
Abstract
Fibrillar collagen is an abundant extracellular matrix (ECM) component of interstitial tissues which supports the structure of many organs, including the skin and breast. Many different physiological processes, but also pathological processes such as metastatic cancer invasion, involve interstitial cell migration. Often, cell movement takes place through small ECM gaps and pores and depends upon the ability of the cell and its stiff nucleus to deform. Such nuclear deformation during cell migration may impact nuclear integrity, such as of chromatin or the nuclear envelope, and therefore the morphometric analysis of nuclear shapes can provide valuable insight into a broad variety of biological processes. Here, we describe a protocol on how to generate a cell-collagen model in vitro and how to use confocal microscopy for the static and dynamic visualization of labeled nuclei in single migratory cells. We developed, and here provide, two scripts that (Fidler, Nat Rev Cancer 3(6):453–458, 2003) enable the semi-automated and fast quantification of static single nuclear shape descriptors, such as aspect ratio or circularity, and the nuclear irregularity index that forms a combination of four distinct shape descriptors, as well as (Frantz et al., J Cell Sci 123 (Pt 24):4195–4200, 2010) a quantification of their changes over time. Finally, we provide quantitative measurements on nuclear shapes from cells that migrated through collagen either in the presence or the absence of an inhibitor of collagen degradation, showing the distinctive power of this approach. This pipeline can also be applied to cell migration studied in different assays, ranging from 3D microfluidics to migration in the living organism.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Cell-collagen culture
- 3D and 4D imaging
- Cell migration
- Nuclear shape
- Morphometric analysis
- Nuclear irregularity index
1 Introduction
Cell migration in three-dimensional (3D) extracellular matrix (ECM) plays a pivotal role in physiology, such as in development, tissue maintenance, and immunity of living organisms, but also in pathological processes, as during cancer progression [1]. Collagens, the most abundant fibrous proteins of the interstitial ECM, are organized into fibrils and bundles of particular stiffness that often are packed into dense architectures that leave little space for migrating cells [2]. Cell migration into these three 3D environments therefore often requires the morphological adaptation of the cell body, including its nucleus, which forms the largest and stiffest cell organelle. The ability of the cell nucleus to deform is determined by chromatin compaction state, expression levels and functionality of nuclear lamina and LINC (linker of nucleoskeleton and cytoskeleton) proteins, the presence of vimentin, actin nucleation, and actin-myosin contractility, and will change with ECM stiffness via a functional ECM-adhesion-actin-LINC-nucleus force axis [3,4,5]. All these determinants provide the nucleus with a certain stiffness that will control to which extent cells can deform to enable transmigration through small pores [6,7,8]. The shape of the cell nucleus therefore often serves as a marker for the adaptability of the cell to these determinants during physically confining migration, and several typical nuclear shapes have been previously described [6] (Fig. 1a). To simulate the various characteristics of the ECM observed in vivo, such as scaffold stiffness or pore dimensions, migration is often studied within reconstituted collagen-based hydrogels in tissue culture models [6, 9,10,11]. Using dense collagen lattices, we have shown that migrating tumor cells expressing matrix metalloproteinases (MMPs) create proteolytic track-like paths of exactly the diameter of the moving cell [12]. Upon MMP inhibition, cells negotiated this confined space by adaptation of cell and nuclear shape, serving as a suitable cell-collagen model to study the extent of nuclear shape change.

