
Abstract
The discovery of a reversible transition in the helical sense of a double-helical DNA was initiated by the first synthesis in 1967 of the alternating sequence poly[d(G-C)]. In 1968, exposure to high salt concentration led to a cooperative isomerization of the double helix manifested by an inversion in the CD spectrum in the 240–310 nm range and in an altered absorption spectrum. The tentative interpretation, reported in 1970 and then in detailed form in a 1972 publication by Pohl and Jovin, was that the conventional right-handed B-DNA structure (R) of poly[d(G-C)] transforms at high salt concentration into a novel, alternative left-handed (L) conformation. The historical course of this development and its aftermath, culminating in the first crystal structure of left-handed Z-DNA in 1979, is described in detail. The research conducted by Pohl and Jovin after 1979 is summarized, ending with an assessment of “unfinished business”: condensed Z*-DNA; topoisomerase IIα (TOP2A) as an allosteric ZBP (Z-DNA-binding protein); B–Z transitions of phosphorothioate-modified DNAs; and parallel-stranded poly[d(G-A)], a double helix with high stability under physiological conditions and potentially also left-handed.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
- Left-handed DNA
- CD left-handed DNA
- R–L transition of poly[d(G-C)]
- Z*-DNA
- TOP2A
- Parallel-stranded psRR-DNA
- Phosphorothioate-modified Z-DNA
1 Introduction
This account has been written at the request of the editors of this volume. I have complied with their kind invitation to relate the historical circumstances of the research initiated at the Max Planck Institute of Physical Chemistry (MPIpc) in Göttingen, Germany, on the “R–L transition,” which preceded by 11 years the publication in 1979–1980 of the first left-handed “Z-DNA” and B-DNA crystal structures. Unfortunately, my partner in the research, Fritz Pohl (“Fritz”), suffered an untimely, tragic death in 1994 at age 55. Thus, while I have tried to accurately reconstruct happenings of more than half a century ago—by relying on my lab books, publications, correspondence, and memory—the product undoubtedly contains errors and inconsistencies. My only defense is that they are unintentional due to incomplete records and the absence of Fritz’s insights and contributions to what would have been a mutual effort. The reader will hopefully forgive an inordinate degree of autobiographical detail, in my judgment required to provide a perspective of the evolving field of molecular biology during the 1960s–1970s.
2 Postdoctoral Experience with Arthur Kornberg at Stanford
I arrived in Palo Alto, California, in the fall of 1964, fresh out of medical school in Baltimore, to embark on what was to be a 3-year postdoc with Arthur Kornberg (“Arthur”) (Fig. 1) in the Department of Biochemistry of Stanford University Medical School. I was not unaware of nucleic acids and their properties. My introduction to DNA had been in 1956, as a freshman at Caltech in the chemistry class taught by Linus Pauling. He would describe features of the Watson–Crick (W–C) model of B-DNA, announced 3 years earlier, while extracting seemingly endless pairs of chains out of a small black bag. Pauling was an enthusiast of the W–C model, despite the criticism that had been leveled at his own (fundamentally flawed) alternative proposal for B-DNA.Footnote 1 At Stanford I participated in the extensive research activities focused on the general field of DNA biochemistry and associated technology development. A key project was a new purification and the biochemical, biophysical, and functional characterization of the “Kornberg enzyme,” E. coli DNA polymerase I (Pol I). Part of this effort was the introduction of polynucleotide celluloses (PTCs) as solid-state primers and templates for polymerases [2]. Despite their demonstrable utility for this and other uses, for example, in the discovery of DNA ligase by fellow Kornberg postdoc Nick Cozzarelli (later a guru of DNA topology and Chief Editor of PNAS), I failed in 1966 to successfully employ these early solid-support reagents in three successive projects that most readers (e.g., those <40 years in age) will find incomprehensibly archaic: [1] search for a putative distinct (i.e., separate from Pol I) enzyme responsible for the presumed 3′-5′ mode of lagging strand synthesis; [2] sequencing DNA; and [3] demonstration of indisputable “biological activity” of a synthetic DNA.

My two mentors, Arthur Kornberg (left) and Manfred Eigen (right; 1967). (The photo of Arthur is taken with permission from Springer Nature from Ref. [4]. The photo of Manfred was kindly provided by Ruthild Oswatitsch-Eigen.)
In the first project, I tried to isolate a bacterial enzyme activity that would extend a 5′-ppp terminated Tn sequence hybridized to PTC-An. This experiment necessarily failed since such an enzyme activity does not in fact exist.Footnote 2 In the second project, I proposed to use PTC as a primer template for sequencing one of the 5′-protruding “sticky” ends of lambda phage DNA, extended (“tailed”) by terminal transferase (TdT; DNA nucleotidylexotransferase).Footnote 3 I mention this because of my initial, control experiment, in which I attempted to “sequence” an oligonucleotide, 5′-d(G-C)4–3′, synthesized and provided by a frequent collaborator of the Kornberg lab, the extraordinary, “multi-p” (prescient, perseverant, prodigiously productive, profoundly gracious) scientist, Gobind Khorana. The expectation was that the hybridized configuration

should have led to the incorporation of G and the remaining residues complementary to the template oligonucleotide, thereby confirming the trial “sequencing.” Unfortunately, this control experiment, my first experience with an alternating d(G-C) sequence, failed, probably due to the formation of a thermodynamically favored three-stranded hybrid occluding the incorporation site for G. Discouraged, I abandoned the effort, especially since Ray Wu, working in the sister lab of Dale Kaiser, was succeeding in sequencing the ends of lambda DNA using a simpler primer extension protocol.Footnote 4
The concerted emergence of cloning, automated DNA synthesis and sequencing, and the application of biophysical principles revolutionized the biological sciences. I had had the enormous privilege of interacting at Stanford with Arthur as a demanding yet generousFootnote 5 supervisor. But it was also my great fortune to count on the mentoring and “intellectual companionship” of other, exceptional student and postdoc colleagues and faculty “heavyweights,” particularly biophysicist Robert Baldwin (“Buzz”) and fluorescence spectroscopist biochemist Lubert Stryer and also the biochemists Bob Lehman and Paul Berg. Buzz and Lubert were responsible for my going to Göttingen as an “Established Investigator” of the American Heart Association.Footnote 6 Buzz had been previously in Göttingen on a sabbatical with Manfred Eigen (“Manfred”) (Fig. 1), the originator of the theory and practice of ultrarapid chemical kinetic relaxation methods, e.g., the “temperature-jump,” and thought it would be advantageous for me to learn and apply these techniques before taking up a position I had been offered and in the meantime accepted in the Biology Department of MIT.Footnote 7
3 My Transition from Stanford to Göttingen, Germany
I arrived in the small town of Göttingen,Footnote 8 Germany, in July, 1967, to work at the Max Planck Institute of Physical Chemistry (MPIpc). The institute (Fig. 2) was located on the grounds of one of the world’s first aerodynamic research institutes operating a wind tunnel, not far from David Hilbert’s Institute of Mathematics or from the Institute of Physics of the University of Göttingen, the birthplace of modern quantum physics. I soon realized that I had transferred from one very high-powered scientific institution in the USA to an equally dynamic one in Germany, reflecting the initiative and intellectual virtuosity of 40-year-old Manfred, the Institute Director. Three months after my arrival, he was awarded a share of the 1967 Nobel Prize in Chemistry. But one perceived the unique scientific heritage of Göttingen in other ways; for example, Werner Heisenberg occasionally came by, and we often had lunch in the company of Otto Hahn.Footnote 9

Max Planck Institutes in Göttingen. Left: MPI of Physical Chemistry (1967). Not having been issued keys for the building, we used the windows at night to gain access to the labs and temperature-jump equipment with discharge voltages of up to 50 KV. Right: MPI for Biophysical Chemistry at its inauguration in 1971. It was merged with the MPI for Experimental Medicine in January 2022, yielding the MPI for Multidisciplinary Sciences
Installed in the MPIpc, I (an Argentine-American) shared a large lab with three colleagues who became lifelong friends and collaborators; each went on to very distinguished careers: Ernst Grell, a Swiss; Israel Pecht, the first scientific postdoc to Germany from Israel (the Weizmann Institute) after WWII; and Rudolf Rigler (“Rudolf”), a Swede-Austrian. We were an international cooking pot of young, hungry, iconoclastic scientists, and despite Manfred’s role as “master cook,” he didn’t stir the pot very often, having turned his attention to quantitative treatments of biological evolution.Footnote 10 Yet neither he nor the rest of us ignored the hot topics in molecular biology which at that time included allosterism, synthesis and sequencing of DNA and RNA, the identity and operation of the genetic code, DNA and RNA structure/function relationships, gene regulation in normal and in disease states, and receptor-dependent signaling in the nervous system. The resident and visiting scientific staff of the institute performed research in these areas since elucidating the underlying binding and conformational transitions was a challenge ideally matched to the new kinetic technologies with high (ns–μs–ms) temporal resolution. Experimental results and theoretical schemes were thrashed out in rapid-fire German at the notorious “Teestunde” (tea hour) sessions and the annual Winter Ski seminars in Austria and Switzerland.
My self-assigned goal was to determine the kinetics of binding and conformational transitions of complexes of the E. coli DNA polymerase I with its substrates, dNTPs and DNA. Additions to the lab, such as a scintillation counter, chromatography columns, and micropipettes, were made so as to purify polymerase starting from the paste of 2–3000 liters of bacterial culture imported from Iowa (recombinant DNA was still in the future). The intermediate and purestFootnote 11 fractions of the polymerase were to be essential tools in the work initiated with Fritz Pohl later in 1967. At the same time, I began acquiring expertise in the theory and practice of rapid chemical kinetics, including devising a fluorescent T-jump apparatus with Rudolf.Footnote 12
4 Fritz Pohl, a Visionary
Fritz, born in Graz, Austria, in 1939, obtained a degree in physics and in 1964 joined Manfred’s team at the MPIpc, first as a postdoc and then as a research associate. He developed a passion for molecular chirality and applied spectroscopic methods for studying the kinetics and thermodynamics of transitions of proteins and nucleic acids subjected to variations in solution conditions. Fritz was not only a gifted experimentalist; he was also very competent with the theoretical issues, particularly when novel insights and approaches were required. In 1967, Fritz developed a T-jump method to study the reversible denaturation of proteases in water and mixed solvents. A review of “cooperative conformational” transitions of globular proteins appeared in 1972. But already much earlier, he had turned his attention to salt-dependent transitions in DNA. On October 12, 1967, Fritz submitted a note (in German) to the journal Naturwissenschaften featuring the difference ORD (native, heat-denatured forms) of calf thymus and T4 bacteriophage DNA in 0.2 and in 6 M NaClO4 [6]. Fritz attributed the inversion of Cotton effects (Fig. 3, left) to a reversal in the helical sense of the DNA from right to left, subject to the assumption that base stacking was being preserved under both conditions.Footnote 13 This remarkable, short note also proposed how such a reversal in helical handedness might be involved in DNA synthesis, recombination (Fig. 3, right) transcription, and packing in chromosomes. The closing sentence is worth quoting: “The proposed model is one conceivable extension of the existing one (he is referring to W-C B-DNA, 1953), but is neither confirmed nor excluded by direct experiment.”

