
Abstract
Open or accessible chromatin typifies euchromatic regions and helps define cell type-specific transcription programs. DNA replication massively disorders chromatin composition and structure, and how accessible regions are affected by and recover from this disruption has been unclear. Here, we present repli-ATAC-seq, a protocol to profile accessible chromatin genome-wide on replicated DNA starting from 100,000 cells. In this method, replicated DNA is labeled with a short 5-ethynyl-2′-deoxyuridine (EdU) pulse in cultured cells and isolated from a population of tagmented fragments for amplification and next-generation sequencing. Repli-ATAC-seq provides high-resolution information on chromatin dynamics after DNA replication and reveals new insights into the interplay between DNA replication, transcription, and the chromatin landscape.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
- repli-ATAC-seq
- DNA replication
- Transcription
- Accessible chromatin
- Chromatin assembly
- Nucleosome-free regions
1 Introduction
Transcriptionally active chromatin is characterized by an open structure that permits entry of transcription factors and RNA polymerases to sites of transcription initiation, such as promoters and enhancers [1]. Such sites are characterized by nucleosome depletion, which renders DNA more accessible to transcription machinery [2]. These regions are important regulatory features of the chromatin landscape and, like the transcription programs they reflect, are cell type-specific [3]. These “accessible” regions can be profiled genome-wide using a number of strategies, including DNase-seq, MNase-seq, and FAIRE-seq [4]. An attractive alternative to these methods, which are labor-intensive and require large amounts of input material, is ATAC-seq, or assay for transposase-accessible chromatin [5]. This method profiles accessible regions in high resolution from low amounts of starting material and, with deep sequencing, can also inform on nucleosome positioning and occupancy, especially in organisms with smaller genomes.
DNA replication compromises the chromatin landscape by evicting proteins from DNA, temporarily disrupting chromatin structure [6]. This includes nucleosomes, which are evicted prior to replication fork passage and rapidly reassembled on the new DNA strands. Recycled parental histones are distributed largely symmetrically between daughter strands [7, 8], and in parallel, newly synthesized histones are deposited to restore nucleosome density [9]. Following replication, the chromatin landscape is extensively remodeled and modified to restore the prereplicative state [6].
DNA replication proceeds in a regulated, cell type-specific manner, with euchromatic regions replicated early in S phase and heterochromatic regions replicated late in S phase [10]. However, this process is inherently heterogeneous, driven by stochastic replication initiation from a large number of possible initiation zones (themselves found in accessible regions). This heterogeneity in the replication program, and its duration across many hours in most cell types, makes it impossible to use standard cell synchronization methods, including cell sorting and drug-based approaches, to address the transient changes in chromatin dynamics that occur in the wake of replication in high resolution.
To investigate how these local effects manifest genome-wide, multiple groups have developed methods that combine metabolic labeling of DNA with next-generation sequencing approaches [6]. Additionally, by chasing the DNA label, restoration of chromatin accessibility after replication can be investigated.
These novel methods have mainly adapted MNase-seq to investigate nucleosome positioning and occupancy in organisms with small genomes, including Saccharomyces cerevisiae [11,12,13] and Drosophila melanogaster [14]. In addition, analysis of subnucleosomal MNase fragments was used to assess transcription factor (TF) occupancy post-replication in D. melanogaster [14].
