamycin) – subsequently named tanespimycin. This drug was progressed to the clinic because it maintained the anticancer effect of geldanamycin but was better tolerated in animals and has an acceptable therapeutic index. In collaboration with colleagues at the US National Cancer Institute (NCI) we showed that in contrast to geldanamycin – but similar to the bioreductive quinones mitomycin C and EO9 (see above) – there is a clear correlation between sensitivity to tanespimycin and NQO1 expression in the NCI collection of 60 human cancer cell lines (the so-called ‘NCI 60’ panel), with greater sensitivity seen in those lines with high NQO1 and lower sensitivity in those with low expression, including lines with a polymorphism resulting in reduced enzyme activity and stability (Kelland et al. 1999). We confirmed a causal relationship in an isogenic pair model either lacking NQO1 or exhibiting high expression. The strong dependence of cancer cell sensitivity on NQO1 of tanespimycin in contrast to geldanamycin was explained by our demonstration in the same paper that 17-AAG is a much better substrate for NQO1 than geldanamycin. Subsequent work by the Ross lab and others showed that the reduced hydroquinone form of tanespimycin is a more potent binder of HSP90 than the parent quinone 17-AAG itself (Guo et al. 2005).
I would like to make three observations about our discovery of NQO1 as a mechanism-based biomarker of tanespimycin sensitivity. The first point is that although we emphasized in several publications that NQO1 expression should be considered when comparing the sensitivity of different cancer cell lines to tanespimycin in vitro, this predictive biomarker relationship is generally ignored. Note, however, that although important in cell culture, NQO1 is likely to be less important in animal (and human) studies due to the metabolism of tanespimycin to 17-amino-geldanamycin – which we showed behaves like geldanamycin in being independent of NQO1 (Kelland et al. 1999). The second point is that the NQO1 effect is, of course, absent from non-quinone HSP90 inhibitors and not relevant for these (ref. (Kelland et al. 1999) and see later). The third point is that it was pleasing when our demonstration of NQO1 as a predictor of cancer cell sensitivity to tanespimycin – that we made using the NCI-60 cell panel – was subsequently confirmed as one of the strongest predictive genetic-pharmacology relationships in the first two major publications on the much bigger panels of hundreds of cancer cell lines that are now commonly used for such correlative analysis and predictive biomarker discovery (Barretina et al. 2012; Garnett et al. 2012). We discussed these findings in a commentary (Workman et al. 2012).
In our first above-mentioned publication on tanespimycin (Kelland et al. 1999) we demonstrated depletion of CRAF and mutant p53 as representative client proteins in cancer cells and the increased expression of HSP72 as a representative heat shock gene product that is upregulated in an HSF1-dependent manner following HSP90 inhibition. Following earlier work on geldanamycin and another natural product radicicol by others, we showed in human colon cancer models that tanespimycin inhibits oncogenic signal transduction in the MAP kinase and PI3 kinase pathways through depletion of HSP90 clients such as CRAF and AKT, resulting in cytostasis and apoptosis (Hostein et al. 2001). Similar findings were made by other labs, including the Neal Rosen group, showing for example that tanespimycin induces G1 cell cycle arrest, apoptosis and morphological and functional differentiation in breast cancer cells and cell cycle arrest and antitumour activity in prostate cancer models, both associated with depletion of the highly sensitive oncogenic HSP90 client protein HER2/ERBB2, steroid hormone receptors and other client proteins (Münster et al. 2001; Basso et al. 2002; Solit et al. 2002). Both the Rosen team and also the Richard Marais lab in collaboration with our group discovered independently that many mutant forms of BRAF, including the common V600E mutant that is the key oncogenic driver of melanoma and other cancers, are more highly dependent on HSP90 and are more rapidly depleted by 17-AAG treatment (Grbovic et al. 2006; da Rocha Dias et al. 2005) – an effect subsequently seen with other mutant/wild type protein pairs.
Our lab was an earlier adopter of gene expression microarrays which we used to profile large-scale transcriptional changes in response to HSP90 inhibition (Clarke et al. 2000) and we also used mass spectrometry-based proteomics to profile broad changes in cellular proteins at scale (Maloney et al. 2007). Studies from many labs have detailed the depletion by proteosomal degradation of multiple oncogenic HSP90 client proteins following inhibition of the molecular chaperone. An online listing of the very large number of client proteins and other HSP90 interacting proteins is maintained by Didier Picard (HSP90 Interactors n.d.). A large-scale study from the Lindquist lab provided a systematic and quantitative survey – using the LUMIER assay system – of human kinases, transcription factors, and E3 ligases with respect to interaction with HSP90 and its cochaperone CDC37 (Taipale et al. 2012). This comprehensive analysis confirmed and enhanced our appreciation that many kinases interact with HSP90 but with different binding affinities – concluding (1) that kinase client binding specificity is dictated by CDC37 and (2) that the extent of depletion after HSP90 inhibition is greatest for strong binders and least for weak binders. A further conclusion was that the interaction of kinases with the HSP90/CDC37 system is dictated by the thermodynamic stability of their kinase folds. HSP90 client kinases are intrinsically less stable than non-client kinases. This finding helped us to understand why the ability to act as an HSP90 client protein cannot be predicted from the DNA or protein sequence. The study also revealed that relatively few transcription factors interact with HSP90, in contrast to the many E3 ligases and the majority of kinases (Taipale et al. 2012).
A number of protagonists supported the concept of progression of HSP90 inhibitors, including our own lab and those of Len Neckers, Neal Rosen, Ed Sausville, Gabriella Chiosis and Francis Burrows. For example, in ref. (Workman et al. 2007) a group of us reviewed the potential advantages of HSP90 inhibitors, especially the simultaneous and combinatorial degradation of multiple oncogenic client proteins, resulting in the blockade of multiple oncogenic pathways and antagonism of all of the pathological hallmark traits of malignancy – with cancer selectivity being achieved by exploiting cancer dependencies and vulnerabilities, including both oncogene addiction and the stressed state of malignant cells. We also argued that multiple downstream effects of HSP90 inhibitors should make the development of resistance more difficult compared to drugs exhibiting more limited effects. Of course the other side of the coin is the potential for significant toxicity, even though many of the affected proteins and pathways are more important to cancer compared to normal cells.