Fritz Pohl first invokes the existence of a transition of right-handed DNA to an alternative left-handed conformation at high salt concentration (Figure adapted from Ref. [6]). Left: difference ORD (25°–95°) of T4 bacteriophage DNA as a function of salt concentration. Right: model for strand exchange between two DNA molecules in a segment bridging left-handed (L) and right-handed (R) helical regions; a junctional structure was also incorporated
5 Birth of Poly[d(G-C)] in Göttingen
In view of the above and my background in “matters DNA,” Fritz and I engaged in a lively interchange of ideas, which quickly led to a working relationship and an enduring friendship (Fig. 4).

The author (left) and Fritz Pohl (right) at the MPIpc in 1967. Note the chiral positions. I will not reveal whether Fritz or I (or both) favored the left orientation
The challenge was to extend the suggestive experimental findings of Fig. 3 to better defined DNAs and a protocol in which the features expected of an intramolecular R(ight)–L(eft) transition would be observable and unambiguous: titratability, cooperativity, reversibility, and concentration independence. I had come to Göttingen with a rich assortment of synthetic DNAs, which constituted attractive samples because of their sequence uniformity, especially in the case of self-complementary dinucleotide sequences such as poly[d(A-T)]. This DNA, however, did not exhibit a perceptible transition between distinct ORD/CD spectra in high salt. It was/is also of low helical stability and capable of adopting alternative topological states such as hairpins and cruciforms. The obvious alternative to poly[d(A-T)] was poly[d(G-C], expected to have much higher inherent stability as a double helix. Unfortunately there was a fundamental problem with this choice: poly[d(G-C] had not been reported in the literature and was thus not available, either from research labs or commercially. The reason was that in contrast to poly[d(A-T)], neither poly[d(G-C] nor poly[d(I-C] had arisen spontaneously as a product of de novo (i.e., template-independent) reactions of the known DNA polymerases with dGTP (or the alternative dITP) + dCTP.
Being the birthplace after WWII of the Max Planck Society—the renaissance of the former Kaiser Wilhelm Society—Göttingen also housed a sister Max Planck Institute for Experimental Medicine (MPIem). Under the leadership of Fritz Cramer, the MPIem enjoyed international recognition in the field of nucleic acid chemistry and biochemistry, especially of RNAs. Eigen and Cramer organized periodic molecular biology symposia, attracting the luminaries in structural and molecular biology of the time to Göttingen from institutions such as the Laboratory of Molecular Biology (LMB) in Cambridge, England; the Institut Pasteur in Paris; and the Weizmann Institute in Israel and many other European, Asian, and US addresses. In the MPIem were two gifted, productive chemists sharing a passion for the element sulfur as a replacement for oxygen in the bases and sugar–phosphate backbone of nucleic acids. Karl-Heinz Scheit introduced the thioketo substitution into thymine and demonstrated that ds4TTP could function as a substrate in the enzymatic synthesis of DNA, e.g., poly[d(A-s4T)] [9]. At the same, Fritz Eckstein created the chiral phosphorothioate (PS) modification of the DNA backbone, substituting sulfur for one (or both) of the two nonbonding oxygens of the phosphate group [10, 11].Footnote 14 Both individuals and both of their innovations would play a very significant role in the work that Fritz Pohl and I would undertake with the “R–L transition” of poly[d(G-C)].
Despite certain misgivingsFootnote 15 the quest for a way to synthesize poly[d(G-C)] continued. During the late 1960s, the Kornberg and Khorana labs—with Robert Wells (“Bob”) as a chief protagonist—described in numerous publications the use of chemically synthesized deoxyribopolynucleotides with repeating short nucleotide sequences as templates for bacterial DNA polymerases. Unfortunately, in my hands, the d(G-C)4 oligonucleotide described earlier was inactive as a template. However, the synthesis of poly[d(G-T)·d(C-A)] and its separation into individual strands in alkaline CsCl gradients had been reported in 1965. We (Karl-Heinz Scheit and I) exploited this information and devised a rather elaborate scheme to synthesize poly[d(G-C)] by using poly[d(G-T)·d(C-A)] as a template and replacing dTTP with ds4TTP as an initial step (Fig. 5). The first reactions (in November 1967) went well, and the product, poly[d(s4T-G)·d(A-C]], was subjected to reductive amination of the thioketo group to an amino group, resulting in the conversion of s4T to m5C. Strand separation in CsCl yielded the desired poly[d(G-m5C)]. This DNA then served as a template, albeit a poor one,Footnote 16 for the synthesis of poly[d(G-C)], which after expansion (Fig. 5) was used in our first experiments in 1968 demonstrating the R–L transition. Unbeknownst to us, Bob Wells and his colleagues were also after poly[d(G-C)] at that time, and in 1972 they published its synthesis and characterization; poly[d(I-C)] was also featured [16]. Bob generously supplied us with these materials, I believe in 1970 ± 1, for use as templates and in comparison experiments. The new polynucleotides also became commercially available. It had turned out that poly[d(G-C)], after all, was not a biohazard.

Strategy (unpublished) for synthesizing poly[d(G-C)] in 1967; see text for more details
6 Salt-Dependent “R–L Transition” of Poly[d(G-C)]
In 1968 and 1969, the experiments with the new poly[d(G-C)] were yielding interesting results, and their interpretation was facilitated by parallel studies of the equilibria and kinetics of nucleic acid helix–coil transitions by Dietmar Pörschke, Manfred’s PhD student, as well as by the labs of institute “alumni” Buzz Baldwin and Don Crothers. We described the work at meetings and seminars. Our presentation at the winter’s meeting of the German Society of Biological Chemistry in 1970 was entitled (my translation from the German) “Kinetics of an ionic strength-dependent structural transition of synthetic DNA.” The published abstract [17] described a reversible, cooperative inversion of a Cotton effect (280, 300 nm) as the concentration of salt (NaCl, NH4Cl, NaClO4) was increased to 2–3 M. The reaction was temperature independent over 20–40 °C, occurred at neutral pH, and was first order with a time constant of 102–103 s. The large activation energy of both the forward (kf) and reverse (kb) rate constants was indicative of a concerted participation of several bases; kf was independent of the concentration in contrast to kb, which was highly dependent. The reaction was interpreted as an all-or-none interconversion between a right-handed double helix (R) and a left-handed double helix (L):
This abstract, and not our universally cited paper in 1972, was the first publication asserting the existence of a left-handed double-helical conformation of DNA, interconvertible in solution with right-handed Watson–Crick B-DNA. In 1971, during a sabbatical at the University of Bristol in England, Fritz made a presentation at the first European Biophysics Congress, “Isomerization of a double-stranded DNA,” which was then published [18]. The abstract stated:
The observations suggest a delicate energetic balance governing different conformations of double-stranded nucleic acids in solution which is influenced by the base sequence and the interactions with other molecules or ions. A possible cation binding site for a L-form of poly d(pur-pur) is proposed.
The related polymers poly[d(I-C)] and poly[d(G)]·poly[d(C)] did not undergo the transition. Fritz proposed that the O2 oxygens of cytosines of adjacent base pairs and two oxygens of the corresponding 3′ phosphate groups acted as equatorial ligands for a cation (Na+). However, the model did not seem to fit the left-handed double helix proposed by Bob Wells and colleagues for poly[d(I-C)], and deduced from fiber diffraction data and CD [19].
7 Birth of the Max Planck Institute for Biophysical Chemistry (1971)
I returned from a visit to Stanford, in April, 1971, accompanied by Donna Arndt, until then a postdoc in Paul Berg’s lab working on features of protein translation and SV40 cell biology. I had somehow persuaded her to join me in marriage and to further pursue her scientific career in Germany. One important selling point was the promise of life in a fourteenth-century castle, Schloss BerlepschFootnote 17 (Fig. 6).