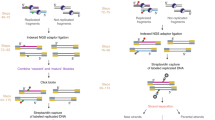
Here we describe replication-coupled ATAC-seq, or repli-ATAC-seq [15] (Fig. 1), a novel method established in mammalian cells to profile accessible chromatin genome-wide on replicated DNA. Repli-ATAC-seq produces genome-wide accessibility profiles with thorough coverage of open chromatin sites, which show high signal and generate clear peaks even in large genomes. By filtering in silico specifically for subnucleosomal-length fragments, repli-ATAC-seq can inform on transcription factor occupancy in replicated chromatin. The protocol described here was developed in mouse embryonic stem cells cultured in serum; cell culturing and lysis conditions may require testing in other cell types. This procedure, which can be completed in one day, starts from 100,000 asynchronous, cultured cells labeled with the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) for as little as 10 min, enabling high-resolution profiling of the nascent chromatin landscape. We recommend using low-binding plasticware throughout the protocol to minimize sample loss. To follow chromatin accessibility throughout chromatin maturation, we include a pulse/chase EdU labeling strategy. To compare baseline accessibility between samples, an option to include native D. melanogaster S2 cell chromatin as an internal spike-in control is described. Although classic ATAC-seq in asynchronous cells can be informative for comparison, we recommend that analyses compare nascent repli-ATAC-seq datasets to fully mature datasets to control for any technical differences introduced in the Click biotinylation and streptavidin pulldown steps. Repli-ATAC-seq offers a novel approach to address the interplay between DNA replication and transcription and provides the means to profile DNA replication-induced changes in rare and dynamic cell populations.

Schematic of repli-ATAC-seq protocol. The cell type of interest is pulsed with EdU and harvested. If using spike-in, freshly harvested, 100% EdU-labeled D. melanogaster S2 cells are mixed with the cells of interest prior to nuclei isolation and lysis. DNA is digested with Tn5 transposase and EdU+ DNA fragments are isolated through Click biotinylation and streptavidin conjugation. These fragments are then amplified and sequenced using next-generation sequencing. (Adapted from Ref. [15])
2 Materials
2.1 Equipment
-
1.
Cell culture hood.
-
2.
Hemacytometer or automated cell counter such as Countess (ThermoFisher).
-
3.
Microcentrifuge.
-
4.
Thermomixer.
-
5.
Magnetic 1.5 mL tube rack.
-
6.
Rotator with side movement.
-
7.
Qubit 2.0 Fluorometer (Life Technologies).
-
8.
BioAnalyzer (Agilent).
-
9.
Thermocycler.
2.2 Reagents
-
1.
Cell culture reagents (cell type-specific).
-
2.
1X Phosphate-buffered saline (PBS).
-
3.
Trypsin.
-
4.
PCR-grade H2O.
-
5.
100% ethanol.
-
6.
1.5 mL LoBinding tubes.
-
7.
LoBinding pipette tips.
-
8.
Illumina Tagment DNA Enzyme and Buffer Kit (Illumina).
-
9.
Qiagen MinElute PCR Purification Kit (Qiagen).
-
10.
EdU, 20 mM stock dissolved in DMSO and aliquoted at −20 °C (ThermoFisher).
-
11.
Click-IT cell reaction buffer kit (ThermoFisher).
-
Click-IT cell buffer additive (Component C) (80 mg): Dissolve in 400 μL deionized H2O (100X) and store in 200 μL aliquots at −20 °C for up to 1 year.
-
-
12.
THPTA, 50 mM stock in PCR-grade H2O (Sigma).
-
13.
Picolyl-Azide-PEG4-Biotin, 100 mM stock dissolved in DMSO and stored at 4 °C (Jena Bioscience).
-
14.
Qiagen MinElute Reaction Cleanup kit (Qiagen) (optional, only for making unbound libraries).
-
15.
Agencourt AMPure XP beads (Beckman Coulter).
-
16.
Myone T1 streptavidin beads (ThermoFisher).
-
17.
NEB Next High-Fidelity 2X PCR Master Mix (NEB).
-
18.
Qubit HS assay (ThermoFisher).
-
19.
Agilent HS DNA kit (Agilent).
-
20.
Triton-X 100.
-
21.
Tween 20.
-
22.
10 mM Tris–HCl pH 7.5.
2.2.1 For Thymidine Chase to Study Chromatin Maturation
Thymidine, 10 mM stock dissolved in deionized H2O and aliquoted at −20 °C (Sigma).
2.2.2 For D. melanogaster Spike-in
-
1.
D. melanogaster S2 cells.
-
2.