Working with our collaborators and supported by the US National Cancer Institute and what is now Cancer Research UK, it was exciting for us when we took tanespimycin into a first-in-human clinical trial in the Drug Development Unit of the Institute of Cancer Research and our hospital partner the Royal Marsden. To enable pharmacokinetic-pharmacodynamic (PK-PD) studies, we validated a protein biomarker signature of HSP90 inhibition, comprising depletion of HSP90 client proteins CRAF and CDK4 together with increased expression of HSP72 (Banerji et al. 2005a). CRAF and CDK were selected as biomarkers that are depleted, because of their applicability for measurement in peripheral blood lymphocytes (PBLs) and multiple cancers. In addition, HSP72 represents an ‘up’ biomarker that is robustly induced upon HSP90 inhibition across a wide range of biological contexts. It should be noted that HSP72 is a more sensitive biomarker of HSP90 inhibition, occurring at lower tanespimycin concentrations and doses; however, depletion of client proteins, which requires higher exposures, is more likely to be indicative of therapeutic effect. Evidence of HSP72 induction in the absence of client protein depletion is not sufficient to conclude that the degree of target modulation will have a reasonable chance of seeing a therapeutic effect. The combined biomarker signature of HSP72 induction and client protein depletion was used in our hypothesis-testing, PK-PK biomarker-led Phase I study of tanespimycin (Banerji et al. 2005b) that applied the Pharmacological Audit Trail (PhAT) framework that my colleagues and I developed for use in nonclinical and clinical studies to link target modulation to biological effects (Banerji and Workman 2016; Workman 2003). PK properties were consistent with target coverage and the PD biomarkers demonstrated HSP90 inhibition in PBLs and tissue biopsies. Our Phase I clinical study of tanespimycin provided proof-of-concept that HSP90 could be inhibited at doses that were tolerated by cancer patients. Dose-limiting side-effects were defined as diarrhoea and liver toxicity. Two patients with metastatic melanoma had stable disease for 15 and 49 months respectively; of note is that these patients’ tumours had a V600E BRAF and a G13D NAS mutation, respectively, and thus both cancers would have contained active forms of the HSP90 client protein BRAF (Banerji et al. 2008).
Tanespimycin progressed to a large number of Phase II studies and showed its most promising activity in combination with the HER2 antibody trastuzumab in patients with HER2-positive metastatic breast cancer who were progressing on trastuzumab (Modi et al. 2011). The trial definitively showed objective responses according to Response Evaluation Criteria in Solid Tumors (RECIST) in these patients and the overall response rate was 22% with a clinical benefit rate of 59%. The effectiveness of tanespimycin in HER2-driven cancers makes mechanistic sense given that HER2 is one of the most highly sensitive HSP90 client proteins to HSP90 inhibition. However, despite its promise the clinical development of tanespimycin as a cancer therapy was terminated by the company for nonclinical, potentially commercial reasons such as costly production/formulation and patent expiry concerns (Arteaga 2011; Neckers and Workman 2012; Garcia-Carbonero et al. 2013). Subsequently, early clinical trials were carried out with additional geldanamycin analogues – specifically alvespimycin which is less dependent on NQO1 (see earlier) and the soluble stabilized hydroquinone form of tanespimycin, retaspimycin, but these have not progressed further. Factors that have been considered as limitations for tanespimycin and its analogues include insufficient depletion of key client proteins, the requirement for activation by NQO1, and side-effects – including the liver toxicity that may have been related to the quinone moiety underdoing bioreductive activation, as discussed above (see also ref. (Neckers and Workman 2012)).
Nevertheless, the proof of concept and promising activity in breast cancer shown by tanespimycin stimulated research to discover second generation, wholly synthetic, non-quinone inhibitors that might overcome the limitations of the geldanamycin class. Two particular new classes initially led the way, namely (1) the purine inhibitors based on PU-3 from Chiosis and colleagues at Memorial Sloan Kettering leading to the clinical candidate PU-H71 and others from Conforma/Biogen (Chiosis et al. 2001; Speranza et al. 2018) and (2) the resorcinol-type compounds exemplified by ganetespib from Synta Pharmaceuticals (Ying et al. 2012) and luminespib from ICR/Vernalis (Eccles et al. 2008; Sessa et al. 2013). Numerous other structurally diverse inhibitors were subsequently discovered and many progressed to the clinic (Travers et al. 2012).
Our own drug discovery research on HSP90 began with our identification by high-throughput screening at the ICR of the diaryl pyrazole resorcinol CCT018159 (Cheung et al. 2005; Sharp et al. 2007a). Improvements on this chemical starting point were enabled by the crystal structure of HSP90 solved by our collaborator Laurence Pearl and colleagues (Prodromou et al. 1997) which revealed precisely how CCT018159 is bound at the nucleotide site in the N-terminal domain of HSP90, mimicking the binding mode of the resorcinylic natural product radicicol (Roe et al. 1999). Our structure-based optimization based on the screening hit CCT018159 yielded more potent, advanced leads and chemical tools, specifically the potent resorcinylic pyrazole/isoxazole analogues VER-49009 and VER-50589 (Sharp et al. 2007b) and in turn resulted in our intravenously administered clinical candidate, the 4,5-diaryl isoxazole resorcinol luminespib (Eccles et al. 2008; Brough et al. 2008). In addition, our collaboration between ICR and Vernalis yielded the oral back-up clinical candidate NVP-BEP800 from a 2-aminothieno[2,3-d]pyrimidine series, derived from a fragment-based and in silico hit-finding approach and optimized by structure-based design (Brough et al. 2009). Both series were licenced to Novartis.
Luminespib was the first of the second generation non-geldanamycin HSP90 inhibitors for which full results of a phase I study were reported (Sessa et al. 2013). In this Phase I clinical trial of intravenous luminespib led by Udai Banerji, the main side-effects were diarrhoea, asthenia/fatigue, anorexia, atrial flutter and visual symptoms and the recommended phase II dose (RP2D) was 70 mg/m2. Application of the PhAT showed that the plasma concentrations of luminespib that were achieved were consistent with those that produced therapeutic effects in a range of human tumour xenograft models. In addition, evidence of target modulation was obtained in peripheral blood mononuclear cells (dose-dependent HSP72 induction) and tumour tissue (HSP70 induction and depletion of client protein AKT in two ER-positive patients at 70 mg/m2). There were no radiological responses, which was attributed potentially to the fact that patients were not molecularly prioritized based on client protein status. However, dose-dependent metabolic responses were observed in a number of patients as measured by a reduction of 18F-fluorodeoxyglucose uptake by PET scan. Based on these Phase I data, Phase II studies were initiated with the dose of 70 mg/m2 in breast, gastric and non-small cell lung cancers with appropriate HSP90 client protein dependencies.
Evidence of activity of luminespib was observed in HER2-positive and ER-positive breast cancer (Schroder et al. 2011; Kong et al. 2016). A Phase II trial with luminespib in NSCLC included patient cohorts with EGFR-mutant, ALK-rearranged and KRAS-mutant disease (Felip et al. 2018). The overall response rate of 13% was modest and no objective responses were observed in patients with oncogenic KRAS mutation. In contrast, the most favourable outcome was seen in patients with oncogenic ALK rearrangement, for whom the objective response rate was 32%, although the median progression-free survival was less than 3 months and no activity was detected against brain metastases, which is a common problem in patients with ALK-rearranged NSCLC. Overall, it was concluded that luminespib had a manageable safety profile and is active in patients with both ALK rearrangement and EGFR mutations, including the gatekeeper mutant T790M, which exhibited resistance to EGFR tyrosine kinase inhibitors. Of interest was the activity of luminespib in a Phase II trial in NSCLC patients harbouring oncogenic EGFR exon 20 insertion mutants who at the time were refractory to available EGFR tyrosine kinase inhibitors, as also seen in nonclinical models (Piotrowska et al. 2018).