Nuclear shape descriptors. (a) Test shapes resembling typical nuclear shapes of migrating cells. The hourglass-like shape represents a shape of both mononucleated cells in network-like confinement and a simple form of the typical multilobular nuclear shape of neutrophils. (b) Overview of the formulas used by Fiji/ImageJ to calculate the indicated shape descriptors. (c) Schematic illustration of the components used in the calculation of the nuclear irregularity index (NII) in the Fiji macro. Image adapted from [16]. (d) Quantification of each test shape using the indicated shape descriptors. Red dotted line representing the NII value of 2.2146 for a circle matches the calculated value from the circular test shape
To quantify the impact of various determinants on nuclear deformation and their shape changes during 3D cell migration and differentiation, the application of nuclear morphometric analysis tools onto 2D shapes such as nuclear diameter, aspect ratio (AR), circularity, or roundness (Fig. 1b), readily available in Fiji ImageJ software, have proven to be very useful [6, 13, 14]. In addition to AR and circularity, other descriptors exist, such as the ratio of maximal and minimal radius and bounding box descriptors for static shapes. All these shape descriptors have been integrated into the nuclear irregularity index (NII) [15] (Fig. 1c; see Note 1). Whereas for shapes captured at high resolution (Fig. 1a) the different single descriptors appear similarly powerful in indicating changes (Fig. 1d), the combination of shape descriptors into the NII has been shown most powerful when shapes were captured at lower resolution, often necessary for live-cell imaging [15, 16]. We therefore prefer the NII over particular single shape descriptors, which can also be used to describe nuclear shape changes over time [16].
In this chapter, we describe procedures for the preparation of collagen-based cell cultures (Fig. 2), their visualization (Fig. 3), and the segmentation and quantification of nuclear shapes from migrating cells, employing a custom-made script within the Fiji ImageJ software (Fig. 4). Using the matrix metalloproteinase inhibitor GM6001 in our experiments, we present images from tumor cells after migration in collagen by proteolytic and proteolysis-independent strategies (Fig. 5). Finally, using a second custom-made script, we demonstrate the application of dynamic nuclear morphometrics during cell migration (Fig. 6).

Preparing a 3D cell-collagen culture for imaging. (a) Procedure of cell-collagen mixture preparation, with indicated pH values and indicator colors at each pipetting step. (b) The cell-collagen mixture was pipetted into either large well plates as a dome-like drop gel (top), or into small well plates (bottom), where the well was then half-filled and covered with culture medium. Right; prior to imaging, drop gels were transferred into a microscopy chamber using an angled spatula (top), or cell-collagen cultures were imaged directly (bottom)

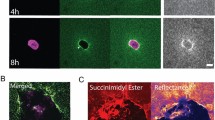
Three- and four-dimensional imaging of cell-collagen cultures. (a) Left, schematic depiction of image acquisition from a 3D cell-collagen culture as z-stacks with a 1–2 μm step size (red lines) and particular thickness (left double arrow). Right, z-stacks were generated at one or several set time points (here referred to as T1–T3). (b, c) Tumor cells with fluorescently labeled nuclei migrating in a collagen matrix were monitored as z-stacks from either a PFA-fixed sample (b) or a live sample at 37 °C (c) by confocal microscopy. (b) Sequence of successive focal planes, with indicated increasing depth in μm, and the resulting collapsed view of the cell nuclei (right). Phal, phalloidin; cyan arrowheads point to tracks in the collagen matrix that have been generated by proteolytically moving cells. (c) Time sequence of cells moving in collagen, with indicated time points in min. Maximal projections of nuclear signal are either merged with transmission signal to visualize cell bodies (top) or reflection signal to visualize collagen (bottom). Upper right, overview of the nuclei after 40 min, with positions of nuclei at 0 min outlined in yellow, and with movement trajectories depicted as blue dotted lines. Lower right, overlay of nuclear signal from a selected nucleus (white rectangle) of a moving cell during 40 min. All image bars, 50 μm, except from inset in (C)(10 μm)