Shields and Sang M3 Insect Medium (Sigma).
-
3.
KHCO3 (Sigma).
-
4.
Yeast Extract (Sigma).
-
5.
Bactopeptone (BD).
-
6.
Fetal Calf Serum (GE Hyclone).
-
7.
Penicillin/Streptomycin (GIBCO).
-
8.
Cell incubator at 25 °C.
2.3 Buffers
-
1.
Buffer A without protease inhibitors: 10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.05% Triton-X. Store filtered and without Triton-X at 4 °C for long-term use and add Triton-X just before use.
-
2.
Lysis buffer: 10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.3% Igepal. Store without Igepal at 4 °C for up to 24 h before use and add Igepal just before use. Discard any remaining buffer after using.
-
3.
Tagmentation Stop Buffer (TSB): 50 mM Tris pH 8, 10 mM EDTA pH 8, 1% SDS. Store at RT.
-
4.
2× B & W buffer: 10 mM Tris–HCl pH 7.5, 1 mM EDTA, 2 M NaCl, 0.1% Tween 20.
-
5.
1× B & W buffer: 5 mM Tris–HCl pH 7.5, 0.5 mM EDTA, 1M NaCl, 0.05% Tween 20.
-
6.
Elution Buffer: 10 mM Tris–HCl pH 8.5.
-
7.
EBT Buffer: 10 mM Tris–HCl pH 8.5, 0.05% Tween 20.
3 Methods
3.1 EdU Labeling
-
1.
Seed 2 × 106 mESCs per sample on a 10 cm gelatinized dish in 10 mL of appropriate media and grow them for 24 h at 37 °C, 5% CO2 (see Note 1).
-
2.
After preparing reagents (see Note 2), begin EdU labeling.
Option 1: Nascent Repli-ATAC-seq
-
1.
Replace culturing media with 10 mL EdU media (final concentration: 20 μM) in the corresponding dish, swirl to disperse, and incubate at 37 °C for 10 min (see Notes 3 and 4).
-
2.
After 10 min, aspirate media and wash twice in 10 mL warmed 1X PBS.
-
3.
Add 2 mL trypsin and return to 37 °C for 1–2 min. Take the cold media out of the fridge and put it under the hood.
-
4.
Check under a microscope for cell detachment. When cells are detached, add 6 mL of COLD media to stop trypsin digestion.
-
5.
Transfer cells to a 15 mL Falcon tube.
-
6.
Take 10 μL of the cell suspension for counting using a hemacytometer or an automated cell counter.
-
(a)
Goal: 100,000 cells for repli-ATAC-seq per sample.
-
(a)
-
7.
Transfer 100,000 cells into a low-binding 1.5 mL tube. Spin cells at 500 × g for 5 min at 4 °C. From this step, keep the tubes on ice (see Note 5).
Option 2: Mature Repli-ATAC-seq
-
1.
Replace culturing media with 10 mL EdU media (final concentration: 20 μM) in the corresponding dish, swirl to disperse, and incubate at 37 °C for 10 min (see Notes 3 and 4).
-
2.
After 10 min, aspirate media and wash twice in 10 mL warmed PBS.
-
3.
Add 10 mL warmed media with 10 μM thymidine to the plate. Return to incubator for the desired maturation time.
-
4.
After the desired maturation interval (see Note 6), aspirate media and wash twice in 10 mL warmed PBS.
-
5.
Add 2 mL trypsin and return to 37 °C for 1–2 min. Take the cold media out of the fridge and put it under the hood.
-
6.
Check under a microscope for cell detachment. When cells are detached, add 6 mL of COLD media to stop trypsin digestion.
-
7.
Transfer cells to a 15 mL Falcon tube.
-
8.
Take 10 μL of the cell suspension for counting using a hemacytometer or an automated cell counter.
-
(a)
Goal: 100,000 cells for ATAC-seq per sample.
-
(a)
-
9.