Ganetespib has progressed all the way to a Phase III study (known as (GALAXY-2) comparing the HSP90 inhibitor plus the taxane docetaxel versus docetaxel alone in advanced NSCLC (Pillai et al. 2017). The trial showed that there was no significant difference in median overall survival for the two arms (10.9 months for the combination versus 10.5 months with docetaxel alone. Of interest, ganetespib appeared to have lower ocular toxicity than luminepsib at the respective doses used, potentially related to the higher exposure of the retinal epithelium to the hydrophilic luminespib compared to the more hydrophobic ganetespib (see ref. (Piotrowska et al. 2018). A weakness of the trial design was that the patients were not molecularly stratified, for example to include only ALK-rearranged and EGFR-mutant. It is also possible that once or twice a week dosing may not give sufficient depletion of key oncogenic client proteins. A commentary on the study (Pillai and Ramalingam 2018) suggested that newer orally administered HSP90 inhibitors could be trialled in molecular stratified patients with NSCLC, particularly if it was possible to dose the drug so as to maintain continued suppression of the relevant HSP90 client oncoproteins. However, the authors pointed out that there are now a range of kinase inhibitors and immunotherapies in NSCLC, together with chemotherapy combinations, which may potentially decrease the interest in pursuing HSP90 inhibitors in this setting at this time (Pillai and Ramalingam 2018).
In a retrospective review of 158 patients treated with various HSP90 inhibitors (tanespimycin, alvespimycin, retaspimycin, ganetespib and CNF2024) at Memorial Sloan Kettering Cancer Center, sufficient tissue to allow biomarker assessment was available for 51 patients and it is noteworthy that 13 of 16 responses strongly correlated with HER2-positive status (Jhaveri et al. 2016). The authors concluded that their findings were consistent with the preclinical data demonstrating that HER2 is the most sensitive client protein of HSP90 inhibition and suggested that this biomarker requires prospective validation in larger studies.
Overall, the experience to date with HSP90 inhibitors in the clinic is that doses can be given that provide evidence of pharmacological inhibition of HSP90. However, greater reliance on HSP72 as a PD biomarker and relatively little information on depletion of oncogenic client proteins, which requires higher exposures, means that it remains unclear whether in the trials described above the key client oncoproteins have been depleted to a sufficient extent and for adequate time to reveal the full therapeutic potential of HSP90 inhibitors. Concerning side-effects, the first-generation geldanamycin analogues exhibited liver toxicity that is potentially related to the quinone moiety – whereas ocular effects, typified by reversible night blindness, blurred vision, and flashing lights, were more of a limitation with non-geldanamycin inhibitors, to a degree that varies between the different second generation inhibitors and may relate to physical properties and retinal exposure.
In a review of HSP90 inhibitors in 2015, we focused on potential approaches to maximize the therapeutic potential of these agents (Butler et al. 2015). Possibilities included optimizing dosing and schedule through use of PD biomarkers; dissecting and exploiting the complex molecular and cellular response to HSP90 inhibition, including effects of co-chaperones; and use of combinatorial drug strategies, including the application of a one-two punch approach – employing a combination of drugs that directly inhibit the function of a key client protein (eg. kinase activity using a kinase inhibitor) together with an HSP90 inhibitor to deplete the overall protein level. Of interest in this regard is our observation that ATP-competitive protein kinase inhibitors can, as single agents, deplete the levels of the target kinase by blocking protein kinase recruitment to the HSP90-CDC37 system (Polier et al. 2013). In support of the kinase-HSP90 inhibitor one-two punch idea, it has been shown that inhibition of HSP90 by the resorcinol drug onalespib delays the emergence of resistance to BRAF kinase inhibition and overcomes resistance to dual BRAF and MEK inhibition in melanoma models (Smyth et al. 2014). In addition, relatively low level HSP90 inhibition blocks the emergence of resistance to anti-oestrogens in breast cancer models (Whitesell et al. 2014). Note also that we have shown that onalespib blocks mRNA splicing of androgen receptor variant 7 in prostate cancer cells (Ferraldeschi et al. 2016). This suggests an approach to overcome resistance mediated via the splice variant, in addition to the more conventional HSP90 inhibitor mechanism of proteasomal depletion of the wild type receptor.
Efforts continue to design novel classes of HSP90 inhibitors with distinct properties. One example is the identification of inhibitors with different HSP90 paralog selectivity profiles (Huck et al. 2019). Another is a series of inhibitors based on the novobiocin structure that bind at or close to the HSP90 C-terminal dimerization domain, potentially involving a putative second nucleotide-binding pocket, from which some analogues are reported to have reduced propensity to induce the heat shock response (Neckers et al. 2018 and see Chap. 9). In addition, we need to remain alert to the identification of cancers not considered previously that might be responsive to HSP90 inhibitors. A possible example is our recent demonstration of the promising activity of HSP90 inhibitors in nonclinical models of bile duct cancers known as cholangiocarcinomas – for which microRNA 21 (miRNA21) appears to mediate resistance by decreasing levels of the DnaJ Heat Shock Protein family (Hsp40) member B5 (DNAJB5) (Lampis et al. 2018). It was suggested that miRNA21 could be a potential marker of sensitivity for HSP90 inhibitor sensitivity in these hard-to-treat cancers (Lampis et al. 2018).
3 Therapeutic Targeting of HSP90 Co-Chaperones, HSP70 and HSF1
It was mentioned above that it may be possible to exploit the complex molecular and cellular response to HSP90 inhibition to enhance therapeutic effectiveness or provide alternative therapeutic targeting. Our lab (often in collaboration with the Pearl group) and others have explored the consequences of depleting co-chaperones such as CDC37 (Smith et al. 2009, 2015), AHA1 (Holmes et al. 2008) and the E3 ubiquitin ligase Cullin-5 (Samant et al. 2014). We showed that depletion of CDC37 sensitizes cancer cells to HSP90 inhibitors and leads to client kinase depletion – but greatly reduced inhibition of binding of CDC37 to HSP90 inhibition does not. Indeed, rather surprisingly, we demonstrated that CDC37 is able to stabilise kinase clients by a mechanism that is not dependent on a substantial direct interaction between CDC37 and HSP90, but nevertheless requires HSP90 activity. These results indicate that pharmacological inhibition of CDC37-HSP90 binding is unlikely to be effective for cancer therapy (Smith et al. 2015). We also showed that knockdown of the HSP90 ATPase-promoting co-chaperone AHA1 decreases client protein activation and increases cancer cell sensitivity to the HSP90 inhibition (Smith et al. 2009). In our work with Cullin-5 (CUL5), we again obtained somewhat surprising results in that knockdown of this E3 ligase modulates multiple molecular and cellular responses to HSP90 inhibition in human cancer cells (Smith et al. 2015). CUL5 was found to be required for degradation of a number of HSP90 clients after treatment with an HSP90 inhibitor. Unexpectedly, silencing CUL5 also slows the earlier loss of HSP90 client protein activity while also delaying co-chaperone dissociation from inhibited HSP90-client complexes. In addition, depleting CUL5 decreased the sensitivity of cancer cells to three distinct HSP90 inhibitors, across four tumour types that are driven by different protein kinases. These findings show that the role of E3 ligases in the pharmacology of HSP90 inhibitors is more complex than previously considered, and not simply concerned with client protein degradation. Pharmacological modulation of E3 ligases is of course of interest. In addition CUL5 may be a potential biomarker for sensitivity to HSP90 inhibitors.