Image processing for the quantification of nuclear shape parameters. Flowchart along the arrows indicates the developed image processing pipeline executed by the Fiji macro. Top, a multichannel image z-stack (left) was collapsed (middle) and the channel with nuclear signal was selected (right). Middle, after applying auto-thresholding based on fluorescence intensity (right), nine nuclei were selected based on particle size (60-pixel infinity; see yellow outlines; middle) and, lastly, visually controlled for cell integrity (left). ROI, region of interest. Cyan arrowheads, condensed DNA fragments from apoptotic cells, which were excluded from analysis by size exclusion (no outline). Bottom, nuclear area and indicated shape descriptors were calculated for the segmented cell nuclei (1–9). Blue box, thin line, shape descriptions that are summed up to NII, thick line

Quantification of nuclear shapes after cell migration in 3D collagen. For the calibration of nuclear shape parameters, circle-shaped beads in collagen and cell-collagen cultures were imaged at the same resolution by confocal microscopy. (a, c) Maximum projections of fluorescein (FITC)-coated beads (left), and HT1080 tumor cells migrating in collagen in the absence (middle) or presence (right) of MMP inhibitor GM6001. H2B-mCh, histon 2B-mCherry; Phal, phalloidin. Cyan arrowheads, cell-derived collagenolytic tracks in the collagen matrix. White arrowheads, nuclei with hourglass-shaped deformations; empty arrowheads, nuclei with protrusions; asterisks, nuclear fragments; M, mitotic nuclei. Cells in (a) are cropped-out examples from C, dotted rectangles). (b) Table depicting values of selected shape descriptors for a bead, nondeformed, and deformed nucleus, as well as the ratio between deformed nucleus and bead. AR, aspect ratio; Circ., circularity. (d, e) Aspect ratio, circularity, and NII, calculated from overlays of nuclear shapes in (c). Each dot depicts a bead or nucleus; red lines, medians. Dotted blue lines depict the value of a perfect circle. Dotted black rectangles in (e) indicate a distinct subpopulation. For all experiments, 12–47 shapes per condition were analyzed. *** p < 0.001; ** p < 0.01; * p < 0.1; ns, nonsignificant (Mann-Whitney t-test)

Quantification of nuclear shape changes during cell migration in 3D collagen. For this approach, the individual NII values derived from the Fiji macro and their differences between time steps were calculated. (a) Maximal projections of tumor cells migrating in collagen at indicated time points. Top, outlines of nuclei in magenta; bottom, trajectories of followed nuclei in yellow. White numbers, cells further analyzed in (c) and (e), red numbers, cells excluded from analysis. (b) Schematic depiction of the different NII values per cell and time step produced by the macro. NII(T1) corresponds to time point 1, NII(T2) to time point 2, etc. (c) NII values over time, as determined from the outlines in (a) (also see Supplemental Movie 1). (d) Schematic depiction of calculation of the mean NII change (mean ΔNII) per cell over time. (e) Mean ΔNII per cell calculated from (c)
2 Materials
2.1 Embedding of Single Cells
-
1.
Cell culture plates (see Note 2).
-
2.
Incubator with controlled environmental conditions (humidified atmosphere, 37 °C, 5% CO2).
-
3.
Migratory cells (adherent cells, such as fibroblasts, mesenchymal or epithelial tumor cells, or cells from suspension culture, such as T lymphocytes, neutrophils, etc.), optionally pre-labeled (see Note 3).
-
4.
Culture medium: Appropriate growth medium for a given cell type, 10% fetal calf serum (FCS), 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U/mL penicillin, 100 mg/mL streptomycin.
-
5.
For adherent cell culture: Phosphate-buffered saline (PBS): 136.9 mM sodium chloride (NaCl), 2.7 mM potassium chloride (KCl), 8.0 mM sodium hydrogen phosphate (Na2HPO4), 1.9 mM potassium dihydrogen phosphate (KH2PO4), pH 7.2–7.4.
-
6.
For adherent cell culture: 2 mM ethylenediaminetetraacetic acid (EDTA) in PBS.
-
7.
10× Minimum Essential Medium Eagle (MEM) with pH indicator.
-
8.
0.5 M sodium hydroxide solution (NaOH).
-
9.
Acidified collagen type I stock solution (rat tail origin; Corning; lot-dependent concentration, e.g., 10.80 mg/mL; see Note 4).
2.2 Fixation and Labeling
-
1.
Fresh 4% paraformaldehyde (PFA) in 0.1 M Sorensen’s phosphate buffer, pH 7.4.
-
2.