Transfer 100,000 cells into a low-binding 1.5 mL tube. Spin cells at 500 × g for 5 min at 4 °C. From this step, keep the tubes on ice (see Note 5).
Option 3: D. melanogaster Spike-in
-
It may be useful to include an internal control of accessibility on replicated DNA. This can be done by EdU-labeling cultured D. melanogaster S2 cells for 40 h (see Note 7) prior to labeling repli-ATAC-seq dishes and adding 100 labeled cells to trypsinized and aliquoted mESCs prior to lysis:
-
1.
Culture D. melanogaster S2 cells in suspension following general procedures as described by the Drosophila Genomics Resource Center [16].
-
2.
Replace culturing media with EdU media (final concentration: 10 μM) in the corresponding dish, swirl to disperse, and incubate at 25 °C for 40 h. Time the EdU labeling such that S2 cells will be fully labeled at the time of labeling nascent and any mature repli-ATAC-seq samples.
-
3.
Transfer cells to 50 mL tubes and spin at 300 × g for 5 min.
-
4.
Aspirate supernatant and wash cells in an equivalent volume of ice-cold 1X PBS.
-
5.
Repeat wash.
-
6.
Take 10 μL of the cell suspension for counting using a hemacytometer or an automated cell counter.
-
(a)
Goal: 100 cells per repli-ATAC-seq sample.
-
(a)
-
7.
Transfer 100 fresh, EdU-labeled S2 cells into tubes containing cells prepared in parallel for repli-ATAC-seq. Place tubes on ice and proceed to cell lysis.
-
1.
3.2 Cell Lysis
-
1.
To prepare, aliquot 995 μL of Buffer A into a 1.5 mL tube and add 5 μL of 10% Triton-X 100 to the aliquot. Invert to mix. For steps 2 and 3, work in a 4 °C room.
-
2.
Carefully remove media (see Note 8) from labeled cells and add 200 μL of cold 1X Buffer A with Triton-X. Pipet up and down 5 times to resuspend, being careful to avoid creating bubbles.
-
3.
Incubate on ice for 7 min. Lay tube on ice in the cold room, but avoid burying the tube in the ice (see Note 9).
-
4.
Pellet nuclei by spinning at 1300 × g for 5 min at 4 °C and carefully remove lysis buffer as in step 2, here using a P200 set to 198 μL and gel-loading tips to aspirate all supernatant.
-
5.
Add 100 μL of cold 1X lysis buffer and pipet up and down 10 times to resuspend, being careful to avoid creating bubbles.
-
6.
Split sample into two 1.5 mL low-binding tubes each containing 50 μL lysate (equivalent to 50,000 cells) each.
-
7.
Vortex samples for 10 s on medium-high strength.
-
8.
Incubate on ice for 15 min at room temperature (RT) (bury tubes in ice).
-
9.
After incubation, vortex tubes for 10 s at medium-high strength again. Pellet nuclei by spinning at 600 × g for 10 min at 4 °C and carefully remove lysis buffer as in steps 2 and 4, here using a P200 set at 48 μL and gel-loading tips to aspirate all supernatant.
3.3 Transposase Digestion
-
1.
Combine 2.5 μL 2X TD buffer and 2.5 μL transposase (TDE1) (from Illumina kit) per tube or as a master mix and aliquot into tubes on ice.
-
2.
Pipet up and down 10 times to resuspend. Vortex on medium-high strength for 10 s.
-
3.
Incubate for exactly 30 min at 37 °C in a thermocycler shaking at 1200 rpm.
-
4.
After incubation, combine the two transposase digestions from each sample into one 1.5 mL low-binding tube (final volume: 10 μL).
-
5.
Add 90 μL TSB to the combined digestions (final volume: 100 μL).
-
6.
Purify with Qiagen MinElute PCR Purification Kit. To elute, add 80 μL PCR-grade H2O to columns and incubate for 5 min at RT. Spin at maximum speed for 1 min and then re-elute samples by adding the eluate back onto its respective column and incubating for a further 5 min at RT. Spin at maximum speed for 1 min and proceed to Click biotinylation (total volume, accounting for loss: approximately 78 μL) (see Note 10).