Inhibition of the heat shock response or components of it (other than HSP90 discussed above) is an alternative approach to cancer therapy in the proteostasis area. The heat shock response was of course famously discovered by Feruccio Ritossa when he observed the effect of the inadvertent increase in a lab incubator temperature on the ‘puffing’ of polytene chromosomes in the salivary glands of Drosophila, which he then linked to new RNA synthesis (Ritossa 1962, 1996). The heat shock response was subsequently shown to be mediated by heat shock transcription factor HSF1 – the major transcriptional regulator of the eukaryotic heat shock response and the primary mediator of transcriptional responses to proteotoxic stress – which binds to Heat Shock Elements in heat shock-regulated genes (Rabindran et al. 1991; Anckar and Sistonen 2011). Studies by Voellmy and colleagues showed that the heat shock response can be activated by the release of HSF1 from the inhibitory stress-sensitive complex it forms with HSP90 (Zou et al. 1998). This can be triggered by unfolded proteins binding to HSP90. The mechanisms are, however, quite complex and certainly involve trimerization, nuclear accumulation and post-translational modification of HSF1, leading to activation of induction of a fraction of heat shock genes by increasing RNA polymerase II release from promoter-proximal pause (Mahat et al. 2016).
The Whitesell lab published an important study on the effect of HSP90 inhibitors in mouse embryo fibroblasts derived from Ivor Benjamin’s HSF1 knockout mice as compared with wild type controls (Bagatell et al. 2000). They showed that HSF1 knockout cells exhibited significantly greater sensitivity to the effect of HSP90 inhibitors, which was linked to failure to activate the cytoprotective heat shock response in knockout cells in contrast to wild type cells; there was no change in response to the cytotoxic agents doxorubicin or cisplatin. Also to note is that, in the same paper, the authors demonstrated that treatment with tanespimycin induced the expression of the representative heat shock protein HSP72 both in normal mouse tissues and human tumour xenografts – an effect subsequently used as part of the pharmacodynamic biomarker signature for HSP90 inhibition in drug discovery and clinical trials (see above).
Given the challenges of inhibiting the HSF1 transcription factor directly, our lab set out to investigate whether inhibiting key components of the heat shock response (other than HSP90) could exert selective anticancer activity and also whether this approach could sensitize cancer cells to HSP90 inhibitors (Powers et al. 2008). HSP70 isoforms are known to contribute to tumorigenesis, for example through their anti-apoptotic activity and their involvement as co-chaperones for HSP90. We showed that silencing the expression of either HSP72 – a well-studied heat shock inducible gene – or heat shock cognate 70 (HSC70) in human cancer cell lines has no effect on the activity of HSP90 in chaperoning client proteins or on cell proliferation. In contrast, simultaneous combinatorial depletion of both of these isoforms induces the proteasome-dependent degradation of HSP90 client proteins, causes G1 cell-cycle arrest and triggers extensive apoptosis – the latter to a much greater extent than pharmacological HSP90 inhibition – all in the absence of HSP90 inhibitors. In contrast, combinatorial silencing of the two HSP70 isoforms in non-tumorigenic cell lines does not cause comparable growth arrest or induction of apoptosis, indicating a potential therapeutic window for this combinatorial approach. The need for HSC70 inhibition in addition to HSP72 was explained by the ability of HSC70 to replace HSP72 in the heterochaperone complex with HSP90. Sensitization to HSP90 inhibitors was also seen in cancer cells. Similar effects of dual knockdown of HSP72 and HSC70 have also been confirmed by others.
These findings provided validation for the discovery of HSP70 inhibitors. HSP70 proteins are members of the actin family of ATPases for which the ATP site is much more challenging to drug than HSP90 and which has delivered very little success in the discovery of high affinity ligands. One challenging feature is the flexibility of the ATP site, which undergoes numerous conformational changes. The Vernalis team identified novel adenosine-derived ligands through structure-based design and found selectivity towards the GRP78 isoform (Williamson et al. 2009). In our efforts to design HSP70 inhibitors we have also adopted structure-based design strategies (Cheeseman et al. 2016; Jones et al. 2016). In particular, our ICR team took the approach of exploiting protein conformational change to optimize adenosine-based inhibitors of HSP70 (Cheeseman et al. 2016). We provided evidence that such adenosine-derived HSP70 ligands have the potential to bind to the protein with a novel mechanism of action, which involves the stabilization by desolvation of an intramolecular salt-bridge that in turn induces a conformational change in HSP70, leading to high affinity ligands. We also demonstrated that through the application of this mechanism, adenosine-derived HSP70 inhibitors can be optimized in a rational manner. We concluded that improved understanding of the flexibility of HSP70 and the impact of this on the affinity of ligands should contribute to better assay design and enhanced efficiency of inhibitor optimization. We also carried out a comprehensive fragment-based exploration of an HSP70 family enzyme, resulting in the discovery of an amino-quinazoline fragment which we then elaborated to produce novel ATP site binders that exhibited physicochemical properties distinct from previously known adenosine-based HSP70 inhibitors (Jones et al. 2016). In addition, my ICR colleagues discovered an irreversible inhibitor of HSP72 that unexpectedly targets lysine-56 and enhanced this series of targeted covalent inhibitors using rational design and kinetic optimization to yield Compound 18 (Pettinger et al. 2017, 2019). Other groups have also reported HSP70 family inhibitors, in particular Gabriella Chiosis and Jason Gestwicki (Rodina et al. 2013; Gestwicki and Shao 2019). However, to date HSP70 inhibitors have not progressed to the clinic.
Recent attention has turned to the HSF1 transcription factor as a potential target for cancer therapy (Whitesell and Lindquist 2009; De Billy et al. 2009). Although the available structure of the DNA binding domain indicates that it would be extremely tough to drug, HSF1 has been strongly validated, especially by key work in the Lindquist lab, as a critical protein involved in supporting oncogenesis and the tumour state (Dai et al. 2007). Other important work by the Lindquist group showed that HSF1 drives a transcriptional programme that is distinct from heat shock and which supports the malignancy of human cancers; moreover, HSF1 expression is prognostic in several clinical cancers (Mendillo et al. 2012). In addition to cell autonomous effects, HSF1 supports the reprogramming of tumour stroma, contributing to malignancy (Scherz-Shouval et al. 2014).