Schloss Berlepsch. See footnote 17 for details
Another selling point for a career/life in Göttingen was the conception, construction, and inauguration (in 1971) of a flamboyant new Max Planck Institute for Biophysical Chemistry (MPIbpc) (Fig. 2), the realization of Manfred’s concept and dream to merge the disciplines of chemistry, physics, and biology for exploring the fundamental principles of life forms and their evolution. He somehow felt that I would fit into this scheme and asked me in 1968 whether I would consider joining the new institute (but first the MPIpc) as a Scientific Member of the Max Planck Society and Director of a new Department. Despite some hesitation (e.g., I would have to give up my childhood dream of being at MIT and would have to deal with German bureaucracy without mastery of the language), I accepted the offer to be considered for the position. The rather elaborate election/appointment procedures culminated in 1969 with the creation of a Department of Molecular Biology. It endured until my reaching emeritus status in 2007 and continued afterward as an Emeritus Laboratory of Cellular Dynamics.
Setting up the new labs and a research program in the new institute was a challenge, yet Donna and I managed, traversing the 30 km between the Schloss and the Institute back and forth every day. But in 1971, there was also scientific business to transact at the Schloss. Fritz returned from his sabbatical sometime in 1971 and joined our Department. It was partly in the spacious library of the castle (Fig. 6), surrounded by numerous incunabula and sometimes under candlelight, that he and I hammered out the first, lengthy paper on the R–L transition. The adjoining hall of armored knights provided additional inspiration.
The title of our definitive draft went something like “The salt-dependent transition of poly[d(G-C)] from a right-handed to a left-handed double helix.” The text included numerous references and discussions of contemporary proposals for potentially left-handed as well as right-handed structures based on fiber diffraction data; the DNA alphabet soup (A, B, C, D, E, etc.) was already extensive yet still growing. We asked Manfred for his appraisal of the paper, his criticism and advice. While he was (and had been) very positive about the experiments and results, he recommended against placing emphasis on a putative but unknown left-handed structure we were assigning to the high-salt conformation. True enough, Fritz and I had no proof, but it was our call, our decision to make. This we did and submitted the manuscript to the Journal of Molecular Biology in October, 1971; the publication appeared in 1972 [20]. In it, the word “left” was absent from the title and occurred only twice in the text.Footnote 18 In retrospect, the decision to back off from what we considered to be a plausible, defensible interpretation of the data was wrong, injudicious. Had we stuck to our guns, the field of “left-handed DNA” might have advanced more rapidly.
The most imposing and widely reproduced figure in the paper is that of the “inverted CD” spectrum near 290 nm of the “L form” of poly d[G-C)] in high salt (Fig. 7, left). This has become the CD signature of Z-DNA, although Fig. 7, right, of a spectrum reported in 1985 by the Tinoco group [8], revealed the much larger (~15x) differential CD signal of B-DNA and Z-DNA (difference between their respective maximal absolute values) in the vacuum UV at <220 nm, a region technically unavailable to us in 1968. Had we reported this result at that time, it might not have required a decade to obtain a crystal structure.

CD spectra of poly[d(G-C)] in the B, L(Z), and A forms. Left: B form in 0.2 M NaCl, pH 7.2, 25 °C; Z form after addition of NaCl to 3.9 M. (Adapted from Fig. 2 of Ref. [20]). Right: B form in 0.01 M Na phosphate, pH 7, 22 °C; Z form after addition of 2 M NaClO4; A form in 80% trifluoroethanol, 0.67 mM Na phosphate, pH 7. (Adapted from Fig. 1 of Ref. [8])
In the meantime, we sought to bolster our assertion that the L form retained a stacked base-paired helical structure by comparing the binding of the fluorescent intercalator ethidium bromide (EtBr) to the two DNA forms. The mechanism(s) of intercalation was a hot topic at the time, due to notable protagonists such as Jean LePecq and Hank Sobell. My first postdoc, Guillermo Ellenrieder, had been studying the rapid kinetics of EtBr binding to a series of oligo[d(A-T)] duplexes, and an obvious extension was to assess the kinetics and extent of reaction with poly[d(G-C)] under low- and high-salt conditions. A key question was whether the base pairs were still stacked in high salt such that intercalation could still be accommodated. In the first experiment performed in February, 1970, I added NaCl up to 3.9 M to a solution of DNA and EtBr, leading to a 35% reduction in fluorescence during a slow first-order reaction (time constant 23 min); the emission parameters (spectrum, polarization) remained constant. This and further experiments revealed that the salt-induced R–L transition was being inhibited and reversed by EtBr. In the ensuing publication [24], the binding reaction and conformational changes were followed by fluorescence and absorption spectroscopy. The remarkable conclusions were that the system operated via a concerted allosteric mechanism characterized by imperceptible binding to the L form and a 40th(!) power dependence on the free dye concentration, one of the highest degrees of cooperativity found in the biochemical literature.
8 Interim Period: 1972 Up to When Z-DNA Appeared in 1979
Fritz and I were well aware of the potential significance of our findings, yet we did not proceed systematically with investigations supplying proof of concept.Footnote 19 Nonetheless, Fritz did continue to generate and publish important biophysical information relevant to the R–L transition: [1] thermodynamic characterization of the helix–coil transition of oligo[d(G-C)] with the determination that the stability of a G·C base pair is close to that of 2 A·T base pairs [25]; [2] laser Raman scattering of the R and L forms and their interconversion [26]; [3] chain length dependence of the hypochromicity of short double helices [27]; and [4] assignment by NMR (1H and 31P chemical shifts) of the dinucleotide repeat structural element of the L form, a collaboration with Dinshaw Patel [28]. The attempt was made to correlate the results with the “alternating B-DNA” conformation proposed by Klug and coworkers for poly[d(A-T)] [29]. It is interesting that Fritz was ready to abandon the left-handed attribution to the L form at this juncture, shortly before the crystal structures appeared. For my part, I continued to work on the ethidium binding project, including the use and interpretation of relaxation kinetics, but was also immersed in the rather, broad research program of the new Department. This included further research on DNA polymerase and other protein DNA interactions—some based on the very useful spectroscopic properties of poly[dA-s4T] (Karl-Heinz Scheit had joined the Department as a senior group leader)—as well as other topics in cell biology and biophysics. The R–L transition was not forgotten but it was relegated to a back burner. Nonetheless, Fritz and I sounded out colleagues whenever the opportunity arose. At a celebrated biophysics summer school convoked by Manfred in 1968 at the incomparable Schloss Elmau in Bavaria, we consulted with Francis Crick about the R–L transition, but, to my recollection, did not receive much of a reaction. As Chris Calladine and Horace Drew wrote in their intriguing, inspiring volume Understanding DNA [30]:
Earlier solution studies by Fritz Pohl and Tom Jovin using circular dichroism methods had suggested that alternating C-G sequences such as CGCG might be either right-handed or left-handed, depending on the salt concentration, but only few crystallographers and other specialists had taken them seriously. (my italics)
Alex Rich (“Alex”) was certainly aware of our work, but I did/do not know whether he “took it seriously.” In 1977, Israel Pecht, Peter Richter, and I organized a symposium in celebration of Manfred’s 50th birthday, “Dynamics and Regulation of Evolving Systems,” again at Schloss Elmau. A Who’s Who of biophysics and molecular biology was in attendance, including Alex, who delivered a lecture entitled “Molecular structure and biological function of transfer RNA in contemporary and evolving biochemical systems.” I cannot recall (but that does not mean that they did not occur) public or private discussions about the ongoing crystallographic efforts of his lab, although by all later accounts they must have been well advanced at the time. Without a doubt, we discussed our work on the R–L transition with him. In addition the postdoc mentioned in footnote 5 had been Alex’s PhD student and was presumably serving as a conduit between the MPIbpc and MIT.Footnote 20
9 Z-DNA (Accompanied by B-DNA) Is Revealed and Proliferates (1979–)
The publications in 1979–1980 [31,32,33] from the Alex Rich lab at MIT (Andrew Wang and colleagues) and from the Richard Dickerson lab at Caltech (Horace Drew and colleagues) of the crystal structures of left-handed (a “surprise”) and right-handed (27 years after 1953!) double-helical DNA were epochal milestones in the history of biological “Wissenschaft.” The correspondence between left-handed d(C-G)n Z-DNA—with its zigzag sugar–phosphate backbone and dinucleotide repeat motif—and our 1972 L form of poly[d(G-C}] was proclaimed and then confirmed [34], and two seminal findings were announced: the first specific binding to Z-DNA of a protein, dsRNA adenosine deaminase, and identification of its Zα-binding domain [35], and the structural details (base pair disruption and base extrusion) of a B-Z DNA junction [36]. In fact, the emergence of Z-DNA in 1979–1980 launched a widespread, wide-ranging scientific effort, one which persists to this day. Thus, of great interest is a recent publication detailing the in silico search for Z-DNA−/Z-RNA-binding proteins in the complete PDB structure database and the AlphaFold2 protein models [37]. The study yielded 14 and 185 candidate proteins, respectively, suggesting that Z-DNA/Z-RNA recognition may indeed be a key feature of numerous cellular processes.
Fritz and I were delighted and intrigued by the unique structural features of Z-DNAFootnote 21 and what it implied about the previous work. The emergence of information and speculation about possible biological function(s) was rapid and extensive. In 1982, Alex organized a symposium at Cold Spring Harbor Laboratory, “Structures of DNA,” 1/7 of which was devoted to the topic “The Handedness of DNA.” Jim Watson wrote a foreword for the published proceedings, of which the following is an excerpt:
The double helix is deceptively simple. When first found in 1953, it appeared so beautifully clear that for a brief period it seemed that by mere visual inspection we must learn all its mysteries. Now almost 30 years later, DNA structure is no longer a child’s game, and those who play with it must be both experienced and of the courage to seek elegance among the almost overwhelming perturbations of its basic double-helical configuration. Not only can DNA be overcoiled or undercoiled, all under strict enzymatic control, it can turn to the left as well as to the right (my italics). These complexities are not laboratory artifacts but, in fact, provide the molecular underpinnings for the successful functioning of our genetic material, further progress in more firmly establishing the various forms of DNA is likely to be essential for the future of much biological research.
Both Fritz [39] and I [40] made presentations at the meeting that were published in the proceedings. Mine (with review character and many authors) summarized a substantial effort in our lab devoted to exploring the biophysical properties and the biological occurrence and possible biological repertoire of Z-DNA, an effort directed primarily by Donna Arndt-Jovin. We published other reviews in 1983 [41] and in 1987 [42], and a series of papers appeared up to 1996.Footnote 22 Let me identify a few “successes,” including some that contributed to the “Z” structural family and its dictionary: the demonstration and naming of Z*-DNA, an associated form of Z-DNA [43] (more about this below), and the demonstration and naming of “Z-RNA,” a collaboration with Kathy Hall and Ignacio “Nacho” Tinoco Jr. [44]. Other studies included [1] generation of antibodies and demonstration of binding to living tissue, e.g., polytene chromosomes; [2] biophysical studies directed at the thermodynamics, kinetics, topology, and spectroscopy of Z-DNA; [3] exploration of sequence, base, and phosphate modification space; [4] transcription of Z-DNA; and [5] recognition of topoisomerase IIA as a Z-DNA binding protein (more about this below). At the same time, we were also engaged in nucleic acid research unrelated to left-handed DNA, dealing with other “noncanonical” conformationsFootnote 23 such as triplexes, quadruplexes, exotic “triad” DNA (a double helix with a base triad per helical rise [48]), and parallel-stranded DNA (more about this below).
In Konstanz, Fritz continued to actively contribute to the field. He wrote to me on the last day of 1981: “You are right about Z-DNA becoming a hot topic and I admire the way Alex Rich pushes the whole topic.” Fritz and his associates [1] generated and applied anti-Z-DNA antibodies to identify and isolate [49] supercoiled plasmids expressing Z-targets; [2] searched for Z-loci in the E. coli genome [50]; and [3] demonstrated binding of anti-Z-DNA antibodies to form V DNA [51]. More biophysical studies included calculations of CD spectra [7] and the energetics and dynamics of the B–Z transition [52]. Nonetheless, Fritz’s activities became increasingly focused on another challenge, sequencing DNA. He developed novel instrumentation and was a pioneer in establishing European consortia for large-scale automated sequencing. In 1990, Fritz and his three sons started a small DNA sequencing company (GATC GmbH) which together with Fritz’s academic lab grew to the most productive contributor to the European consortia of genome projects and the Gene Alliance.
10 Unfinished Business
I will now briefly discuss issues that arose during our “R–L” research 25–40 years ago whose potential relevance, in my estimation, seems to have survived the passage of time and merit reconsideration.
-
Z*-DNA, a Cross-linked, Condensed Form of Z-DNA.
In 1982, we reported the B-to-Z transition of poly[d(G-C)] under the synergistic influence of very low Mg2+ concentrations and the cosolvent EtOH [43], or by use of the first row transition metal ions such as Mn2+ [53]. The product was designated Z*-DNA because it was sedimentable (although the solutions were not turbid), could support binding of various DNA-specific drugs, and served as a template for transcription. These studies were generalized to other DNAs and a variety of solution conditions leading to such condensed species [40, 41] (Table 1).
It was suggested that such condensed–associated properties of Z*-DNA could fulfill important structural and functional roles in the organization and dynamics of chromatin and chromosomes and in the genetic rearrangement and recombination of viral and other DNAs (Fig. 8). An extensive study of the structure, stability, and morphology of Z*-DNA in 1988 emphasized its unique properties [54]. Z*-DNA may also be a component of interaction sites with cytoskeletal elements such as intermediate filaments [55]. It would seem advantageous to undertake studies of such condensed states within the context offered by current knowledge of the “Z world” players: “flipon” loci and Z-DNA-binding proteins (ZBPs and their DNA and RNA targets) [56,57,58]. Emphasis could be placed on elucidating the structures of the condensed states and their protein complexes by use of higher-resolution microscopies (EM, cryo-EM, super-resolution fluorescence) and improved bioinformatic and molecular modeling tools currently available and to establishing relevance to cellular mechanisms of physiological and disease states—infection, immune response, cancer—already linked to Z-DNA [59, 60]. Modeling the lateral association of Z-DNA helices (e.g., the ZII variant) mediated by multivalent cations and hydration state modifiers known to promote the left-handed conformation would be very instructive.
-
Eukaryotic Topoisomerase IIα (TOP2A) Is an Allosteric ZBP.