3.4 Click Biotinylation
-
1.
Prepare THPTA–CuSO4 premix by mixing 1 μL 50 mM THPTA and 0.1 μL 100 mM CuSO4 per sample in a separate 1.5 mL low-binding tube.
-
2.
Prepare 10X buffer additive by mixing 1 μL 100X buffer additive and 9 μL PCR-grade H2O per sample in a separate tube.
-
3.
Set up the Click reaction by adding the reagents to the purified DNA in the following order: 10 μL 10X Click-IT buffer, 0.5 μL 100 mM picolyl-azide-PEG4-biotin, 1.1 μL THPTA–CuSO4 premix, 10 μL 10X buffer additive (see Note 11).
-
4.
Incubate for 30 min at RT.
-
5.
During incubation, equilibrate AMPure beads at RT for 30 min prior to use. Keep AMPure beads at RT to use after Library Amplification (see Subheading 3.6).
-
6.
To purify DNA, add 55 μL equilibrated AMPure beads to each sample (0.55:1 bead ratio).
-
7.
Mix thoroughly by vortexing.
-
8.
Incubate the tube(s) at RT for 10 min to bind large, unwanted DNA fragments to the beads.
-
9.
During incubation, prepare another silconized 1.5 mL tube with 245 μL AMPure beads.
-
10.
During incubation, prepare 400 μL of 80% ethanol per sample.
-
11.
During incubation, warm a thermoblock to 37 °C.
-
12.
Place the tube(s) on the magnet to capture the beads. Incubate until the liquid is clear.
-
13.
Carefully remove the supernatant and transfer it to the corresponding prepared tube containing AMPure beads (3:1 final ratio). Discard tube(s) containing used beads (see Note 12).
-
14.
Incubate tube(s) at RT for 10 min to bind the desired DNA fragments to the beads.
-
15.
Place the tube(s) on the magnet to capture the beads. Incubate until the liquid is clear.
-
16.
Carefully remove and discard the supernatant.
-
17.
Keeping the tube(s) on the magnet, add 200 μL of freshly prepared 80% ethanol. On the rack, turn the tubes 180°, forcing the beads through the ethanol to the opposite wall of the tube.
-
18.
Incubate the tube(s) on the magnet at RT for ≥30 s.
-
19.
Carefully remove and discard the ethanol.
-
20.
Repeat steps 17–19 once. Try to remove all residual ethanol without disturbing the beads, using a P10 pipette if necessary.
-
21.
Dry the beads at RT for 1–2 min. Caution: Avoid overdrying of the beads, as it may result in dramatic yield loss.
-
22.
Remove the tube(s) from the magnet. Resuspend the beads in 52 μL of Elution Buffer.
-
23.
Put the tube(s) with lid(s) open in a warmed thermoblock at 37 °C. Cover with a top of a tip box or a piece of aluminum foil to prevent contamination of open tubes.
-
24.
Incubate for 5–10 min to elute DNA and evaporate residual ethanol.
-
25.
Place the tube(s) on the magnet to capture the beads. Incubate until the liquid is clear.
-
26.
Carefully transfer 50 μL of the supernatant to a new low-binding tube.
3.5 Streptavidin Pulldown
-
1.
Resuspend the stock of Myone T1 streptavidin beads by vortexing.
-
2.
Pipet 20 μL of bead suspension per sample into a 1.5 mL DNA low-binding tube. Pellet the beads using a magnetic rack (≥30 s). Remove and discard the supernatant.
-
3.
Remove the tube from the magnetic rack and add 200 μL of 1X B & W buffer. Mix by pipetting. Place the tube back to the magnetic rack to pellet the beads. Remove and discard the supernatant.
-
4.
Repeat 1X B & W buffer wash 3 times.
-
5.
Resuspend washed streptavidin beads in 50 μL 2X B & W buffer per sample.