Recognising the druggability challenge posed by HSF1, we carried out a mechanism-based phenotypic screen to identify small-molecule inhibitors of the HSF1-mediated heat shock response; through this we discovered the bisamide amide class of inhibitors that exhibit a potent and selective effect on the HSF1 pathway in intact cancer cells (Cheeseman et al. 2017). We optimized this series, in particular to enhance intrinsic solubility, resulting in the advanced lead compound CCT251236 which showed therapeutic activity in models of human ovarian cancer and multiple myeloma (Cheeseman et al. 2017; Fok et al. 2018). Using chemical proteomics and validated by biophysical methods of surface plasmon resonance and x-ray crystallography, we identified the putative co-transcriptional regulator pirin as a molecular target that may be responsible for the anti-migration properties of CCT251236 (Cheeseman et al. 2017). Moreover, we designed and synthesized a pirin degrader (CCT367766) which showed potent and specific depletion of pirin in cancer cells; this can be used to demonstrate in-cell target engagement with pirin (Chessum et al. 2018). Meanwhile we continue to search for additional molecular targets of CCT251236 while progressing a clinical candidate towards first-in-human studies.
4 The Importance of Chemical Probes
CCT251236 has been rated positively as a ‘chemical probe’ for pirin at the Chemical Probes Portal (The Chemical Probes Portal n.d.). High-quality chemical probes are important reagents, used alongside genetic and biochemical tools, for probing biology and disease mechanisms and validating drug targets (Workman and Collins 2010; Frye 2010). A number of small-molecules discussed herein and also other compounds are important chemical probes or ‘tools’ in the chaperone/proteostasis area (Gestwicki and Shao 2019; Shrestha et al. 2016). To be useful, and not misleading, high-quality chemical probes must exhibit essential features or ‘fitness factors’ – particularly high potency and selectivity towards the desired target, together with cellular permeability and evidence of modulation of the target in cells (Workman and Collins 2010; Frye 2010). Unfortunately, the literature is full of examples of the use of chemical agents that lack the characteristics of high quality chemical probes and may even act broadly as chemically reactive compounds or behave as detergents or aggregators. Compounds such as apoptozole and 2-phenylethynesulfonamide (also known as PES and pifithrin-μ) have been used as chemical probes for HSP70 proteins but have subsequently been shown to be very non-specific (Evans et al. 2015; Schlecht et al. 2013).
My colleagues and I at ICR have provided chemical probes for use by the community, and have also contributed to guidelines for best practice in the selection and use of chemical probes for biological and biomedical research (Workman and Collins 2010; Arrowsmith et al. 2015; Blagg and Workman 2017). We are now hosting the Chemical Probes Portal – a public resource providing expert peer-viewed recommendations for chemical probes (Arrowsmith et al. 2015). We have also developed a public resource called Probe Miner (Antolin et al. 2018) that is complementary to the Portal in providing objective, quantitative, data-driven assessment of chemical probes, building on our canSAR knowledgebase (Coker et al. 2019). In addition, we recently provided an overview of the public resources available to help in the selection of chemical probes – rather than relying on the literature, general search engines and vendor catalogues that are biased in various ways and lack annotation – and we also included advice on how to navigate the various resources to make an informed choice of chemical probes (Antolin et al. 2019).
5 Concluding Remarks and Future Outlook
I hope this personal view has provided an insight into how findings in basic research on proteostasis networks can provide chemical probes and drug candidates for use in oncology research and cancer treatment. I also very much hope that the HSF1 pathway inhibitor will fulfil its potential in showing clinical activity in ovarian cancer – the hard-to-treat disease that cruelly took Sue Lindquist from us way too early.
Change history
24 September 2020
The original version of the book was revised: Chapter 11 is now available open access under a CC BY 4.0 license and the copyright holder has been changed to ‘The Author(s)’.
References
Anckar J, Sistonen L (2011) Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem 80:1089–1115
Antolin AA, Tym JE, Komianou A, Collins I, Workman P, Al-Lazikani B (2018) Objective, quantitative, data-driven assessment of chemical probes. Cell Chem Biol 25(2):194–205.e5
Antolin A, Workman P, Al-Lazikani B (2019) Public resources for chemical probes: the journey so far and the road ahead. Future Med Chem [Online ahead of print]
Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frederiksen M, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KV, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Müller S, O’Hagan RC, Overington JP, Owen DR, Rosenberg SH, Roth B, Ross R, Schapira M, Schreiber SL, Shoichet B, Sundström M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Willson TM, Workman P, Young RN, Zuercher WJ (2015) The promise and peril of chemical probes. Nat Chem Biol 11:536–541
Arteaga CL (2011) Why is this effectiveHSP90 inhibitor not being developed in HER2+ breast cancer? Clin Cancer Res 17:4919–4921
Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S (2000) Benjamin IJ Whitesell L. Induction of a Heat Shock Factor 1-dependent stress response alters the cytotoxic activity of Hsp90-binding agents. Clin Cancer Res 6:3312–3318
Banerji U, Workman P (2016) Critical parameters in targeted drug development: the pharmacological audit trail. Semin Oncol 43:436–445
Banerji U, Walton M, Raynaud F, Grimshaw R, Kelland L, Valenti M, Judson I, Workman P (2005a) Pharmacokinetic-pharmacodynamic relationships for the heat shock protein 90 molecular chaperone inhibitor 17-allylamino, 17-demethoxygeldanamycin in human ovarian cancer xenograft models. Clin Cancer Res 11:7023–7032
Banerji U, O’Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M, Walton M, Lakhani S, Kaye S, Workman P, Judson I (2005b) Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino,17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol 23:4152–4161
Banerji U, Affolter A, Judson I, Marais R, Workman P (2008) BRAF and NRAS mutations in melanoma: potential relationships to clinical response to HSP90 inhibitors. Mol Cancer Ther 7:737–739
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483:603–607
Basso AD, Solit DB, Munster PN, Rosen N (2002) Ansamycin antibiotics inhibit Akt activation and cyclin D expression in breast cancer cells that overexpress HER2. Oncogene 21:1159–1166
