
Abstract
The triggers as well as etiologies for Acute exacerbation of idiopathic pulmonary fibrosis (AE-IPF) are not known. AE-IPF is defined as an “acute, clinically significant respiratory deterioration characterized by evidence of new widespread alveolar abnormality typically less than 1 month’s duration. The underlying pathologic insult is classically described as diffuse alveolar damage. Ideally, infection is excluded by BAL as in the case presentation, but the severity of hypoxemia and the desire to avoid endotracheal intubation may preclude the performance of this procedure. Supportive care is the mainstay of therapy as there are no proven therapies, although corticosteroids, cytotoxic agents and anti-coagulation have all been suggested as possible treatments. The mortality is high, particularly once invasive ventilation has been instituted.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
Case Presentation
A 65 year-old gentleman with Idiopathic Pulmonary Fibrosis (IPF) diagnosed 2 years prior by imaging and surgical lung biopsy is evaluated for acute on chronic shortness of breath. Previously he was able to perform his activities of daily living with supplemental oxygen administered by nasal cannula at 4 liters per minute. One day prior to presentation his breathlessness rapidly escalated, with inability to ambulate or bathe, prompting emergent evaluation. He had no fever, sick exposures, recent travel or immobilization, change in his baseline cough, or lower extremity edema. He is on pirfenidone in addition to oxygen for his condition.
Upon evaluation in the emergency room he is noted to require 100% oxygen to maintain his oxygen saturation, although saturation continued to fall with minimal exertion. Arterial blood gas showed a pH of 7.50, PCO2 of 44, PaO2 of 65 with saturation of 91% on 100% oxygen by non-rebreather facemask. His hypoxemia continued to worsen and he required invasive mechanical ventilation. He was admitted to the intensive care unit (ICU).
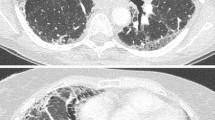
On evaluation, beta naturetic peptide and echocardiogram were normal and he was without signs of volume overload. White blood cell count was elevated to 14,000 per mm3 (normal 4000–10,000 per mm3). A computed tomography of the chest was completed and lung windows are shown in Fig. 30.1 alongside his baseline CT scan. Contrast to enhance the pulmonary vasculature showed no evidence of venous thromboembolism. Bronchoscopy is completed and bronchoalveolar lavage (BAL) was negative for infectious organisms including respiratory virus polymerase chain reaction (PCR).

(a) Baseline CT scan of patient with basal predominant interlobular septal thickening and traction bronchiectasis and mild (lower right lung) honeycombing typical of IPF. (b) CT scan of the same patient after presentation for acute on chronic dyspnea and worsened hypoxemia
Question
What is the likely diagnosis?
Answer
The patient has most likely suffered an acute exacerbation of IPF (AE-IPF).
Principles of Management
The triggers as well as etiologies for AE-IPF are not known. The 2007 criteria proposed by Idiopathic Pulmonary Fibrosis Network (IPFnet) defined acute exacerbation of IPF as acute clinical worsening (<30 days) in a patient with known or newly diagnosed IPF with acceleration of dyspnea and/or hypoxemia and new radiologic changes, typically ground glass opacities, on a background of fibrotic disease (example Fig. 30.1) [1, 2]. The revised criteria in 2016 did not set a definite 30-day duration of onset of symptoms but defined AE-IPF as an “acute, clinically significant respiratory deterioration characterized by evidence of new widespread alveolar abnormality typically less than one month’s duration” [3]. Common mimics like fluid overload due to heart failure and infections should be excluded although this is not straightforward in some patients. The underlying pathologic insult is classically described as diffuse alveolar damage (DAD) [4], the histologic finding of acute respiratory distress syndrome (ARDS), which has been superimposed on usual interstitial pneumonia. However, biopsy of the lung to prove DAD is rarely ever pursued due to the morbidity and mortality of the procedure. Common concomitant symptoms mimic a viral lower respiratory tract infection with fever, malaise, flu like symptoms and cough; though these symptoms are not needed to make the diagnosis [1,2,3]. Ideally, infection is excluded by BAL as in the case presentation; however given the worsening hypoxemia in addition to typical poor baseline pulmonary health, a BAL may precipitate a life-threatening impairment in gas exchange. If a BAL cannot be completed, treatment for typical bacterial organisms or hospital acquired organisms are employed presumptively, as applicable. If structural lung disease exists (as most patients usually have traction bronchiectasis) it may be worthwhile to treat Pseudomonas on an empiric basis, though evidentiary support for this is lacking.