-
6.
Add 50 μL resuspended streptavidin beads to each sample (final B & W concentration 1X). Mix by pipetting.
-
7.
Incubate tubes for 30 min at RT on a tube rotator. Ensure beads are continually in suspension.
-
8.
Spin tubes briefly. Pellet beads on a magnetic rack. Remove the supernatant (see Note 13).
-
9.
Wash beads with 200 μL 1X B & W buffer and mix by pipetting.
-
10.
Pellet the beads using a magnetic rack (≥30 s). Remove and discard the supernatant.
-
11.
Repeat steps 9 and 10 3 times, waiting 1 min off the magnetic rack between washes. Perform washes on maximum 4–6 reactions at a time to avoid overdrying the beads.
-
12.
Wash beads as in steps 9 and 10 twice with 200 μL EBT Buffer.
-
13.
Wash beads as in steps 9 and 10 once with 200 μL 10 mM Tris–HCl pH 7.5.
-
14.
Pellet the beads on a magnetic rack and carefully remove all supernatant.
-
15.
Resuspend the beads in 10 μL PCR-grade H2O, transfer to a 0.2 mL low-binding tube, and keep on ice. Proceed to Library Amplification (see Note 14).
3.6 Library Amplification (on Beads)
-
1.
Set up the PCR reaction by adding the following reagents to bead-bound DNA in 0.2 mL tubes: 1.25 μL 25 μM Primer 1, 1.25 μL 25 μM Primer 2 (see Note 15), 12.5 μL NEB Next High-Fidelity 2X PCR Master Mix.
-
2.
Vortex to mix and spin down briefly.
-
3.
Amplify libraries using the following conditions: 72 °C, 5 min; 98 °C, 30 s; 12 cycles of: 98 °C, 10 s; 63 °C, 30 s; 72 °C, 30 s; 4 °C hold.
-
4.
Add 25 μL PCR-grade H2O to each library (final volume: 50 μL).
-
5.
To purify libraries, add 80 μL equilibrated AMPure beads to each sample (1.6:1 bead ratio).
-
6.
Mix thoroughly by vortexing.
-
7.
Incubate the tube(s) at RT for 10 min to bind DNA fragments to the beads.
-
8.
During incubation, prepare 400 μL of 80% ethanol per sample.
-
9.
During incubation, warm a thermoblock to 37 °C.
-
10.
Place the tube(s) on the magnet to capture the beads. Incubate until the liquid is clear.
-
11.
Carefully remove and discard supernatant.
-
12.
Keeping the tube(s) on the magnet, add 200 μL of freshly prepared 80% ethanol.
-
13.
Incubate the tube(s) on the magnet at RT for ≥30 s, turning the tubes 180° to ensure all beads pass through the ethanol.
-
14.
Carefully remove and discard the ethanol.
-
15.
Repeat steps 12–14 once. Try to remove all residual ethanol without disturbing the beads, using a P10 pipette if necessary.
-
16.
Dry the beads at RT for 1–2 min. Caution: Avoid overdrying of the beads, as it may result in dramatic yield loss.
-
17.
Remove the tube(s) from the magnet. Resuspend the beads in 12 μL of Elution Buffer.
-
18.
Put the tube(s) with open lids in a warmed thermoblock at 37 °C. Cover with a top of a tip box or a piece of aluminum foil to prevent anything from falling into the open tubes.
-
19.
Incubate for 5–10 min to elute DNA and evaporate residual ethanol.
-
20.
Place the tube(s) on the magnet to capture the beads. Incubate until the liquid is clear.
-
21.
Carefully transfer 10 μL of the supernatant to a new 1.5 mL low-binding tube.
3.7 Quality Control
Prior to sequencing, perform quality control of repli-ATAC-seq libraries by quantifying library concentration with Qubit and checking library fragment length distribution using an Agilent BioAnalyzer or an equivalent fragment analyzer (Fig. 2).