In case a 6–24-well plate is used for hydrogel formation (see Fig. 2b), and the cell-collagen culture requires labeling, use a 48-well, flat-bottom culture plate or the imaging chamber for staining in a low volume.
-
3.
A round, angled end laboratory spatula.
-
4.
Permeabilization and blocking solution: 0.1% Triton X-100, 0.1% bovine serum albumin (BSA) in PBS.
-
5.
A stock concentration (e.g., 5 mg/mL) of 4′,6-diamidino-2-phenylindole (DAPI) solution in water.
-
6.
Alternatively, or in addition, primary antibodies directed against nuclear or nuclear envelope epitopes (e.g., histone 2B (H2B), lamin A/C, emerin, etc.) and secondary antibodies can be used, as well as phalloidin for F-actin staining, all fluorescently tagged.
2.3 Imaging and Analysis
-
1.
Imaging chamber suitable for fluorescence imaging (not required if a 48–96 imaging well plate is used for cell-collagen gel formation; see Fig. 2b).
-
2.
An inverted confocal microscope.
-
3.
For live-cell imaging, microscope stage should be surrounded by a chamber containing above indicated controlled conditions applied for cell culture.
-
4.
CCD camera mounted onto the microscope and connected to a computer with frame grabber software installed.
-
5.
Free Fiji ImageJ software installed on a computer.
-
6.
Optional: Installed TrackMate Fiji plugin available at https://imagej.net/plugins/trackmate.
-
7.
Custom-made 1. Fiji script for the analysis of static nuclear shapes: “Nuclear shape analysis.ijm” provided at https://github.com/RTC-Microscopy/Nuclear-Shape-Analysis, can be downloaded as a zip file together with other relevant content by clicking on a green, roll down “Code” button.
-
8.
Custom-made 2. Fiji script for the analysis of dynamic nuclear shape changes: “Nuclear shape change analysis.ijm” provided at https://github.com/RTC-Microscopy/Nuclear-Shape-Analysis, downloadable as described in the previous step.
3 Methods
3.1 Embedding of Cells in Rat Tail Collagen
-
1.
Detach adherent cells with EDTA (2–10 min, 37 °C) or collect suspension cells. Wash cells once or twice in PBS and resuspend (e.g., 1.5 × 105 tumor cells, or 3 × 105 leukocytes) in 500 μL culture medium.
-
2.
From here onward, work on ice. For the generation of a 1.7-mg/mL rat tail collagen lattice, here calculated from a collagen stock solution of 10.80 mg/mL (see Note 5), add NaOH (6.8 μL) to 10× MEM (50 μL) in a small sterile tube and add water (286 μL). Indicator color changes from yellow (acidic) to purple (alkaline; Fig. 2a).
-
3.
Add cell-free collagen stock solution (157 μL) and mix until homogenous light orange color is obtained. Avoid air bubbles while mixing.
-
4.
Add cell suspension in medium (500 μL) to the collagen mix (together now 1 mL) and mix until homogenous bright pink to orange color is reached. Again, avoid generating air bubbles. Now, the pH should be 7.3–7.5 (Fig. 2a; see Note 6).
-
5.
Add the cell-containing collagen solution into a well plate (Fig. 2b; see Note 7).
-
6.
Place the well plate into the incubator for collagen polymerization and equilibration of the gas conditions.
-
7.
After 1–2 min turn the plate upside down.
-
8.
Repeat step 7 four times (see Note 8).
-
9.
Allow the collagen in the newly formed collagen culture to fully polymerize (10–20 min).
-
10.
Cover the collagen culture with warm and previously equilibrated medium, providing a final pH of 7.4–7.5. This medium can contain factors, such as chemokines, antibodies, or pharmacological inhibitors.
-
11.
If cells are monitored after migration, allow cell migration in the collagen culture for 2–48 h in the incubator before fixation (see Note 9).
-
12.
If cells are monitored during migration, transport the cell-collagen culture in a pre-warmed or warmth-preserving box to the microscope and continue as described in Subheading 3.3.
3.2 Fixation and Labeling (Optional)
-
1.