Other conditions that need to be excluded are pneumothorax, pulmonary hypertension, left ventricular failure from systolic or diastolic dysfunction, pulmonary embolism, or ARDS of other known causes (e.g. sepsis, pancreatitis, trauma, pneumonia). Diagnostic specificity of AE-IPF is further limited due to unavailability of specific serologic markers. Cross-sectional imaging is used to rule out small pneumothorax not seen as well as to confirm radiologic changes, which are typically ground glass in appearance, but dense infiltrates can also be seen. A CT pulmonary angiogram has the advantage of evaluating for pulmonary embolus in patients without renal impairment.
In summary, AE-IPF is diagnosed when a patient with IPF has acute worsening of dyspnea and/or hypoxemia as well as new CT opacities after exclusion of the conditions in Table 30.1. Diffuse alveolar damage is the typical underlying histopathology.
Incidence, Risk Factors and Prognosis
Based on prospective trials of IPF the incidence of acute exacerbation in IPF is approximately 2–14% per year [5,6,7]. Acute exacerbations are often devastating with a median survival of usually about 4 months [8]. If an IPF patient requires mechanical ventilation, mortality has been reported to be 78–86% [9, 10]. Some authors have proposed that mechanical ventilation in IPF patients is futile and these patients should not receive this intervention [11, 12]. Others have argued, and shown, that even short-term survival allows a window for pulmonary transplantation [10]. Additionally, it is noted that mortality is not absolute, and thus not “futile” in all cases. Due to the poor prognosis, an accurate diagnosis of AE-IPF is of utmost importance to exclude reversible causes; however, this is often challenging.
The risk factors for AE-IPF are not well known. It remains unclear if the acute worsening of IPF is simply an accelerated progression of the underlying interstitial lung disease (ILD) or a maladaptive response to an external trigger such as micro aspiration of gastric contents, trauma, mechanical stretch or infections. It is likely that AE-IPF is the result of interactions between genetic and environmental stimuli in a significant fraction of patients with IPF.
Pathologic insult
The classical description of pathologic insult due to acute exacerbation of IPF is diffuse alveolar damage [4], which is the same as ARDS, but here is superimposed on the pathological findings of idiopathic pulmonary fibrosis: usual interstitial pneumonia.
Corticosteroids
As idiopathic pulmonary fibrosis is a rare condition, and acute exacerbations occur spontaneously and abruptly, large prospective randomized trials evaluating treatment are lacking. Expert consensus recommends treatment with corticosteroids, though recommended dosages have not been established [13]. Dosing ranges of methylprednisolone from 1 mg/kg up to “pulse” doses of 1000 mg per day [1]. It is our practice to use 2 mg/kg/day of methylprednisolone in divided doses daily similar to what has been studied in ARDS [14], given the similar underlying histopathologic insult.
Supportive care
Supportive care is essential for treatment of idiopathic pulmonary fibrosis given the lack of evidence-based therapies. Endotracheal intubation and invasive mechanical ventilation is sometimes needed, but noninvasive ventilation can be attempted in the appropriate candidate (see below). While evaluation for conditions listed in Table 30.1 is suggested, this is not always possible in a given patient. Presumptive treatment with antibacterial agents, diuretics in conjunction with systemic corticosteroids are typically administered unless significant contraindications exist.
Palliative care
Acute exacerbation of idiopathic pulmonary fibrosis can often be a terminal event. Patients and family should be made aware of the poor prognosis to make appropriate decisions about possibly limiting intensive care interventions. Ideally, goals of care planning would occur in the outpatient setting prior to clinical worsening. In patients who are not transplant candidates, palliative care is often a valid choice [11].
Ensure the correct diagnosis of IPF as underlying ILD
The diagnosis of acute exacerbation of IPF is in the domain of the intensivist. However diagnosis and management of idiopathic pulmonary fibrosis is usually the realm of the pulmonologist. Ensuring that the patient has an accurate diagnosis of idiopathic pulmonary fibrosis is critical to determining prognosis for the underlying condition. Fibrotic lung disease associated with collagen-vascular disease, such as polymyositis, has also been associated with acute exacerbations, and may have a better prognosis [15]. Many pulmonologists who are less familiar with interstitial lung diseases often incorrectly attribute all types of lung fibrosis to idiopathic pulmonary fibrosis [16]. Given the disparate outcomes, accurate discrimination of IPF from other fibrotic lung diseases is critical. Clues on CT scan suggesting the diagnosis of IPF are basal, subpleural predominance of interlobular septal thickening with honeycombing and traction bronchiectasis (see Fig. 30.1) [13]. In the absence of AE-IPF, evidence of extensive ground glass infiltrates and nodules on CT imaging typically argues against IPF [13]. Clinical findings should include a conspicuous absence of inhalational exposures and rheumatologic conditions. “Velcro” rales are typically present on physical exam.