Functional states and constructs involving Z*-DNA. Upper panel: equilibrium between R(B) and L(Z) states + intermediates in variable states of aggregation mediated by environmental factors, enzymatic activities, and ligand binding (ions, small molecules, proteins, including ZBPs). (Adapted from Fig. 1 of Ref. [41]). Lower panel: potential interactions in chromosomes mediated by Z*-DNA. Straight lines, R(B) form; jagged lines, L(Z) form. (a, b) Cross-linked and parallel arrays; (c, d) displacement of inherent R–L equilibrium (constant K) by ligands with specificity for R and L or junction J; (e) catalysis of R–L transition by a topoisomerase with some properties of a putative “conformase” [41]; (f) chromomere model with Z* DNA-stabilized base and loops incorporating facultative Z-forming segments. (Adapted from Fig. 20 in Ref. [41])
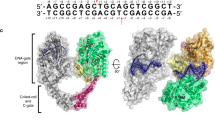
The properties of twisted topologies are key to understanding the roles of left-handed DNA [61, 62]. The generation of negatively supercoiled DNA (nscDNA) upstream of processive enzymes mediating transcription and replication is a general phenomenon leading to problematical DNA looping, entanglement and catenation. Local transitions of susceptible sequences to the left-handed Z conformation are also thermodynamically favored in nscDNA. The resolution of such biochemically “toxic” topological structures requires the action of topoisomerases. In 1993, our lab reported the isolation of a protein from Drosophila cells and embryos with a 100-fold greater affinity for Z-DNA than for B-DNA [63]. We established its identity with the known cellular topoisomerase IIα (TOP2A) and demonstrated a preferential binding to curved over linear DNA [64]. A further intriguing finding was that GTP irreversibly inactivated the enzyme while at the same time increasing its affinity for Z-DNA five- to tenfold. When bound to linear Z-DNA, TOP2A was unable to relax separate supercoiled DNA, although intramolecular relaxation of highly negatively supercoiled minicircles with Z-forming sequences proceeded efficiently [65]. It was proposed that the sensitive Z-DNA recognition by TOP2A could be important in targeting the enzyme to structural motifs of chromatin organization and to sites of local supercoiling as in replication and gene expression during embryogenesis and early development. These properties gave rise to an allosteric scheme proposed in 1993 and augmented here (Fig. 9) with the notion of TOP2A functioning as a ZBP that stabilizes a given state of a topological (sub)domain before or after it (and/or other molecules) exerts enzymatic activity. Such “Z-clamp” complexes could also serve as recognition targets and/or as blocks (“Z-barriers”), for example, of translation by DNA-bound proteins.

Conception of TOP2A as an allosteric ZBP. TOP2A binds to B-DNA and Z-DNA segments via “ATP clamps” [67, 69]. Relaxation proceeds directly from B-DNA loci but indirectly from Z-DNA loci. The affinity of TOP2A for Z-DNA sites is much higher than for B-DNA and increases even further under the allosteric influence of bound GTP, which also inhibits catalysis and thus DNA relaxation. These sites thus constitute potential “Z-clamps” and “Z-barriers” (see text). (Adapted from Fig. 11 of Ref. [63]. See also Fig. 12)
From the above it follows that TOP2A already in 1993 exhibited some (but not all!) the properties of a “DNA conformase” formulated by us in 1983 [41] and by Rich et al. in their seminal 1984 review of Z-DNA [66]. Yet although there have been extensive structural analyses of TOP2A and its complexes by crystallography [67, 68] and other techniques, none have featured Z-DNA. Thus, it is imperative to perform a crystallographic study of TOP2A ± GTP bound to Z-DNA(s) and to establish whether its function in the cell is indeed regulated via GTP in a manner linked functionally to GTPases, for example, the Ras superfamily, so prominent in growth signaling and tumorigenesis. Inasmuch as topoisomerases are the main targets of anticancer drug discovery, it can be anticipated that structural elucidation of the Z-DNA-related complexes will stimulate the design of novel agents.
-
Phosphorothioate (PS) Labeled DNA Avoids or Embraces the Z State.
Whereas the enzymatic synthesis of PS polynucleotides is chiral and can only target OR (Rp diastereomer), chemical synthesis allows substitution of either the OR or the OS (RS diastereomer) or both [11]. In a collaboration with Fritz Eckstein, poly[(d(G-C)] was synthesized with the Rp phosphorothioate (PS) modifications, d[Gp(S)C] and d[Cp(S)G] [11, 40, 41]. Models of the left-handed ZI and ZII conformations [70] are shown in Fig. 10.

Stereopair representations of phosphorothioate derivatives of poly[d(G-C)] with the RP diastereomers of the two constituent dinucleotides (left). Color scheme: bases, black; sugar–phosphate backbone, blue; S of d[Gp(S)C], green; S of d[Cp(S)G], red. Both ZI and ZII configurations [70] are depicted. Note the rotation of the dGpC phosphate group out of the minor groove in the ZII form. (Adapted from a figure generated by Reinhard Klement in 1983)
The dCpG and dGpC phosphodiester linkages in Z-DNA have different torsion angles which, together with the respective syn and anti glycosidic configurations and corresponding sugar puckers of the G, C deoxynucleosides, define the repeating dinucleotide unit characteristic of Z-DNA. The PS substitutions have served to assign the resonances of the corresponding 31P-NMR spectra of Z-form poly[d(G-C)] to the two steps of the dinucleotide repeat [40]. In the experiments referred to above, the stereospecific substitutions of oxygen with sulfur had profound effects upon the B–Z transition of poly[d(G-C)]: [1] The incorporation of the sulfur into the Rp dGpC step greatly potentiated the Z form, whereas the corresponding substitution in the Rp dCpG step completely blocked the B–Z transition under all conditions tested. With both substitutions present, the Z form was achieved, although less readily; [2] In a related study of oligonucleotides [71], both the sequence and the diastereomer proved to be important. Unmodified and Sp dGpC substituted d(G-C)4 molecules did not convert to the Z form, whereas the Rp substituted molecules did. Conversely, unmodified and Sp dCpG substituted d(C-G)4 molecules did convert, while the Rp substituted molecules did not. The PS [40, 72] as well as corresponding methylphosphonate substitutions [73] also alter thermal stability selectively. In studies of all-Rp and all-Sp substituted octamers [74], a key influence was attributed to hydrogen bonding of G-N2 to the 5′- OR phosphate in order to initiate the B–Z transition and to the G-N2 to 3′- OR water bridge to stabilize the Z form (Fig. 11). Both interactions would presumably be blocked or hindered in Rp-d[Cp(S)G]. Ultrahigh resolution has been recently achieved in cryo neutron crystallography of the left-handed Z-DNA duplex [d(CGCGCG)]2 [75]. Water molecules are resolved bridging pairs of cytosine N4 amino groups or pairs of guanine O6 keto groups on the convex surface, while in the minor groove transverse hydration patterns link both cytosine O2 oxygens and guanine N2 amino groups to backbone phosphates.