Blagg J, Workman P (2017) Choose and use your chemical probe wisely to explore cancer biology. Cancer Cell 32:9–25
Brough PA, Aherne W, Barril X, Borgognoni J, Boxall B, Cansfield JE, Cheung KM, Collins I, Davies NGM, Drysdale MJ, Dymock B, Eccles SA, Finch H, Fink A, Hayes A, Howes R, Hubbard RE, James K, Jordan AM, Lockie A, Martins V, Massey A, Matthews T, McDonald E, Northfield CJ, Pearl LH, Prodromou C, Ray S, Raynaud FI, Roughley SD, Sharp SY, Surgenor A, Walmsley DL, Webb P, Wood M, Workman P, Wright L (2008) 4,5-Diaryl isoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem 51:196–218
Brough PA, Barril X, Borgognoni J, Chene P, Davies NGM, Davis B, Drysdale MJ, Dymock B, Eccles SA, Garcia-Echeverria C, Fromont C, Hayes A, Hubbard RE, Jordan AM, Jensen MR, Massey A, Merrett A, Padfield A, Parsons R, Radimerski T, Raynaud FI, Robertson A, Roughley SD, Schoepfer J, Simmonite H, Sharp SY, Surgenor A, Valenti M, Walls S, Webb P, Wood M, Workman P, Wright L (2009) Combining hit identification strategies: fragment-based and in silico approaches to orally active 2-aminothieno[2,3-d]pyrimidine inhibitors of the Hsp90 molecular chaperone. J Med Chem 52:4794–4809
Brunton VG, Workman P (1993) Cell-signaling targets for antitumour drug development. Cancer Chemother Pharmacol 32:1–19
Brunton VG, Steele G, Lewis AD, Workman P (1998) Geldanamycin-induced cytotoxicity in human colon-cancer cell lines: evidence against the involvement of c-Src or DT-diaphorase. Cancer Chemother Pharmacol 41:417–422
Butler LM, Ferraldeschi R, Armstrong HK, Centenera MM, Workman P (2015) Maximizing the therapeutic potential of HSP90 inhibitors. Mol Cancer Res 13:1445–1451
Cartwright CA, Meisler AI, Eckhart W (1990) Activation of the pp60c-src protein kinase is an early event in colon carcinogenesis. Proc Natl Acad Sci U S A 87:558–562
Cheeseman MD, Westwood IM, Barbeau O, Rowlands M, Dobson S, Jones AM, Jeganathan F, Burke R, Kadi N, Workman P, Collins I, van Montfort RL, Jones K (2016) Exploiting protein conformational change to optimize adenosine-derived inhibitors of HSP70. J Med Chem 59:4625–4636
Cheeseman MD, Chessum NE, Rye CS, Pasqua AE, Tucker MJ, Wilding B, Evans LE, Lepri S, Richards M, Sharp SY, Ali S, Rowlands M, O’Fee L, Miah A, Hayes A, Henley AT, Powers M, Te Poele R, De Billy E, Pellegrino L, Raynaud F, Burke R, van Montfort RL, Eccles SA, Workman P, Jones K (2017) Discovery of a chemical probe bisamide (CCT251236): an orally bioavailable efficacious pirin ligand from a Heat Shock Transcription Factor 1 (HSF1) phenotypic screen. J Med Chem 60:180–120
Chessum NEA, Sharp SY, Caldwell JJ, Pasqua AE, Wilding B, Colombano G, Collins I, Ozer B, Richards M, Rowlands M, Stubbs M, Burke R, McAndrew PC, Clarke PA, Workman P, Cheeseman MD, Jones K (2018) Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766). J Med Chem 61(3):918–933
Cheung KM, Matthews TP, James K, Rowlands MG, Boxall KJ, Sharp SY, Maloney A, Roe SM, Prodromou C, Pearl LH, Aherne GW, McDonald E, Workman P (2005) The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg Med Chem Lett 15:3338–3343
Chiosis G, Timaul MN, Lucas B, Munster PN, Zheng FF, Sepp-Lorenzino L, Rosen N (2001) A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol 8:289–299
Clarke PA, Hostein I, Banerji U, Di Stefano F, Maloney A, Walton M, Judson I, Workman P (2000) Gene expression profiling of human colon adenocarcinoma cells following inhibition of signal transduction by 17-allylamino-17-demethoxyl-geldanamycin, an inhibitor of the hsp90 molecular chaperone. Oncogene 19:4125–4133
Coker EA, Mitsopoulos C, Tym JE, Komianou A, Kannas C, Di Micco P, Villasclaras Fernandez E, Ozer B, Antolin AA, Workman P, Al-Lazikani B (2019) canSAR: update to the cancer translational research and drug discovery knowledgebase. Nucleic Acids Res 47(D1):D917–D922
da Rocha Dias S, Light Y, Friedlos F, Springer C, Workman P, Marais R (2005) Oncogenic B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-AAG. Cancer Res 65:10686–10691
Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130:1005–1001
De Billy E, Powers MV, Smith JR, Workman P (2009) Drugging the heat shock factor 1 pathway: exploitation of the critical cancer cell dependence on the guardian of the proteome. Cell Cycle 8:3806–3808
Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K, Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A, Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B, Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood M, Wright L, Workman P (2008) NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res 68:2850–2860
Evans LE, Cheeseman MD, Yahya N, Jones K (2015) Investigating apoptozole as a chemical probe for HSP70 inhibition. PLoS One 10:e0140006
Felip E, Barlesi F, Besse B et al (2018) Phase 2 study of the HSP-90 inhibitor AUY922 in previously treated and molecularly defined patients with advanced non-small cell lung cancer. J Thorac Oncol 13:579–587
Ferraldeschi R, Welti J, Powers MV, Yuan W, Smyth T, Seed G, Riisnaes R, Hedayat S, Wang H, Crespo M, Nava Rodrigues D, Figueiredo I, Miranda S, Carreira S, Lyons JF, Sharp S, Plymate SR, Attard G, Wallis N, Workman P, de Bono JS (2016) Second-generation Hsp90 inhibitor onalespib blocks mRNA splicing of androgen receptor variant 7 in prostate cancer cells. Cancer Res 76:2731–2742
Fitzsimmons SA, Workman P, Grever M, Paull K, Camelier R, Lewis AD (1996) Reductase expression across the National Cancer Institute tumor cell line panel: correlation with sensitivity to mitomycin C and EO9. J Natl Cancer Inst 88:259–269
Fok JHL, Hedayat S, Zhang L, Aronson LI, Mirabella F, Pawlyn C, Bright MD, Wardell CP, Keats JJ, De Billy E, Rye CS, Chessum NEA, Jones K, Morgan GJ, Eccles SA, Workman P, Davies FE (2018) HSF1 is essential for myeloma cell survival and a promising therapeutic target. Clin Cancer Res 24:2395–2407
Frye SV (2010) The art of the chemical probe. Nat Chem Biol 6:159–161
Garcia R, Parikh NU, Saya H, Gallick GE (1991) Effect of herbimycin A on growth and pp60c-src activity in human colon tumor cell lines. Oncogene 6:1983–1989
Garcia-Carbonero R, Carnero A, Paz-Ares L (2013) Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol 14:e358–e369
Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH (2012) Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483:570–575
Gestwicki JE, Shao H (2019) Inhibitors and chemical probes for molecular chaperone networks. J Biol Chem 294:2151–2216
Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, Rosen N (2006) V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci U S A 103:57–62
Guo W, Reigan P, Siegel D, Zirrolli J, Gustafson D, Ross D (2005) Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinoneoxidoreductase1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res 65:10006–10015