If the diagnosis is in doubt, consultation with a pulmonary specialist with experience in interstitial lung disease is recommended.
Exacerbation during surgical procedures
Often, patients with IPF undergo surgical biopsy to establish a diagnosis of IPF. Additionally, patients with IPF are at higher risk of lung cancer which may require surgical treatment. Thoracic surgery for lung cancer resection or surgical lung biopsy can precipitate an acute exacerbation of IPF [17,18,19,20]. Interestingly the insult is often radiologically worse in the nonoperative lung [19]. This is theorized to be due to the ventilator-associated lung injury from excessive stretch from single lung ventilation during the operation to de-gas the operative lung. Some have suggested restrictive intraoperative fluid management may minimize post-operative AE-IPF risk, but this has not been prospectively validated [17]. AE-IPF after non-pulmonary operation has been reported, but rarely [21].
Evidence Contour
Noninvasive ventilation
Given the poor prognosis of patients who require mechanical ventilation, some have suggested that noninvasive ventilation would be a good strategy for patients with clinical deterioration and idiopathic pulmonary fibrosis. Small, retrospective studies have shown improved outcomes in AE-IPF patients supported with noninvasive positive pressure ventilation (NIPPV) [22,23,24], however a selection bias may account for the better prognosis as patients who can be successfully supported with NIPPV are likely less ill. Of the patients in these studies who failed NIPPV, mortality was reported as 85–100% [22,23,24].
High flow nasal cannula (HFNC):
High flow nasal cannula has been shown to have salutatory affects in idiopathic pulmonary fibrosis patients without an acute exacerbation, specifically decreased minute ventilation, respiratory rate, capillary carbon dioxide were seen [25]. Additionally, small increases in airway pressure were reported, suggesting a partial “PEEP” affect [25].
A recent multi-center open label trial that compared high flow oxygen therapy with standard therapy and non-invasive ventilation, in patients with non-hypercapnic hypoxic respiratory failure of various etiologies, reported non-significant improvement in rates of intubation, but a significant improvement in 90 day mortality in favor of high flow therapy [26].
In a retrospective study of patients with AE-IPF, Ito and colleagues demonstrated a decreased mortality rate and need for mechanical ventilation or NIPPV in the epoch after implementation of high flow nasal, suggesting significant benefit of HFNC in AE-IPF [27].
Randomized, prospective data does not exist for high flow nasal cannula use in AE-IPF. Anecdotally, we have used high flow oxygen therapy with great success in patients with IPF and acute exacerbations and would recommend routine use.
Ventilator settings
The optimal ventilator settings for acute exacerbation of IPF are not known, however to the extent possible, we adhere to lung protective with low tidal volume ventilation similar to ARDS. Unfortunately, the need for mechanical ventilation in a patient experiencing acute exacerbation portends poor prognosis due to high mortality (reported mortality of 78–86% if intubation required) [9, 10]. Therefore, noninvasive methods that support and permit safe oxygenation should be employed whenever possible. Patient and families should be educated about the poor outcomes of mechanical ventilation prior to pursuing it.
Higher levels of PEEP have been associated with higher mortality in single retrospective analysis [28]. A selection bias for patients with worse hypoxemia requiring higher levels of mean airway pressure is one explanation; however multiple variables were accounted for in the analysis. It may be true that patients with acute exacerbation of IPF have a different physiology and those with ARDS where higher levels of PEEP are felt to be beneficial.
Anticoagulation
A single, unblinded prospective study has shown a benefit in patients admitted with clinical worsening of idiopathic pulmonary fibrosis treated with anticoagulation [29]. Anticoagulation was initiated at the time of clinical worsening, which may or may not have been an acute exacerbation. Warfarin was used in the outpatient setting and low molecular weight heparin was used if the patient was admitted, such as with AE-IPF. All patients were treated with corticosteroid as well. In the subset of patients who had AE-IPF, anticoagulated patients had a lower mortality (18% vs. 71%). However, 30% of patients randomized to the treatment arm dropped out of the study. Additionally, pulmonary embolism was not excluded as a cause of clinical worsening, and may have played a role in some patients [13].