Warm the 4% PFA solution to 37 °C.
-
2.
Remove the culture medium from the cell-collagen cultures and wash each gel with 1× PBS (37 °C).
-
3.
Add pre-warmed 4% PFA solution and ensure submersion of the entire gel. Incubate for 30 min at 37 °C (see Note 10), protected from light.
-
4.
Remove PFA solution and wash 3× 15 min with 1× PBS at room temperature, protected from light.
-
5.
For the cell-collagen gels formed in 6–24-well plate, fill a well in a 48-well plate with 1× PBS (150 μL). Gently scoop the fixed gel with a laboratory spatula into a 48-well plate. If the gels were made directly in a 48- or 96-well plate, omit this step.
-
6.
Carefully exchange PBS with permeabilization and blocking solution (150 μL). Incubate for 1 h at room temperature. Protect from light.
-
7.
Remove the solution and wash 3× 15 min with 1× PBS (150 μL).
-
8.
Add 150 μL of a 5 μg/mL DAPI solution in 1× PBS for 10–60 min at room temperature (see Note 11).
-
9.
Facultatively, prepare a primary antibody solution according to manufacturer’s instructions (see Note 12).
-
10.
Replace 1× PBS with the primary antibody solution (150 μL) and incubate at conditions optimized for the particular antibody, protected from light (see Note 13).
-
11.
Remove the primary antibody solution and wash 5× 15 min with 1× PBS (150 μL).
-
12.
Prepare appropriate secondary antibody solutions according to manufacturer’s instructions. Add the secondary antibody solution and incubate at conditions optimized for the particular antibody, protected from light.
-
13.
Wash 5× 15 min with 1× PBS.
-
14.
Transfer sample to a closed container and store at 4 °C (see Note 14) or to an imaging chamber (Fig. 2b).
3.3 Imaging
-
1.
Place the sample onto the stage of a confocal microscope (in case of live-cell imaging, see Note 15).
-
2.
Using bright-field or fluorescence mode for visual inspection, focus through the 3D sample from its bottom to top and back, and set the chosen “top” and “bottom” z positions in the microscope software (Fig. 3a, left; see Note 16). To capture an entire cell nucleus, at least one additional focal plane above and below the imaged nucleus should be included in the z-stack. The step size between respective focal planes should not exceed 1 μm for imaging of fixed samples or 2 μm for live-cell imaging (Fig. 3a; see Note 17).
-
3.
For live-cell imaging, set suitable time steps to image z-stacks over time (Fig. 3a, right). For migrating fibroblasts or mesenchymal and epithelial tumor cells, 5- or 10-min steps, and for leukocyte movement, 0.5-min steps are advised.
-
4.
Image a region containing several randomly positioned cells within a given imaging window. This applies to both fixed and live samples (Fig. 3b and c).
-
5.
For live-cell imaging, repeat scanning the predefined z-stacks for the set time points. The overall duration of live imaging may vary depending on the cell type and the kinetics of the studied biological phenomenon [see Note 18; Fig. 3c and Electronic Supplemental Movie 1 (on https://github.com/RTC-Microscopy/Nuclear-Shape-Analysis)].
-
6.
Collect and save multichannel z-stacks, optionally collected over several time points.
3.4 Fiji-Based Image Segmentation and Quantification of Single Images
-
1.
Open Fiji software.
-
2.
Install and open the macro: “Nuclear shape analysis.ijm” (see Note 19).
-
3.
Import an image z-stack for analysis (Fig. 4, top row, left; see Note 20).
- 4.
-
5.
Deselect the incorrectly segmented nuclei (Fig. 4, middle left), by clicking on respective outlines and pressing “Delete”.
-
6.
Choose a directory in which the results should be stored.
-
7.
Two files are created:
-
(a)
A zip file “‘Image name’_RoiSet.zip” containing the segmented regions of interest (ROIs) created in steps 4 and 5. This file can be loaded into the ROI manager by dragging the file onto the Fiji window and can be used for further analysis and image processing.