Progression through the B–Z transition. (a) H binding during initial rotation of G ring. (b) Water bridge stabilizing Z conformation. (c) Critical G-N2–OR bond in the W-DNA model of left-handed DNA [76]. (a, b) Reproduced with permission from ACS Publications from Fig. 1 of Ref. [74]. (c) Adapted from Fig. 8 of Ref. [45]
The preferential and/or actual pathway(s) of the B–Z transition remain(s) to be better elucidated. An exhaustive analysis of 13+ postulated mechanisms [77] includes a discussion praising—as does Dickerson in his wide-ranging and still relevant review of DNA “anatomy” [45]—the features of yet another proposed left-handed conformation, Z(WC) DNA or W-DNA (Dickerson designation) [76]. This model emphasizes the G-N2–OR bond (Fig. 11c) but, more importantly, provides a means for circumventing a fundamental steric dilemma of many (most) models of the B–Z transition, the chain sense paradox [45, 76, 77], which in simplest terms states that a simple unwinding of B-DNA does not, cannot, lead directly to the Z-DNA configuration. The feasibility or existence of W-DNA in solution and then in the cell, as an intermediate or an alternative left-handed conformation, remains to be established, including in the context of the PS modifications featured above. There is an additional practical consideration. PS derivatives—augmented, if desired, with other modifications affecting the B–Z transition, e.g., C5-pyrimidine adducts—can today be freely incorporated synthetically in a sequence positional and chiral manner. Such constructs would permit clamping given sequence segments in either a right-handed (transition-inhibited) or a left-handed (transition-promoted) configuration, even independently of external conditions. Such constructs would constitute valuable tools in studies of biochemical and biophysical mechanisms.
-
Parallel-Stranded Poly[d(G-A)], Yet Another Left-Handed DNA?
In 1988, stimulated by the theoretical work of Pattabiraman [78], our lab demonstrated the existence of stable double helices, denoted ps-DNA, formed from d(A)10 and d(T)10 stems joined at their end in such a manner enforcing a parallel orientation [79]. These studies were extended to numerous appropriately selected sequences with normal bases and unaltered sugar–phosphate backbones, which hybridized spontaneously under physiological solution conditions (reviewed in [80]). Base-pairing in sequences based on d(A·T) is reverse Watson–Crick; G·C base pairs are tolerated but destabilizing. We proposed numerous circumstances under which ps-DNAs could arise in combination with other conformational elements and function biologically [81] (Fig. 12). A surprising observation is that while parallel-stranded DNAs (ps-DNA) exhibit biophysical and biochemical properties differing from those of conventional antiparallel-stranded (aps) B-DNA, they are the most stable alternatives to B-DNA under physiological conditions [82]. A 2017 review of ps-DNA provides insights gained from natural sequences and constructs [83].

Models for integration of ps-DNA with other DNA and DNA·RNA structural elements. (a) Combinations of aps and ps regions and (top right) with R (right-handed) and L (left-handed) segments. (b) Mechanism proposed for reverse gyrase generation of positive supercoils involving a ps intermediate 2 leading to strand passage 3 and religation 4. (c) Stabilization of single-stranded genomes by ps helical segments; topological consequences are not depicted. (d) Alternative models for triplex stabilization during transcription of Ig class switching genes: 1, pur–pyr–pur triplex; 2, pyr–pur–pyr triplex stabilized by a ps RNA·DNA duplex with G·G and A·A self-pairs. (Adapted from Ref. [81])
The above presentation will now assume relevance with respect to the “left-handedness” theme by reference to a novel duplex form of DNA we introduced in 1992, denoted psRR-DNA, in which oligopurine strands pair in a parallel orientation under physiological conditions (10 mM MgCl2, neutral pH) [84]. The sequence leading to the greatest stability of psRR-DNA was alternating d(G-A)n, and numerous experimental methods established the parallel strand orientation and stabilization of the double helix via G·G and A·A base pairs. Stereochemical modeling and force-field calculations yielded a right-handed double helix with a trans orientation of the glycosidic bonds, syn for dG and anti for dA (Fig. 13a). For comparison is shown the crystal structure of poly[rA] under acidic conditions, also a ps double helix (Fig. 13b). The original fiber diffraction measurements of this homopurine dsRNA were by Alex Rich (with celebrated collaborators), one of the first new structural determinations after the appearance of W–C DNA in 1953.