A large, double blind prospective trial of warfarin in the treatment of IPF was stopped early due to increased mortality in warfarin arm [30]. No difference in incidence of AE-IPF was observed.
Use of anticoagulation in IPF patients without thromboembolic disease was recommended against by a panel of experts [13]. Anticoagulation specifically used to treat AE-IPF currently does not have sufficient data to support its use.
Cyclophosphamide
Cyclophosphamide has been used in case series for treatment of AE-IPF. Morawiec and colleagues described 10 patients who were treated with cyclophosphamide and pulse dose methylprednisolone during acute exacerbations with 100 and 72% at 1 month and 3 months survival rates, respectively [31]. These results were compared to reported outcomes and not historical controls at the same institution. Lack of randomization significantly limits this utility of this study and a prospective randomized trials are needed prior to wide spread adoption.
Using propensity score analysis, others have failed to show benefit of cyclophosphamide in patients with AE-IPF, but also with low numbers of patients studied [32].
A multicenter study, double blind randomized study of cyclophosphamide in addition to corticosteroids for treatment of AE-IPF is currently planned [33].
Cyclosporine A
Inase and coworkers retrospectively analyzed 13 patients after AE-IPF. All patients received pulse methylprednisolone followed by oral prednisone, and 7 received cyclosporine A titrated A to serum levels of 100–150 in addition to steroids [34]. In the patients treated with cyclosporine A none experienced a re-exacerbation of IPF. All patients with steroids alone died of respiratory failure within 66 weeks, whereas four out of the seven treated with cyclosporine A survived for over 2 years after their exacerbation.
Sakamoto also evaluated the use of cyclosporine A in AE-IPF retrospectively [35]. Similar to Inase, all patients were treated with pulse methylprednisolone followed by prednisone. Two out of eleven patients treated with cyclosporine A died during their initial exacerbation, compared to six out of eleven patients who were treated with steroids alone. Prevention of re-exacerbation was not observed with five patients experiencing repeat exacerbations while on cyclosporine A.
In a relatively large retrospective database review, Aso and colleagues were unable to demonstrate a difference of in hospital mortality in AE-IPF patients treated with cyclosporine A and corticosteroids as compared to corticosteroids alone [36].
Similar to cyclophosphamide, lack of large scale randomized prospective trials limit widespread adoption of cyclosporine A for treatment of AE-IPF.
Other agents
Several other agents have been studied in small scale to treat acute exacerbation of IPF. Rituximab in combination with plasma exchange was used in 11 patients experience AE-IPF in an attempt to target an autoimmune pathway of inflammation [37]. Nine out of the eleven patients responded with improvement in oxygenation, although a few experienced relapse. In some patients, when the above treatment protocol was combined with intravenous immunoglobulin, a sustained improvement in gas exchange without relapse was observed. This study result has not been reproduced in large-scale studies, and has limited clinical application currently since the serologic markers used to determine active auto-immunity are mostly research-based biomarkers and not currently available for clinical use.
Other agents studied for acute exacerbation of IPF have included recombinant thrombomodulin and hemoperfusion with polymixin B immobilized fiber. Recombinant thrombomodulin is used on the premise of its anti-coagulant and anti-fibrinolytic properties. Small studies that have compared recombinant thrombomodulin with conventional therapy for AE-IPF demonstrated a significant mortality benefit compared to conventional arm; with a similar adverse effect profile [38, 39].
Although the above small studies are provocative, routine use cannot be advised until further study.
Restrictive operative fluid balance
Mizuno retrospectively evaluated 52 patients with IPF after pulmonary resection of non-small cell lung cancer and found that higher positive intraoperative fluid balance was associated with AE-IPF after multivariate analysis [17]. Prospective use of restrictive fluid practices towards the prevention of postoperative AE-IPF have not been established.
Lung transplantation
In patients without other medical comorbidities, lung transplantation can be performed in patients with idiopathic pulmonary fibrosis. In patients who are already listed for transplant and develop acute exacerbation, extracorporeal life-support or mechanical ventilation are not contraindications to transplantation; though vigilance of maintaining a robust functional status and avoiding critical care weakness are major challenges.