Holmes JL, Sharp SY, Hobbs S, Workman P (2008) Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allyl amino-17-demethoxygeldanamycin. Cancer Res 68:1188–1197
Hostein I, Robertson D, Di Stefano F, Workman P, Clarke PA (2001) Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res 61:4003–4009
HSP90 Interactors (n.d.) www.picard.ch/downloads/Hsp90interactors.pdf
Huck JD, Que NLS, Sharma S, Taldone T, Chiosis G, Gewirth DT (2019) Structures of Hsp90α and Hsp90β bound to a purine-scaffold inhibitor reveal an exploitable residue for drug selectivity. Proteins 87:869–877
Jhaveri K, Chandarlapaty S, Iyengar N, Morris P, Corben A, Patil S, Akram Towers R, Sakr R, King TA, Norton L, Rosen N, Hudis C, Modi S (2016) Biomarkers that predict sensitivity to Heat Shock Protein 90 inhibitors. Clin Breast Cancer 16:276–278
Jones AM, Westwood IM, Osborne JD, Matthews TP, Cheeseman MD, Rowlands MG, Jeganathan F, Burke R, Lee D, Kadi N, Liu M, Richards M, McAndrew C, Yahya N, Dobson SE, Jones K, Workman P, Collins I, van Montfort RL (2016) A fragment-based approach applied to a highly flexible target: insights and challenges towards the inhibition of HSP70 isoforms. Sci Rep 6:34701
Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P (1999) DT-diaphorase expression and tumor cell sensitivity to 17- allylamino,17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst 91:1940–1949
Kong A, Rea D, Ahmed S, Beck JT, López López R, Biganzoli L, Armstrong AC, Aglietta M, Alba E, Campone M, Hsu Schmitz SF, Lefebvre C, Akimov M, Lee SC (2016) Phase 1B/2 study of the HSP90 inhibitor AUY922 plus trastuzumab in metastatic HER2-positive breast cancer patients who have progressed on trastuzumab-based regimen. Oncotarget 7:37680–33769
Lampis A, Carotenuto P, Vlachogiannis G, Cascione L, Hedayat S, Burke R, Clarke P, Bosma E, Simbolo M, Scarpa A, Yu S, Cole R, Smyth E, Mateos JF, Begum R, Hezelova B, Eltahir Z, Wotherspoon A, Fotiadis N, Bali MA, Nepal C, Khan K, Stubbs M, Hahne JC, Gasparini P, Guzzardo V, Croce CM, Eccles S, Fassan M, Cunningham D, Andersen JB, Workman P, Valeri N, Braconi C (2018) MIR21 drives resistance to Heat Shock Protein 90 inhibition in cholangiocarcinoma. Gastroenterology 154:1066–1079
Mahat DB, Salamanca HH, Duarte FM, Danko CG, Lis JT (2016) Mammalian Heat Shock Response and mechanisms underlying its genome-wide transcriptional regulation. Mol Cell 62:63–78
Maloney A, Clarke PA, Naaby-Hansen S, Stein R, Koopman JO, Akpan A, Yang A, Zvelebil M, Cramer R, Stimson L, Aherne W, Banerji U, Judson I, Sharp S, Powers M, de Billy E, Salmons J, Walton M, Burlingame A, Waterfield M, Workman P (2007) Gene and protein expression profiling of human ovarian cancer cells treated with the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res 67:3239–3253
Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150:549–562
Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, D’Andrea G, Dickler M, Moynahan ME, Sugarman S, Ma W, Patil S, Norton L, Hannah AL, Hudis C (2011) HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res 17:5132–5139
Münster PN, Srethapakdi M, Moasser MM, Rosen N (2001) Inhibition of heat shock protein 90 function by ansamycins causes the morphological and functional differentiation of breast cancer cells. Cancer Res 61:2945–2952
Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18:64–76
Neckers L, Blagg B, Haystead T, Trepel JB, Whitesell L, Picard D (2018) Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones 23:467–482
Pettinger J, Le Bihan YV, Widya M, van Montfort RL, Jones K, Cheeseman MD (2017) An irreversible inhibitor of HSP72 that unexpectedly targets lysine-56. Angew Chem Int Ed Engl 56:3536–3540
Pettinger J, Carter M, Jones K, Cheeseman MD (2019) Kinetic optimization of lysine-targeting covalent inhibitors of HSP72. J Med Chem 62:11383–11398.
Pillai RN, Ramalingam S (2018) Throwing more cold water on Heat Shock Protein 90 inhibitors in NSCLC. J Thorac Oncol 13:473–474
Pillai R, Fennell D, Kovcin V, Ciuleanu T, Ramlau R, Kowalski D, Schenker M, Perin B, Yalcin I, Teofilovici F, Vukovic V, Ramalingam S (2017) Phase 3 study of ganetespib, a heat shock protein 90 inhibitor, with docetaxel versus docetaxel in advanced non-small cell lung cancer (GALAXY-2). J Thorac Oncol 12:S7–S8
Piotrowska Z, Costa DB, Oxnard GR, Huberman M, Gainor JF, Lennes IT, Muzikansky A, Shaw AT, Azzoli CG, Heist RS, Sequist LV (2018) Activity of the Hsp90 inhibitor luminespib among non-small-cell lung cancers harboring EGFR exon 20 insertions. Ann Oncol 29:2092–2020
Polier S, Samant RS, Clarke PA, Workman P, Prodromou C, Pearl LH (2013) ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nat Chem Biol 9:307–312
Powers MV, Clarke PA, Workman P (2008) Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 14:250–262
Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH (1997) Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90:65–75
Rabindran SK, Giorgi G, Clos J, Wu C (1991) Molecular cloning and expression of a human heat shock factor, HSF1. Proc Natl Acad Sci U S A 88:6906–6910
Ritossa FA (1962) New puffing pattern induced by temperature Shock and DNP in Drosophila. Experientia 18:71–573
Ritossa F (1996) Discovery of the heat shock response. Cell Stress Chaperones 1:97–98
Rodina A, Patel PD, Kang Y, Patel Y, Baaklini I, Wong MJ, Taldone T, Yan P, Yang C, Maharaj R, Gozman A, Patel MR, Patel HJ, Chirico W, Erdjument-Bromage H, Talele TT, Young JC, Chiosis G (2013) Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem Biol 20:1469–1480
Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH (1999) Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 42:260–266
Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D (2000) NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact 129:77–97
Samant RS, Clarke PA, Workman P (2014) E3 ubiquitin ligase Cullin-5 modulates multiple molecular and cellular responses to heat shock protein 90 inhibition in human cancer cells. Proc Natl Acad Sci U S A 111:6834–6839
Sartorelli AC (1988) Therapeutic attack of hypoxic cells of solid tumors: presidential address. Cancer Res 48:775–778
Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, Dias-Santagata D, Koeva M, Stemmer SM, Whitesell L, Lindquist S (2014) The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158:564–578
Schlecht R, Scholz SR, Dahmen H, Wegener A, Sirrenberg C, Musil D, Bomke J, Eggenweiler HM, Mayer MP, Bukau B (2013) Functional analysis of Hsp70 inhibitors. PLoS One 8:e78443
Schroder CP, Pedersen JV, Chua S, Swanton C, Akimov M, Ide S et al (2011) Use of biomarkers and imaging to evaluate the treatment effect of AUY922, an HSP90 inhibitor, in patients with HER2+ or ER+ metastatic breast cancer. J Clin Oncol 29(suppl):abstr e11024
Sessa C, Shapiro GI, Bhalla KN, Britten C, Jacks KS, Mita M, Papadimitrakopoulou V, Pluard T, Samuel TA, Akimov M, Quadt C, Fernandez-Ibarra C, Lu H, Bailey S, Chica S, Banerji U (2013) First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res 19:3671–3680