Parallel-stranded DNA and RNA. (a) Model of the psRRG·A double helix adopted by the alternating sequence d(A-G)5. Color coding: sugars, white; phosphates (including 03′ and 05′), red; adenine, cyan; guanine, blue. (Adapted from Fig. 7 of Ref. [84]). (b) Crystal structure of ps-RNA double helix of r(A)11 under acidic conditions and presence of NH4+ ions (yellow, blue) H-bonded to adjacent intra-strand phosphate groups. (Adapted from Fig. 3 of Ref. [86]). (c) CD spectra of d(GA)15 at 7 °C without MgC12 (dashed line) and with 5 mM MgCl2 at 7 °C (solid line) and at 55 °C (dotted line). (Adapted from Fig. 3 of Ref. [84])
At one point (about 1995), molecular modeling of theoretician Vitaly Kuryavyi (“Vitaly”) in our lab (he is now at the Memorial Sloan Kettering Cancer Center) indicated that energetically superior structures for psRR-DNA could be constructed by reversing the helical sense, i.e., to that of a left-handed structure. This exercise was also prompted by the observed inversion of the vacuum UV CD spectra of duplexes stabilized by MgCl2 (Fig. 13c). The phenomenon is qualitatively, although not quantitatively, similar to that exhibited by poly[d(G-C)] (Figs. 7) and is generally regarded as an indicator of “left-handedness” [85]. To investigate the possibility experimentally, we (Elizabeth Jares-Erijman and I) devised a FRET method for establishing the helical sense of small duplexes, validating and calibrating the method with hybrid molecules consisting of two fused helical segments, systematically varied in length, one with B/Z character, d(m5C·G), and the other a psRYA·T DNA [87]. The FRET method was then adapted to psRRG-A DNA. The characterization of the constructed DNAs confirmed their duplex character, as had been true for other psRR-psRY joint molecules [88]. However, the FRET measurements yielded helical parameters that were not totally in accordance with expectation for a left-handed psRR conformation. A more detailed structural model, for example, incorporating repeat substructures, is probably required for this FRET technique. The conclusion is that one must resort to crystallography and/or other high-resolution imaging and spectroscopic techniques in order to establish the helical sense of psRRG-A DNA.
Vitaly has resumed modeling building of a left-handed psRR-DNA double helix and has graciously contributed the following preliminary statement regarding the (successful) outcome. A manuscript is in preparation (Kuryavyi, Rippe, Jovin).
Z-DNA is an antiparallel-stranded duplex composed of W-C nucleotide pairs with locally parallel oriented sugar residues. Co-orientation is a result of the purine dG adopting a syn glycosidic conformation. The stereochemically and thermodynamically favorable connectivity of nucleotide pairs with such a geometry results in regular flipping of the nucleotide pair along the DNA axis and thus two distinct steps in helical progression. At the dCpG step, the C2′ and C3′ of neighboring sugars face each other, and at the dGpC step, the O4′ atoms of the neighboring nucleotides face each other. At the dGpC step, stacking between the bases is pronounced, whereas at the dCpG step, the sugar residue of dC participates in stacking interactions with the guanine base.
Purine residues easily adopt syn and anti conformations and have three edges capable of H-bond formation: Watson Crick (WC) edge, Hoogsteen (H) edge, and sugar (S) edge [89]. This results in a substantial polymorphism of the ps duplex DNA models with alternating A·A and G·G base pairs. As in the double helix with isomorphous A·T and G·C base pairs, there is a possibility for isomorphic trans WC/WC G·G and A·A base pairs. However, the NMR structure of the self-associated d(CGA) trinucleotide has revealed a non-isomorphic geometry, G·G (SE/SE), and A·A (H/H) [90]. The stacking of bases G and A at the dGpA step is remarkable, with almost complete base overlap. However, the stacking is poor at the dApG step for base pairs with such geometries, and there are no X-Ray or NMR experimental data available for a ps dGAG homo-associated duplex motif. The distance between C1′ atoms of two constitutive dGs in SE/SE geometry is shorter than in the C+C base pair 5′ to it (7.5 Å vs. 9.5 Å). And while charge-charge interactions in a hemi-protonated C+C base pair contribute a driving force for self-association, the size similarity does not, inasmuch as the C+C base pair can be replaced by a Gsyn·Ganti trans WC/H base pair [91], with a C1′-C1′ distance Gsyn·G ≈ 11 Å. Our current left-handed model of psRRG-A DNA shares some similarity to stacking features of the dCpG and dGpC steps with those in Z-DNA. Noteworthy, however, are the clusters of Mg2+ ions along the entire axis of the left-handed duplex, as is a preference for an anti glycosidic orientation. The base pairing and duplex nature of left-handed psRRG-A DNA is retained after 100 ns of molecular dynamics computation.
11 Concluding Remarks
This chapter has dealt with many aspects of “nucleic acid polymorphism,” obviously with a focus on the “left-handed.” Stephen Neidle, a very astute observer of and contributor to the field and its evolution, has recently issued a cogent perspective “Beyond the double helix…” [92]. My impression about nucleic acid research is that it sometimes suffers from the “can’t see the forest for the trees” adage but that it is also somewhat like weaving. The major themes constitute the vertical warp threads while the shuttles of the horizontal weft threads represent individual investigators, each contributing in his/her manner to the patterns that emerge upon viewing the collective effort. These patterns can persist, fade away, or resurge. A distinguished colleague maintains that accessing the scientific literature prior to 2000 is not worthwhile. I hope that this chapter serves to refute such an assertion. In fact, I will quote from a scientist who over 2000 years ago made a pronouncement quite relevant to the “R–L transition”:
…where ‘twas left
It comes to be the right, and then again
Returns and changes round unto the left.
Titus Lucretius Carus
Of the Nature of Things (about 60 B.C.)
(Translation W.E. Leonard)
Finally, my eternal thanks to my patient wife and colleague, Donna, without whom little of what I have related would have come to pass. And I am likewise indebted to the Max Planck Society for 52 years of very generous support.
Notes
- 1.
Linus Pauling was a brilliant chemist but human. In his monumental book, The Nature of the Chemical Bond, he chastised Watson and Crick for omitting the third H-bond of the G–C base pair in their 1953 publications and not citing the proposal of a complementarity principle of replication he made together with Max Delbrück in 1940 [1]. Alex Rich was a postdoc with Pauling at Caltech until 1954.
- 2.
The emerging identification of Okazaki fragments and additional polymerases and cofactors of the bacterial DNA replication machinery revealed a mechanism of lagging as well as leading strand synthesis much more complex than anyone had anticipated. Arthur’s son Tom, an accomplished cellist, but interim (later full-time) biochemist while recovering from a hand injury, was responsible for key advances in the field.
- 3.
I introduced TdT, the “Bollum” enzyme, to the Biochemistry Department, much to the displeasure of Arthur, who regarded template-independent polymerases with disdain and who also complained of the stench and butcher-like character of purification procedures starting from kilogram amounts of calf thymus. He overcame his skepticism, especially after the Paul Berg lab down the hall used TdT for tailing DNA as part of the pioneering cloning strategies and experiments that ushered in the new age of molecular biology.
- 4.
This was the first sequence reported in the literature [3] of a significant length (13 bases) of a natural DNA. The third project cited above also deserves a brief mention because it reflected a certain hysteria prevalent at that time (about 1966). Criticisms were being leveled at research involving DNA synthesis in vitro because—as was maintained—one had failed to unambiguously demonstrate “biological activity” of the products or failed to exclude contributions from minute residual contamination with the original “natural” DNAs. Despite three Nobel Laureates (Arthur Kornberg, Josh Lederberg, Gobind Khorana) as “collaborators,” I failed to produce purely synthetic B. subtilis DNA fragments with demonstrable transforming capability. The probable cause was the progressive shortening of the DNA product through the many steps of the procedure, such that the minimum size required for gene expression and function was not maintained. Another postdoc in the lab, Mehran “Mickey” Goulian, later (1970) succeeded in “creating life in a test tube” using single-stranded phiX174 bacteriophage DNA. The media coverage was immense; molecular biology had been vindicated.
- 5.
By way of example, Arthur acquiesced to my requested purchases of prodigious quantities of rather expensive Sephadex G100, sometimes allowed me to beat him at tennis, and on one later occasion, while I was visiting from Germany, lent me his beloved MG convertible to drive down the coast on Highway 1 to LA, during the night, in order to interview a prospective postdoc. Arthur (1918–2007) is acknowledged to have been one of the foremost biochemists of the twentieth century. He was a truly exceptional human being [4].
- 6.
A joke. I had an M.D., no PhD, and only a 3-year postdoc to my credit.
- 7.
Younger readers of this article may disbelieve my assertion that during the late 1960s, postdocs in the better labs were inundated—to the point of annoyance—with job offers from the top institutions.
- 8.
Göttingen is located in the province of Niedersachsen (Lower Saxony), close to the geographical center of reunified Germany. The train station has a sign greeting one to “die Stadt (the town) die Wissen schafft.” This is a play of words around the German compound noun for science, Wissenschaft, which combines Wissen (knowledge) with the verb schaffen denoting the acts of generating, collecting, systematizing. The English “science,” derived from the Latin word for knowledge, “scientia,” lacks the “gathering” function. Göttingen, with only 120,000 inhabitants, has a unique history of scientific progress, being the birthplace of much (most) of modern chemistry, physics, and mathematics. Innumerable (>350) plaques affixed to the outer walls of venerable half-timbered houses feature names such as Gauss, Weber, Dirichlet, von Haller, Courant, Hilbert, Wöhler, Koch, Wallach, Nernst, Wigner, Debye, Windaus, Franck, Born, Heisenberg, Hahn, Debye, Klein, Noether, as well as the Grimm brothers, Goethe, King George II of Great Britain (and Ireland; he was also the Duke of Hannover and a Prince-elector of the Holy Roman Empire), Bismarck, Brentano, Lichtenberg, and Benjamin Franklin. During a 2-week visit in 1766, Franklin intensively consulted with two eminent professors of German constitutional law and history, greatly influencing his federalist theories that found their way into the US Constitution. More than 40 Nobel Laureates have either studied, performed research, or taught in Göttingen.