De novo evaluation of patients with acute exacerbation for pulmonary transplantation is difficult as the typical pre-operative studies, such as colonoscopy, heart catheterization, informed consent, etc. become much more perilous in a patient with severe respiratory failure or on extracorporeal life-support. However, this has been reported successfully [40]. Age limitations for lung transplant vary greatly among centers, and if in doubt, discussion with a transplant center is advised.
Extracorporeal life support (ECLS)
Outcomes of patients with respiratory failure and AE-IPF are poor, as described above. Theoretically, extracorporeal support eliminates ventilator induced lung injury and may lead to improved outcomes. Data to support this assumption does not exist. The largest case series of ECLS in ILD included all subtypes of ILD, with only 3 out of 21 patients having IPF [41]. Despite including potentially reversible causes of respiratory failure (e.g. pneumonia, pneumothorax, connective tissue disease exacerbation), the outcomes remained dismal with a 93% mortality if the patient did not receive a lung transplant. ECLS did allow time for evaluation, listing, and transplant of a few select patients.
In patients who are not listed for transplant nor candidates for pulmonary transplantation, we suggest against extracorporeal life support for AE-IPF given the overall poor prognosis of the condition. We do, however, use ECLS as a bridge to transplant or a bridge to decision in appropriate cases. When in doubt, discussion with and ECLS capable transplant center is advised.
Anti-fibrotic medications
Two anti-fibrotic medications, pirfenidone and nintedanib, have been approved for treatment of IPF. Pirfenidone, an inhibitor of transforming growth factor beta (TGF-β), has pleiotropic effects that regulate important profibrotic cascades, fibroblast proliferation, and collagen synthesis [42]. Nintedanib is a broad-spectrum tyrosine kinase inhibitor that targets several pathways including vascular endothelial growth factor, fibroblast growth factor, and platelet-derived growth factor [7]. Early studies suggested use of pirfenidone lowered the rate of acute exacerbation of idiopathic pulmonary fibrosis [5], though this was not seen on subsequent studies [43]. In a combined analysis, nintedanib significantly reduced the time to adjudicated AE-IPF, but not investigator reported AE-IPF [7]. It is noted that this was a secondary analyses and the studies were not specifically powered to evaluate prevention of AE-IPF.
The initiation of pirfenidone or nintedanib as a treatment specifically for AE-IPF is not currently recommended, but use of nintedanib as an adjunctive treatment of AE-IPF has been reported in a single case report [44]. In those that survive to hospital discharge, antifibrotic medications should be considered to prevent further deterioration in pulmonary function.
References
Hyzy R, et al. Acute exacerbation of idiopathic pulmonary fibrosis. Chest. 2007;132(5):1652–8.
Collard HR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(7):636–43.
Collard HR, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med. 2016;194(3):265–75.
Kondoh Y, et al. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest. 1993;103(6):1808–12.
Azuma A, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171(9):1040–7.
Martinez FJ, et al. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2093–101.
Richeldi L, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
Song JW, et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37(2):356–63.
Kim DS, et al. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27(1):143–50.
Gaudry S, et al. Invasive mechanical ventilation in patients with fibrosing interstitial pneumonia. J Thorac Cardiovasc Surg. 2014;147(1):47–53.
Mallick S. Outcome of patients with idiopathic pulmonary fibrosis (IPF) ventilated in intensive care unit. Respir Med. 2008;102(10):1355–9.
Al-Hameed FM, Sharma S. Outcome of patients admitted to the intensive care unit for acute exacerbation of idiopathic pulmonary fibrosis. Can Respir J. 2004;11(2):117–22.
Raghu G, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Steinberg KP, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354(16):1671–84.
Tachikawa R, et al. Clinical features and outcome of acute exacerbation of interstitial pneumonia: collagen vascular diseases-related versus idiopathic. Respiration. 2012;83(1):20–7.
Flaherty KR, et al. Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis? Am J Respir Crit Care Med. 2007;175(10):1054–60.
Mizuno Y, et al. The importance of intraoperative fluid balance for the prevention of postoperative acute exacerbation of idiopathic pulmonary fibrosis after pulmonary resection for primary lung cancer. Eur J Cardiothorac Surg. 2012;41(6):e161–5.
Shintani Y, et al. Predictive factors for postoperative acute exacerbation of interstitial pneumonia combined with lung cancer. Gen Thorac Cardiovasc Surg. 2010;58(4):182–5.
Kondoh Y, et al. Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir Med. 2006;100(10):1753–9.
Park JH, et al. Mortality and risk factors for surgical lung biopsy in patients with idiopathic interstitial pneumonia. Eur J Cardiothorac Surg. 2007;31(6):1115–9.