Representative BioAnalyzer profiles of a repli-ATAC-seq library (top) and its matched, unbound control library (bottom)
3.8 Sequencing and Analysis
Libraries can be sequenced on an appropriate sequencing platform (repli-ATAC-seq was developed using Illumina NextSeq 500). Paired-end sequencing will provide both positional information and fragment length for all reads; single-end sequencing will provide positional information only. To enable analysis specifically of subnucleosomal fragments, we therefore prefer paired-end sequencing, though this may not be necessary for all research questions. After sequencing, quality assessment using FastQC and adaptor trimming using TrimGalore! or a similar software is recommended. PCR duplicates, reads mapping to the mitochondrial genome, and reads mapping to any relevant sequencing blacklists (e.g., the 10 mm sequencing blacklist [17]) should be discarded, as should reads with a MAPQ score < 20. The remaining reads can be processed using peak-calling and other standard bioinformatic analyses. If D. melanogaster spike-in was employed, map to both the target and D. melanogaster genomes. From the D. melanogaster Binary Alignment Map (BAM) file, calculate the total number of unique reads. Calculate a spike-in normalization factor for each sample by dividing 106 by the total number of unique reads from D. melanogaster. To quantify spiked-in samples by reference-adjusted reads per million (RRPM), calculate the coverage of each bin or region of interest in the target genome by computing the number of unique reads per bin. Then, multiply by the spike-in normalization factor, prior to any log transformation or further manipulation of the data.
4 Notes
-
1.
When setting up repli-ATAC-seq experiments, inclusion of an unlabeled, EdU-negative dish processed in parallel can be useful to ensure the experiment is free from any contamination from unlabeled DNA fragments in repli-ATAC-seq libraries.
-
2.
Prior to beginning EdU labeling, ensure the following reagents are ready and at the appropriate temperature:
-
Microcentrifuge is cooled to 4 °C.
-
Thermomixer is warmed to 37 °C.
-
6 mL of appropriate media per dish is cooled to 4 °C.
-
2 mL trypsin per dish is warmed to 37 °C.
-
20 mL 1X sterile PBS per dish is warmed to 37 °C.
-
Lysis buffer is freshly prepared and kept cold on ice.
-
EdU media is prepared: 10 mL appropriate media with 20 μM EdU per 10 cm dish, prepared and warmed to 37 °C.
-
If generating mature samples (Option 2), prepare 10 mL of appropriate media with 10 μM thymidine per dish and warm to 37 °C.
-
-
3.
For pulse/chase experiments, it is recommended to stagger your EdU labeling such that all dishes are ready for collection at the same time because the first pause point following EdU labeling is after 2 h of processing.
-
4.
The length of EdU labeling may need to be optimized depending on the proliferation rate of the cell type of interest.
-
5.
The remaining cells can be kept short-term on ice and processed, e.g., as FACS controls during the transposase digestion.
-
6.
One cell cycle post-pulse, EdU-labeled loci will replicate again; maturation times should therefore be well below one cell cycle length for the cell type of interest.
-
7.
Labeling S2 cells with EdU for 40 h will ensure genome-wide labeling.
-
8.
It is critical to avoid disturbing or losing the cell pellet. To do this, first pipet out 900 μL supernatant using a P1000, then switch to a P200 set to 98 μL and, using gel-loading tips to pipet from the bottom of the sample while not disturbing the pellet, aspirate the remaining supernatant.
-
9.
During incubation, the remaining, saved cells can be spun 500 × g for 5 min, and resuspended in 300 μL PBS. To fix, add 700 μL ice-cold 100% EtOH dropwise while vortexing on low and save at 4 °C for FACS labeling.
-
10.
Purified DNA can be quantitated for quality control at this stage. It can be kept short-term (1–2 days) at 4 °C or frozen at −20 °C for long-term storage, but library quality seems poorer when this is done. The recommendation is to continue immediately to Click biotinylation.
-
11.