Sharp SY, Boxall K, Rowlands M, Prodromou C, Roe SM, Maloney A, Powers M, Clarke PA, Box G, Sanderson S, Patterson L, Matthews TP, Cheung KM, Ball K, Hayes A, Raynaud F, Marais R, Pearl L, Eccles S, Aherne W, McDonald E, Workman P (2007a) In vitro biological characterization of a novel, synthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors. Cancer Res 67:2206–2216
Sharp SY, Prodromou C, Boxall K, Powers MV, Holmes JL, Box G, Matthews TP, Cheung KM, Kalusa A, James K, Hayes A, Hardcastle A, Dymock B, Brough PA, Barril X, Cansfield JE, Wright L, Surgenor A, Foloppe N, Hubbard RE, Aherne W, Pearl L, Jones K, McDonald E, Raynaud F, Eccles S, Drysdale M, Workman P (2007b) Inhibition of the heat shock protein 90 molecular chaperone in vitro and in vivo by novel, synthetic, potent resorcinylic pyrazole/isoxazole amide analogues. Mol Cancer Ther 6:1198–1211
Shrestha L, Patel HJ, Chiosis G (2016) Chemical tools to investigate mechanisms associated with HSP90 and HSP70 in disease. Cell Chem Biol 23:158–172
Smith JR, Clarke PA, de Billy E, Workman P (2009) Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene 28:157–169
Smith JR, deBilly E, Hobbs S, Powers M, Prodromou C, Pearl L, Clarke PA, Workman P (2015) Restricting direct interaction of CDC37 with HSP90 does not compromise chaperoning of client proteins. Oncogene 34:15–26
Smyth T, Paraiso KH, Hearn K, Rodriguez-Lopez AM, Munck JM, Haarberg HE, Sondak VK, Thompson NT, Azab M, Lyons JF, Smalley KSM, Wallis NG (2014) Inhibition of HSP90 by AT13387 delays the emergence of resistance to BRAF inhibitors and overcomes resistance to dual BRAF and MEK inhibition in melanoma models. Mol Cancer Ther 13:2793–2804
Solit DB, Zheng FF, Drobnjak M, Münster PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB, Scher HI, Rosen N (2002) 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res 8:986–993
Speranza G, Anderson L, Chen AP, Do K, Eugeni M, Weil M, Rubinstein L, Majerova E, Collins J, Horneffer Y, Juwara L, Zlott J, Bishop R, Conley BA, Streicher H, Tomaszewski J, Doroshow JH, Kummar S (2018) First-in-human study of the epichaperome inhibitor PU-H71: clinical results and metabolic profile. Investig New Drugs 36:230–223
Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S (2012) Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150:987–1001
The Chemical Probes Portal (n.d.) https://www.chemicalprobes.org/cct251236
Travers J, Sharp S, Workman P (2012) HSP90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today 17:242–252
Walton MI, Bibby MA, Double JA, Plumb JA, Workman P (1992) DT-diaphorase activity correlates with sensitivity to the indoloquinone EO9 in mouse and human colon carcinomas. Eur J Cancer 28A:1597–1600
Whitesell L, Lindquist S (2009) Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets 13:469–478
Whitesell L, Mimnaugh EG, DeCosta B, Myers CE, Neckers LM (1994) Inhibition of the heat-shock protein hsp90-pp60 (v-Src) heteroprotein complex-formation by benzoquinone ansamycins – essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A 91:8324–8328
Whitesell L, Santagata S, Mendillo ML, Lin NU, Proia DA, Lindquist S (2014) HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc Natl Acad Sci U S A 111:18297–18302
Williamson DS, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Foloppe N, Francis GL, Graham CJ, Howes R, Macias AT, Murray JB, Parsons R, Shaw T, Surgenor AE, Terry L, Wang Y, Wood M, Massey AJ (2009) Novel adenosine-derived inhibitors of 70 KDa heat shock protein, discovered through structure-based design. J Med Chem 52:1510–1513
Workman P (2003) Auditing the pharmacological accounts for Hsp90 molecular chaperone inhibitors: unfolding the relationship between pharmacokinetics and pharmacodynamics. Mol Cancer Ther 2:131–138
Workman P, Collins I (2010) Probing the probes: fitness factors for small molecule tools. Chem Biol 17:561–567
Workman P, Stratford IR (1993) The experimental development of bioreductive drugs and their role in cancer therapy. Cancer Metastasis Rev 12:73–82
Workman P, Burrows F, Neckers L, Rosen N (2007) Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci 1113:202–216
Workman P, Clarke PA, Al-Lazikani B (2012) Patient-predictive panel power. Cancer Cell 21:455–458
Ying W, Du Z, Sun L, Foley KP, Proia DA, Blackman RK, Zhou D, Inoue T, Tatsuta N, Sang J, Ye S, Acquaviva J, Ogawa LS, Wada Y, Barsoum J, Koya K (2012) Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol Cancer Ther 11:475–484
Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94:471–480
Acknowledgements
I acknowledge grant support from Cancer Research UK (CRUK) [C309/A11566] and Wellcome [212969/Z/18/Z] and I am a CRUK Life Fellow. I would like to thank many lab members, colleagues and collaborators for their invaluable contributions and discussions and to Dr. Matt Cheeseman for preparing Figure 1. Like many others, I am also indebted to Sue Lindquist for her outstanding leadership, inspiration and research contributions. This article is a brief personal perspective and I apologize that many excellent publications could not be cited.
Conflict of Interest
PW is Chief Executive, President and an employee of the Institute of Cancer Research (ICR) which has a commercial interest in a range of drug targets, including HSP90 and HSF1. The ICR operates a Rewards to Inventors scheme whereby employees of the ICR may receive financial benefit following commercial licensing of a project. PW received research funding relevant to this article from Vernalis, Astex, AstraZeneca, BACIT and Sixth Element Capital/CRT Pioneer Fund. PW contributed to intellectual property on HSP90 inhibitors that was licenced to Vernalis and Novartis and on HSF1 that was licenced to Sixth Element Capital/CRT Pioneer Fund. PW has been a consultant to Novartis, a consultant/scientific advisory board member for NextechInvest, Storm Therapeutics, Astex Pharmaceuticals and CV6, and holds stock in Chroma Therapeutics, NextInvest and Storm Therapeutics. He is also a Non-Executive Director of Storm Therapeutics and the Royal Marsden NHS Trust and a Board Director of the non-profit Chemical Probes Portal.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this chapter

Cite this chapter
Workman, P. (2020). Reflections and Outlook on Targeting HSP90, HSP70 and HSF1 in Cancer: A Personal Perspective. In: Mendillo, M.L., Pincus, D., Scherz-Shouval, R. (eds) HSF1 and Molecular Chaperones in Biology and Cancer. Advances in Experimental Medicine and Biology, vol 1243. Springer, Cham. https://doi.org/10.1007/978-3-030-40204-4_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-40204-4_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-40203-7
Online ISBN: 978-3-030-40204-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)