Ghatol A, Ruhl AP, Danoff SK. Exacerbations in idiopathic pulmonary fibrosis triggered by pulmonary and nonpulmonary surgery: a case series and comprehensive review of the literature. Lung. 2012;190(4):373–80.
Tomii K, et al. Role of non-invasive ventilation in managing life-threatening acute exacerbation of interstitial pneumonia. Intern Med. 2010;49(14):1341–7.
Yokoyama T, et al. Noninvasive ventilation in acute exacerbation of idiopathic pulmonary fibrosis. Intern Med. 2010;49(15):1509–14.
Gungor G, et al. Why do patients with interstitial lung diseases fail in the ICU? A 2-center cohort study. Respir Care. 2013;58(3):525–31.
Braunlich J, et al. Effects of nasal high flow on ventilation in volunteers, COPD and idiopathic pulmonary fibrosis patients. Respiration. 2013;85(4):319–25.
Frat JP, et al. High-flow oxygen through nasal cannula in acute hypoxemic respiratory failure. N Engl J Med. 2015;372(23):2185–96.
Ito J, et al. Respiratory management of acute exacerbation of interstitial pneumonia using high-flow nasal cannula oxygen therapy: a single center cohort study. J Thorac Dis. 2019;11(1):103–12.
Fernandez-Perez ER, et al. Ventilator settings and outcome of respiratory failure in chronic interstitial lung disease. Chest. 2008;133(5):1113–9.
Kubo H, et al. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005;128(3):1475–82.
Noth I, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(1):88–95.
Morawiec E, et al. Exacerbations of idiopathic pulmonary fibrosis treated with corticosteroids and cyclophosphamide pulses. Eur Respir J. 2011;38(6):1487–9.
Hozumi H, et al. Efficacy of corticosteroid and intravenous cyclophosphamide in acute exacerbation of idiopathic pulmonary fibrosis: a propensity score-matched analysis. Respirology. 2019;24:792–8.
Naccache JM, et al. Study protocol: exploring the efficacy of cyclophosphamide added to corticosteroids for treating acute exacerbation of idiopathic pulmonary fibrosis; a randomized double-blind, placebo-controlled, multi-center phase III trial (EXAFIP). BMC Pulm Med. 2019;19(1):75.
Inase N, et al. Cyclosporin A followed by the treatment of acute exacerbation of idiopathic pulmonary fibrosis with corticosteroid. Intern Med. 2003;42(7):565–70.
Sakamoto S, et al. Cyclosporin A in the treatment of acute exacerbation of idiopathic pulmonary fibrosis. Intern Med. 2010;49(2):109–15.
Aso S, et al. Effect of cyclosporine A on mortality after acute exacerbation of idiopathic pulmonary fibrosis. J Thorac Dis. 2018;10(9):5275–82.
Donahoe M, et al. Autoantibody-targeted treatments for acute exacerbations of idiopathic pulmonary fibrosis. PLoS One. 2015;10(6):e0127771.
Isshiki T, et al. Recombinant human soluble thrombomodulin treatment for acute exacerbation of idiopathic pulmonary fibrosis: a retrospective study. Respiration. 2015;89(3):201–7.
Arai T, et al. Recombinant thrombomodulin for acute exacerbation in idiopathic interstitial pneumonias. Respirology. 2019;24:658–66.
Hoopes CW, et al. Extracorporeal membrane oxygenation as a bridge to pulmonary transplantation. J Thorac Cardiovasc Surg. 2013;145(3):862–7; discussion 867–8
Trudzinski FC, et al. Outcome of patients with interstitial lung disease treated with extracorporeal membrane oxygenation for acute respiratory failure. Am J Respir Crit Care Med. 2016;193(5):527–33.
King TE Jr, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Taniguchi H, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821–9.
Ito Y, et al. Therapeutic effect of nintedanib on acute exacerbation of interstitial lung diseases. Respir Med Case Rep. 2019;26:317–20.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kumar, A., Hadley, R. (2020). Respiratory Failure in a Patient with Idiopathic Pulmonary Fibrosis. In: Hyzy, R.C., McSparron, J. (eds) Evidence-Based Critical Care. Springer, Cham. https://doi.org/10.1007/978-3-030-26710-0_30
Download citation
DOI: https://doi.org/10.1007/978-3-030-26710-0_30
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-26709-4
Online ISBN: 978-3-030-26710-0
eBook Packages: MedicineMedicine (R0)