-
(b)
A text file “‘Image name’_CompleteResults.txt”. This file contains, per numbered cell nucleus (Fig. 4, middle row, middle image), the determined values of the nuclear area, the various shape descriptors (see Fig. 1), the calculated NII values, and the roundness and solidity values, as additional shape descriptors calculated by Fiji (Fig. 4, bottom). This data can now be further processed and analyzed employing suitable software (e.g., GraphPad, Excel spreadsheets, R, etc.).
-
(a)
3.5 Analysis of Nuclear Shapes
-
1.
To test the power of the different shape descriptors, analyze nuclei from cells that have migrated in collagen in the absence or the presence of MMP inhibitor GM6001 (Fig. 5a, c). With GM6001, note the loss of collagen defects and the change into deformed nuclear shapes deviating from the typical roundish or ellipsoid nuclear shape of proteolytic cells (Fig. 5c).
-
2.
Analyze three example shapes (bead, elliptic nucleus, deformed hourglass-shaped nucleus) (Fig. 5b). The corresponding shape quantifications reveal that the broadest range of values and therefore the highest sensitivity is achieved with NII.
-
3.
When groups of objects are analyzed (beads, nuclei; Fig. 5c), plot the respective selected shape descriptors in a graph (Fig. 5d). The circularity analysis fails to detect a difference between circle-shaped beads and nondeformed cell nuclei, while the aspect ratio and NII shape descriptors show a significant difference. However, a difference between cell nuclei in controls and GM6001-treated cells is obvious for all shape descriptors.
-
4.
To better display the analytic power of particular shape descriptors on nondeformed nuclei during proteolytic versus deformed nuclei during non-proteolytic migration, display all individual data points as two-dimensional plots combining each two shape descriptors (e.g., NII and aspect ratio, Fig. 5e). The combination of circularity and aspect ratio does not reveal a population distinct from control cells after treatment with GM6001 (see dotted square). Combining the NII with the aspect ratio or the circularity morphometrics, however, reveals distinct subsets of proteolytically independent migrating cells with highly deformed nuclei. Along the x-axis, when compared to the y-axis, values were 2–3 times more spread (see dotted rectangles), highlighting NII as a potent and sensitive shape descriptor. We therefore recommend the use of NII for the calculation of nuclear shapes during 3D cell migration.
3.6 Fiji-Based Image Segmentation and Quantification of Image Sequences
-
1.
Open Fiji software.
-
2.
Install and open the macro: “Nuclear shape change analysis.ijm” (see Note 19).
-
3.
Open a time stack containing a nuclear staining (see Note 22).
-
4.
Using TrackMate plugin [17], create an image stack of labeled tracks (see Note 23).
-
5.
Run the “Nuclear shape change analysis.ijm” macro.
-
6.
Select the image stack opened in step 3 as the mask image and press “OK.”
-
7.
Select the image stack created by TrackMate in step 4 and press “OK.”
-
8.
Choose a directory in which the results should be stored.
-
9.
The macro generates a spreadsheet containing nuclear shape descriptors including ΔNII (similar to the table in Fig. 4), a color-coded mask of nuclei tracked over time, and a zip file for each segmented and tracked nucleus (see Note 24).
3.7 Analysis of Nuclear Shape Changes over Time
-
1.
To display the changes of nuclear shape from an acquired time-lapse sequence (Fig. 6a) over time, use readouts (e.g., NII) from the generated spreadsheet (Fig. 6b) and visualize as connected, individual NII data points of single nucleus per time step (Fig. 6c).
-
2.
To quantify the changes of nuclear shape over time, use readouts (e.g., ΔNII) from the generated spreadsheet to calculate the mean ΔNII using the formula shown in Fig. 6d.
-
3.
Consequently, visualize nuclear shape changes as mean ΔNII (Fig. 6e), quantitatively describing the changes in nuclear shape during cell migration, with nuclei of highest deformation showing the highest values.
-
4.
To ensure that the moving cells and their dynamically changing nuclear shapes are assigned and segmented correctly throughout the image sequence, load each ROI (generated per cell) into Fiji and verify visually.