- 9.
We were also acutely aware of the thousands of Russian troops just over the nearby border with East Germany.
- 10.
Israel Pecht and I wrote a retrospective of Manfred Eigen upon his death in 2019 [5].
- 11.
The highly purified E. coli DNA polymerase I produced at the MPIpc became one of the first molecular biological offerings of Boehringer Mannheim. I also provided the enzyme to Fred Sanger upon his request—at the time he was developing his method for DNA sequencing.
- 12.
Rudolf went on to invent fluorescence correlation spectroscopy (FCS), a key tool of single-molecule biochemistry and biophysics. Elliot Elson, a graduate student of Buzz Baldwin, was the independent co-inventor of FCS; it has been my great privilege to have shared publications, albeit not about FCS, with both Rudolf and Elliot.
- 13.
- 14.
The PS substitution in oligonucleotides and polynucleotides renders them generally resistant to enzymatic degradation by nucleases. This and other properties have led to its widespread application in basic and applied chemistry and in biomedicine [12]. A recent finding is that PS occurs in nature (bacteria, archaea, etc.) and is found in the human microbiome [13].
- 15.
My search in 1967 for a strategy enabling the synthesis of poly[d(G-C)] was not straightforward. The reader may choose not to believe me, yet I can reveal that an important consideration was whether to attempt a synthesis at all. My (in retrospect naive) hesitation was based on the notion that poly[d(G-C)] might have extraordinary stability and other properties enabling it to irreversibly “take over” the “world” of DNA, first in the test tube, but more generally. Similar considerations had arisen with respect to “polywater,” a (postulated) polymerized form of liquid water that Russian scientists had reported in the late 1960s and which was only debunked in 1973 (some research on polywater had actually been initiated at the MPIpc). The fear had been that polywater would autocatalytically convert and thereby “inactivate” the world’s supply of liquid water. In addition, although the Asilomar Conference on Recombinant DNA convoked by Paul Berg would not take place until 1975, the potential biohazards of biotechnology were already under discussion.
- 16.
It was of course not known to us in 1967 that poly[d(G-m5C)] undergoes the R–L transition at all and even less that it does so with greater facility than poly[d(G-C)] [14]. The interesting question arises as to why this DNA served us as a template for further poly[d(G-C)] synthesis (Fig. 5), inasmuch as the midpoint of the B–Z transition is at 0.6 mM MgCl2 compared to the much higher 6.7 mM of the enzymatic reaction, implying that the template should have been predominantly in the Z form and presumably inactive. It was finally reported in 1987 that poly[d(G-m5C)] is indeed a progressively poorer template as [Mg2+] is increased [15].
- 17.
The Schloss (castle) was/is surrounded by extensive forest and agricultural holdings. Our apartment was equipped with a canopy bed and a piano and our monthly rent was about $80. Families of scientists working at the MPIpc were able to live in the castle as a benefit of Manfred’s friendship with the hereditary owner, the Graf (Count) von Berlepsch. Fritz, his wife Edda, and their three children—Fritz, Peter, and Thomas—lived there until 1970, in the apartment above ours.
- 18.
Yet we used the symbols “L” and “R” throughout and it took little imagination to deduce what they represented. The single letter designations of DNA helical forms are subject to ambiguity. A somewhat mysterious left-handed underwound “L-DNA” has appeared in torsion measurements of single molecules [21], and the same designation has been applied to the L-enantiomers (mirror images) of B-DNA [22]. The term Z-DNA has been subverted as well [23].
- 19.
A litany of what we could/should have attempted: [1] exploration of models of potential left-handed helical forms (would we have come up with G in syn? maybe but doubtful); [2] engagement of crystallographers to elucidate the structure of the L form (although we had ready access to our frequent expert visitors (LMB), there were others close at hand in Göttingen: Wolfram Saenger at the MPIem and, later, George Sheldrick at the University of Göttingen; admittedly, until 1979 no one had crystallized DNA oligonucleotide duplexes); [3] systematic exploration of chemical modifications of the bases and of the solution conditions facilitating the R–L transition; [4] fishing for proteins binding to the L form; [5] drawing fibers of the L form for X-ray diffraction studies; [6] exploring topological implications with closed circular or otherwise constrained DNA; [7] raising and applying antibodies to the L form for use on cytological specimens; and [8] searching for other small and large molecule ligands. All of this would have been possible in 1968 but happened only after 1979 in the collective effort of the scientific community instigated by the crystal structures.
- 20.
I do not wish in any way to imply criticism of Alex Rich, a person and scientist whom I regarded (he died in 2015) with the greatest admiration. But the fact is that some communications regarding ongoing structural studies of the interconversions of helical DNA were unnecessarily limited, impeding what could have been very fruitful collaborations. During the course of preparation of this manuscript, Horace Drew reminded me that I visited him and Dick Dickerson at Caltech in 1978 and encouraged his work on the d(C-G)2 tetramer.
- 21.
We did question, but only sotto voce, the designation “Z-DNA” because in our opinion it emphasized the peculiar disposition of the sugar–phosphate backbone but not the more fundamental feature of the Z(L) form, namely, its “left-handedness.” Judging from current knowledge about proteins binding to Z-DNA [37, 38], they (the proteins) would have agreed with us.
- 22.
During this period we had the privilege of working not only with a number of excellent students (notably Niels Ramsing, Lawrence McIntosh, and Karsten Rippe; I sometimes envision them in a reenactment of Destiny’s Child “Survivor”) but also (too many to name) postdocs, scientists on sabbatical, and/or collaborations based on grants and interlab visitations in Göttingen and outside; the work was reflected in many joint publications. But there were others with whom we shared our exploration of “Z-DNA space,” including Eric Westhof (modeling), Astrid Gräslund (dynamic light scattering), Daniela Rhodes and Aaron Klug (chromatin reconstitution), Etienne Delain (EM), David Kearns (NMR), Bob Wells (antibodies, polynucleotides, sequence searching), Bob Ratliff and Roger Wartell (spectroscopy), Struther Arnott (fiber diffraction), and Alfred Nordheim (after he returned from MIT to Germany, antibodies).
- 23.
It seems that a consensus exists in the field of nucleic acid conformation, specifically of double helices, that not genera (e.g., B-DNA, Z-DNA, etc.) but rather their constituent species (polymorphs identified by crystallography [30, 45], spectroscopy [46], and modeling [42, 47]) dictate biological function. Families of genera might be defined according to the fundamental attributes of [1] helical twist [right-handed (R), left-handed (L)], [2] relative strand orientation [antiparallel-stranded (aps), parallel-stranded (ps)], and base-pairing (sequence). However, an important distinction from the Linnaean taxonomic system is that the polymorphs are interconvertible, crossing the boundaries between genus and family, thereby being defined not merely by their chemical constituents but, and probably most importantly, by the chemical environment: solution components and conditions (ions, small molecules, pH, hydration, temperature, small and large ligands, topological constraints) and enzymatic activities dynamically affecting primary structure via modifications of the bases, sugars, and phosphate groups.
References
Pauling L, Delbrück M (1940) The nature of the intermolecular forces operative in biological processes. Science 92:77–79
Jovin TM, Kornberg A (1968) Polynucleotide celluloses as solid state primers and templates for polymerases. J Biol Chem 243:250–259
Wu R, Kaiser A (1968) Structure and base sequence in the cohesive ends of bacteriophage lambda DNA. J Mol Biol 35:523–537
Fuller RS, Bambara R, Baker T, Funnell B, Wahle E, O’Donnell M, Kaiser D, Skarstad K, Konforti B, Maki S (2008) A tribute to Arthur Kornberg 1918-2007. Nat Struct Mol Biol 15:2–17
Pecht I, Jovin TM (2019) Manfred Eigen (1927–2019). Science 364:33
Pohl FM (1967) Ein Modell der DNS-Struktur Naturwissenschaften 54:616
Richterich P, Pohl FM (1987) Calculation of the CD of oligo (dG-dC): influence of basic optical parameters. Biopolymers 26:231–250
Riazance JH, Baase WA, Johnson WC Jr, Hall K, Cruz P, Tinoco I Jr (1985) Evidence for Z-form RNA by vacuum UV circular dichroism. Nucleic Acids Res 13:4983–4989
Lezius A, Scheit K (1967) Enzymatic synthesis of DNA with 4-thio-thymidine triphosphate as substitute for dTTP. Eur J Biochem 3:85–94
Eckstein F (1966) Nucleoside phosphorothioates. J Am Chem Soc 88:4292–4294
Eckstein F (1985) Nucleoside phosphorothioates. Annu Rev Biochem 54:367–402
Eckstein F (2014) Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther 24:374–387
Sun Y, Kong L, Wu G, Cao B, Pang X, Deng Z, Dedon PC, Zhang C, You D (2020) DNA phosphorothioate modifications are widely distributed in the human microbiome. Biomol Ther 10:1175. (article number)
Behe M, Felsenfeld G (1981) Effects of methylation on a synthetic polynucleotide: the B-Z transition in poly (dG-m5dC) · poly (dG-m5dC). Proc Natl Acad Sci U S A 78:1619–1623
Ramesh N, Shouche YS, Brahmachari SK (1986) Recognition of B and Z forms of DNA by Escherichia coli DNA polymerase I. J Mol Biol 190:635–638
Grant RC, Kodama M, Wells RD (1972) Enzymic and physical studies on (dI-dC)n·(dI-dC)n and (dG-dC)n·(dG·dC)n. Biochemistry 11:805–815
Pohl FM, Jovin TM (1970) Kinetik einer ionenstärkeabhängigen Strukturänderung von synthetischer Desoxyribonucleinsäure. Hoppe Seylers Z Physiol Chem 351:124
Pohl FM (1971) Isomerisation of a double-stranded DNA. In: Broda H et al (eds) First European Biophysical Congress, Verlag der Medizinischen Akademie, Vienna, pp 343–347
Mitsui Y, Langridge R, Shortle BE, Cantor CR, Grant RC, Kodama M, Wells RD (1970) Physical and enzymatic studies on poly d(I-C)·poly d(I-C), an unusual double-helical DNA. Nature 228:1166–1169
Pohl FM, Jovin TM (1972) Salt-induced co-operative conformational change of a synthetic DNA: equilibrium and kinetic studies with poly(dG-dC). J Mol Biol 67:375–396
Marko JF, Neukirch S (2013) Global force-torque phase diagram for the DNA double helix: structural transitions, triple points, and collapsed plectonemes. Phys Rev E Stat Nonlinear Soft Matter Phys 88:062722. (article number)