The order in which the reagents for Click biotinylation are added to samples is important because the addition of the buffer additive starts the Click reaction. It is not recommended to make a master mix of Click-IT reagents.
-
12.
This double size-selection removes fragments larger than approximately 600 bp, since longer fragments are both more difficult to sequence on Illumina platforms and generally less informative for accessibility studies. If these fragments would be informative in specific experiments, a 1.8:1 ratio in a single size-selection could be used to preserve and purify fragments of all lengths.
-
13.
If desired, the supernatant can be removed and saved in a new 1.5 mL tube. This contains tagmented, non-EdU-labeled DNA fragments and can be used to generate “unbound” libraries. Unbound libraries are virtually interchangeable in terms of coverage with standard bulk ATAC-seq libraries, and can complement repli-ATAC-seq libraries. To create unbound libraries, purify supernatant using the MinElute Reaction Cleanup Kit, eluting in 12 μL PCR-grade H2O or EB buffer. Amplify and purify libraries as described in Subheading 3.6, except use 8 cycles of PCR instead of 12 to amplify libraries.
-
14.
Streptavidin-captured DNA can be stored 4 °C for a short time, but it is recommended to proceed directly to PCR after streptavidin capture.
-
15.
Primer sequences are from ref. [5], except that in repli-ATAC-seq the final two nucleotides in each primer are joined by a phosphorothioate bond. Primer 2 is indexed for multiplexing sequencing lanes.
References
Core LJ, Martins AL, Danko CG et al (2014) Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46:1311–1320
Li W, Notani D, Rosenfeld MG (2016) Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet 17:207–223
Boyle AP, Davis S, Shulha HP et al (2008) High-resolution mapping and characterization of open chromatin across the genome. Cell 132:311–322
Furey TS (2013) ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet 13:840–852
Buenrostro JD, Giresi PG, Zaba LC et al (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10:1213
Stewart-Morgan KR, Petryk N, Groth A (2020) Chromatin replication and epigenetic cell memory. Nat Cell Biol 22:361–371
Petryk N, Dalby M, Wenger A et al (2018) MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361:1389–1392
Yu C, Gan H, Serra-Cardona A et al (2018) A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 361:1386–1389
Annunziato AT (2013) Assembling chromatin: the long and winding road. Biochim Biophys Acta 1819:196–210
Marchal C, Sima J, Gilbert DM (2019) Control of DNA replication timing in the 3D genome. Nat Rev Mol Cell Biol 20:721–737
Fennessy RT, Owen-Hughes T (2016) Establishment of a promoter-based chromatin architecture on recently replicated DNA can accommodate variable inter-nucleosome spacing. Nucleic Acids Res 44:7189–7203
Vasseur P, Tonazzini S, Ziane R et al (2016) Dynamics of nucleosome positioning maturation following genomic replication. Cell Rep 16:2651–2665
Gutierrez MP, MacAlpine HK, MacAlpine DM (2019) Nascent chromatin occupancy profiling reveals locus- and factor-specific chromatin maturation dynamics behind the DNA replication fork. Genome Res 29:1123–1133
Ramachandran S, Henikoff S (2016) Transcriptional regulators compete with nucleosomes post-replication. Cell 165:580–592
Stewart-Morgan KR, Reverón-Gómez N, Groth A (2019) Transcription restart establishes chromatin accessibility after DNA replication. Mol Cell 75:284–297.e6
Luhur A, Klueg KM, Roberts J, Zelhof AC (2019) Thawing, culturing, and cryopreserving drosophila cell lines. JoVE 146. https://doi.org/10.3791/59459
ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Stewart-Morgan, K.R., Groth, A. (2023). Profiling Chromatin Accessibility on Replicated DNA with repli-ATAC-Seq. In: Marinov, G.K., Greenleaf, W.J. (eds) Chromatin Accessibility. Methods in Molecular Biology, vol 2611. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2899-7_6
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2899-7_6
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2898-0
Online ISBN: 978-1-0716-2899-7
eBook Packages: Springer Protocols