Hauser NC, Martinez R, Jacob A, Rupp S, Hoheisel JD, Matysiak S (2006) Utilising the left-helical conformation of L-DNA for analysing different marker types on a single universal microarray platform. Nucleic Acids Res 34:5101–5111
Herbert A, Wang A, Jovin TM, Mocarski ES, Pasparakis M, Vasquez KM (2021) Concern over use of the term Z-DNA. Nature 594:333
Pohl FM, Jovin TM, Baehr W, Holbrook JJ (1972) Ethidium bromide as a cooperative effector of a DNA structure. Proc Natl Acad Sci U S A 69:3805–3809
Pohl FM (1974) Thermodynamics of the helix-coil transition of (dG-dC) oligomers. Eur J Biochem 42:495–504
Pohl FM, Ranade A, Stockburger M (1974) Laser Raman scattering of two double-helical forms of poly(dG-dC). Biochim Biophys Acta 335:85–92
Pohl FM (1974) Hypochromicity of short double helices. FEBS Lett 38:202–204
Patel DJ, Canuel LL, Pohl FM (1979) “Alternating B-DNA” conformation for the oligo(dG-dC) duplex in high-salt solution. Proc Natl Acad Sci U S A 76:2508–2511
Klug A, Jack A, Viswamitra M, Kennard O, Shakked Z, Steitz T (1979) A hypothesis on a specific sequence-dependent conformation of DNA and its relation to the binding of the lac-repressor protein. J Mol Biol 131:669–680
Calladine CR, Drew H (1997) Understanding DNA: the molecule and how it works. Academic Press, San Diego
Wang AH-J, Quigley GJ, Kolpak FJ, Crawford JL, Van Boom JH, van der Marel G, Rich A (1979) Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature 282:680–686
Drew H, Takano T, Tanaka S, Itakura K, Dickerson RE (1980) High-salt d(CpGpCpG), a left-handed Z′ DNA double helix. Nature 286:567–573
Wing R, Drew H, Takano T, Broka C, Tanaka S, Itakura K, Dickerson RE (1980) Crystal structure analysis of a complete turn of B-DNA. Nature 287:755–758
Thamann TJ, Lord RC, Wang AH, Rich A (1981) The high salt form of poly(dG-dC)· poly(dG-dC) is left-handed Z-DNA: Raman spectra of crystals and solutions. Nucleic Acids Res 9:5443–5458
Herbert A, Alfken J, Kim Y-G, Mian IS, Nishikura K, Rich A (1997) A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc Natl Acad Sci U S A 94:8421–8426
Ha SC, Lowenhaupt K, Rich A, Kim Y-G, Kim KK (2005) Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Nature 437:1183–1186
Bartas M, Slychko K, Brázda V, Cerven J, Beaudoin CA, Blundell TL, Pečinka P (2022) Searching for new Z-DNA/Z-RNA binding proteins based on structural similarity to experimentally validated Zα domain. Int J Mol Sci 23:768–785
Lee AR, Kim NH, Seo YJ, Choi SR, Lee JH (2018) Thermodynamic model for B-Z transition of DNA induced by Z-DNA binding proteins. Molecules 23:2748. (article number)
Pohl FM (1983) Salt-induced transition between two double-helical forms of oligo(dC-dG). Cold Spring Harb Symp Quant Biol 47(Pt 1):113–117
Jovin TM, van de Sande JH, Zarling DA, Arndt-Jovin DJ, Eckstein F, Fuldner HH, Greider C, Grieger I, Hamori E, Kalisch B, McIntosh LP, Robert-Nicoud M (1983) Generation of left-handed Z-DNA in solution and visualization in polytene chromosomes by immunofluorescence. Cold Spring Harb Symp Quant Biol 47(Pt 1):143–154
Jovin TM, McIntosh LP, Arndt-Jovin DJ, Zarling DA, Robert-Nicoud M, van de Sande JH, Jorgenson KF, Eckstein F (1983) Left-handed DNA: from synthetic polymers to chromosomes. J Biomol Struct Dyn 1:21–57
Jovin TM, Soumpasis DM, McIntosh LP (1987) The transition between B-DNA and Z-DNA. Annu Rev Phys Chem 38:521–558
van de Sande JH, Jovin TM (1982) Z*-DNA, the left-handed helical form of poly[d(G-C)] in MgCl2-ethanol, is biologically active. EMBO J 1:115–120
Hall K, Cruz P, Tinoco I Jr, Jovin TM, van de Sande JH (1984) ‘Z-RNA’–a left-handed RNA double helix. Nature 311:584–586
Dickerson RE (1992) [5] DNA structure from a to Z. Methods Enzymol 211:67–111
Marini M, Legittimo F, Torre B, Allione M, Limongi T, Scaltrito L, Pirri CF, di Fabrizio E (2021) DNA studies: latest spectroscopic and structural approaches. Micromachines 12:1294. (article number)
Moradi M, Babin V, Roland C, Sagui C (2013) Reaction path ensemble of the B-Z-DNA transition: a comprehensive atomistic study. Nucleic Acids Res 41:33–43
Kurayavyi V, Jovin TM (1995) Triad-DNA: a model for trinucleotide repeats. Nat Genet 9:339–341
Thomae R, Beck S, Pohl FM (1983) Isolation of Z-DNA-containing plasmids. Proc Natl Acad Sci U S A 80:5550–5553
Hoheisel JD, Pohl FM (1987) Searching for potential Z-DNA in genomic Escherichia coli DNA. J Mol Biol 193:447–464
Pohl F, Thomae R, DiCapua E (1982) Antibodies to Z-DNA interact with form V DNA. Nature 300:545–546
Pohl FM (1987) Left-handed DNA: energetic and dynamic aspects. In: Structure. Springer, Dynamics and Function of Biomolecules, pp 224–228
van de Sande JH, McIntosh LP, Jovin TM (1982) Mn2+ and other transition metals at low concentration induce the right-to-left helical transformation of poly[d (G-C)]. EMBO J 1:777–782
Chaires JB, Norcum MT (1988) Structure and stability of Z* DNA. J Biomol Struct Dyn 5:1187–1207
Li G, Tolstonog GV, Traub P (2003) Interaction in vitro of type III intermediate filament proteins with Z-DNA and BZ-DNA junctions. DNA Cell Biol 22:141–169
Chiang C, Li Y, Ng SK (2020) The role of the Z-DNA binding domain in innate immunity and stress granules. Front Immunol 11:625504. (article number)
Herbert A (2019) A genetic instruction code based on DNA conformation. Trends Genet 35:887–890
Herbert A (2020) ALU non-B-DNA conformations, flipons, binary codes and evolution. R Soc Open Sci 7:200222. (artice numbert)
Herbert A (2019) Z-DNA and Z-RNA in human disease. Commun Biol 2:2. (article number)
Ravichandran S, Subramani VK, Kim KK (2019) Z-DNA in the genome: from structure to disease. Biophys Rev 11:383–387
Xu W, Dunlap D, Finzi L (2021) Energetics of twisted DNA topologies. Biophys J 120:3242–3252
Kim SH, Jung HJ, Lee I-B, Lee N-K, Hong S-C (2021) Sequence-dependent cost for Z-form shapes the torsion-driven B–Z transition via close interplay of Z-DNA and DNA bubble. Nucleic Acids Res 49:3651–3660
Arndt-Jovin DJ, Udvardy A, Garner MM, Ritter S, Jovin TM (1993) Z-DNA binding and inhibition by GTP of Drosophila topoisomerase II. Biochemistry 32:4862–4872
Bechert T, Diekmann S, Arndt-Jovin DJ (1994) Human 170 kDa and 189 kDa topoisomerases II bind preferentially to curved and left-handed linear DNA. J Biomol Struct Dynam 12:605–623
Glikin GC, Jovin TM, Arndt-Jovin DJ (1991) Interactions of Drosophila DNA topoisomerase II with left-handed Z-DNA in supercoiled minicircles. Nucleic Acids Res 19:7139–7144
Rich A, Nordheim A, Wang AH (1984) The chemistry and biology of left-handed Z-DNA. Annu Rev Biochem 53:791–846
Schmidt BH, Osheroff N, Berger JM (2012) Structure of a topoisomerase II–DNA–nucleotide complex reveals a new control mechanism for ATPase activity. Nat Struct Mol Biol 19:1147–1154
Riccio AA, Schellenberg MJ, Williams RS (2020) Molecular mechanisms of topoisomerase 2 DNA–protein crosslink resolution. Cell Mol Life Sci 77:81–91
Roca J, Wang JC (1992) The capture of a DNA double helix by an ATP-dependent protein clamp: a key step in DNA transport by type II DNA topoisomerases. Cell 71:833–840
Wang A, Quigley GJ, Kolpak FJ, Van Der Marel G, Van Boom JH, Rich A (1981) Left-handed double helical DNA: variations in the backbone conformation. Science 211:171–176
Cosstick R, Eckstein F (1985) Synthesis of d(GC) and d(CG) octamers containing alternating phosphorothioate linkages: effect of the phosphorothioate group on the B-Z transition. Biochemistry 24:3630–3638
Boczkowska M, Guga P, Stec WJ (2002) Stereodefined phosphorothioate analogues of DNA: relative thermodynamic stability of the model PS-DNA/DNA and PS-DNA/RNA complexes. Biochemistry 41:12483–12487
Callahan L, Han F-S, Watt W, Duchamp D, Kézdy FJ, Agarwal K (1986) B-to Z-DNA transition probed by oligonucleotides containing methylphosphonates. Proc Natl Acad Sci U S A 83:1617–1621
Boczkowska M, Guga P, Karwowski B, Maciaszek A (2000) Effect of P-chirality of internucleotide bonds on B-Z conversion of stereodefined self-complementary phosphorothioate oligonucleotides of the [PS]-d(CG)4 and [PS]-d(GC)4 series. Biochemistry 39:11057–11064
Harp JM, Coates L, Sullivan B, Egli M (2021) Water structure around a left-handed Z-DNA fragment analyzed by cryo neutron crystallography. Nucleic Acids Res 49:4782–4792
Ansevin AT, Wang AH (1990) Evidence for a new Z-type left-handed DNA helix: properties of Z (WC)-DNA. Nucleic Acids Res 18:6119–6126
Fuertes MA, Cepeda V, Alonso C, Perez JM (2006) Molecular mechanisms for the B-Z transition in the example of poly[d(G-C)·d(G-C)] polymers. A critical review. Chem Rev 106:2045–2064
Pattabiraman N (1986) Can the double helix be parallel? Biopolymers 25:1603–1606
van de Sande JH, Ramsing NB, Germann MW, Elhorst W, Kalisch BW, Von Kitzing E, Pon RT, Clegg RC, Jovin TM (1988) Parallel stranded DNA. Science 241:551–557
Rippe K, Jovin TM (1992) Parallel-stranded duplex DNA. Methods Enzymol 211:199–220
Rippe K, Kuryavyi VV, Westhof E, Jovin TM (1992) Polymorphism and possible biological functions of parallel-stranded DNA. In: Structural tools for the analysis of protein-nucleic acid complexes. Birkhäuser, pp 81–107
Garcia AE, Soumpasis DM, Jovin TM (1994) Dynamics and relative stabilities of parallel-and antiparallel-stranded DNA duplexes. Biophys J 66:1742–1755
Szabat M, Kierzek R (2017) Parallel-stranded DNA and RNA duplexes - structural features and potential applications. FEBS J 284:3986–3998
Rippe K, Fritsch V, Westhof E, Jovin T (1992) Alternating d(G-A) sequences form a parallel-stranded DNA homoduplex. EMBO J 11:3777–3786
Rich A, Davies DR, Crick FHC, Watson J (1961) The molecular structure of polyadenylic acid. J Mol Biol 3:71–86
Safaee N, Noronha AM, Rodionov D, Kozlov G, Wilds CJ, Sheldrick GM, Gehring K (2013) Structure of the parallel duplex of poly(A) RNA: evaluation of a 50 year-old prediction. Angew Chem Int Ed Engl 52:10370–10373
Jares-Erijman E, Jovin TM (1996) Determination of DNA helical handedness by fluorescence resonance energy transfer. J Mol Biol 257:597–617
Evertsz EM, Rippe K, Jovin TM (1994) Parallel-stranded duplex DNA containing blocks of trans purine-purine and purine-pyrimidine base pairs. Nucleic Acids Res 22:3293–3303
Leontis NB, Stombaugh J, Westhof E (2002) The non-Watson–Crick base pairs and their associated isostericity matrices. Nucleic Acids Res 30:3497–3531
Robinson H, Wang A (1993) 5’-CGA sequence is a strong motif for homo base-paired parallel-stranded DNA duplex as revealed by NMR analysis. Proc Natl Acad Sci U S A 90:5224–5228
Saoji M, Zhang D, Paukstelis PJ (2015) Probing the role of sequence in the assembly of three-dimensional DNA crystals. Biopolymers 103:618–626
Neidle S (2021) Beyond the double helix: DNA structural diversity and the PDB. J Biol Chem 296:100553. (article number)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this protocol

Cite this protocol
Jovin, T.M. (2023). The Origin of Left-Handed Poly[d(G-C)]. In: Kim, K.K., Subramani, V.K. (eds) Z-DNA. Methods in Molecular Biology, vol 2651. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-3084-6_1
Download citation
DOI: https://doi.org/10.1007/978-1-0716-3084-6_1
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-3083-9
Online ISBN: 978-1-0716-3084-6
eBook Packages: Springer Protocols