4 Notes
-
1.
All nuclear shapes are measured from flat, maximal projected images even though all cell nuclei in the experiment consist of particular volumes. Although volume measurements are in principle more correct, 3D segmentations consume time, microscopy hours, and data space. By only quantifying the shape of the projected “shadow” of the nucleus, in addition to assuming isotropy when turning the nucleus around its length axis, we, therefore, underestimate shape differences between experimental conditions but offer a method of higher practicality.
-
2.
In principle, any container suitable for cell culture can be used. If the 3D collagen culture is formed in a 6-, 12-, or 24-well plate, the resulting drop gel can be moved to a suitable container after collagen polymerization. If a 96-well plate is being used, the resulting 3D cell-collagen culture fills half of the well, and removing it from the well is not advisable. We suggest to use a 96-well plate for culture, but with a bottom suitable for fluorescence imaging.
-
3.
Pre-labeling of cells: transfected/transduced with tagged nuclear label, such as H2B-XFP, or labeled with Hoechst dye allowing imaging up to several hours.
-
4.
In principle, collagen isolated from different sources can be used, such as bovine, rat, or pig; available in different stock concentrations and from different providers. For the polymerization protocol, follow the provided manufacturer’s instructions on the respective datasheet.
-
5.
A 1.7 mg/mL rat tail collagen preparation is suitable for cell motility of, e.g., fibrosarcoma tumor cells [16] but also immune cells. Other collagen concentrations can be generated as well [6].
-
6.
If less volume is required, smaller cell-collagen mixtures should be prepared. However, note that only around 80% of the suspension can be pipetted, due to the viscosity of the collagen.
-
7.
100 μL of a cell-collagen suspension is optimal to form a dome-like drop gel in a 12- (but also 6- or 24-) well plate or to fill half a well in a 96-well imaging plate (Fig. 2b).
-
8.
In the process of collagen polymerization, a substantial fraction of the cells will sink to the bottom of the container before the gel is polymerized. To avoid cell accumulation on the bottom, the well plate must be turned repeatedly during collagen polymerization.
-
9.
The time needed for collagen-embedded cells to polarize and migrate depends on the cell type used (usually 2 h for leukocytes; 24-48 h for mesenchymal tumor cells). If a longer culture period is needed, the medium should be replaced every 48 h. Cells with high proteolytic and/or contractile activity may alter the collagen matrix over time, resulting in gel detachment and floating.
-
10.
The fixation time may range from 15 to 60 min, based on the volume of the gel. Note that a longer fixation period may cause higher autofluorescence.
-
11.
Nuclear staining by DAPI does not require membrane permeabilization and blocking. Hence if no antibodies are used, the steps 9–12 from Subheading 3.2. Fixation and labeling can be omitted. Nevertheless, a permeabilization step might improve the quality of nuclear staining.
-
12.
It is of utmost importance to ensure labeling by a primary antibody is not a result of nonspecific staining, and labeling with a nontargeting isotype control should be carried out in parallel.
-
13.
Labeling of several proteins can be carried out simultaneously by utilizing a combination of primary antibodies originating from different host animals and secondary antibodies conjugated to different fluorophores. Incubation conditions depend on the volume of the gel and antibodies used, and prior to serial antibody stainings, the dilutions, incubation periods, and incubation temperatures need to be optimized.
-
14.
For long-term storage, addition of an antimicrobial agent (e.g., 0.04% sodium azide) is required. However, generally, sample staining intensities deteriorate over time, and imaging within 48 h post-staining is advised.
-
15.
In the case of live-cell imaging, cell culture conditions (37 °C, 5% CO2, and humid atmosphere) within the microscope stage surrounding chamber should be stably equilibrated before placing the sample onto the stage. The sample itself should be allowed to accommodate to this environment for approximately 30 min before imaging.
-
16.
When choosing the z-stack positions, avoid the region of highest cell density (the cells which are positioned in the 2D interphase between the bottom of the collagen gel and the glass/plastic bottom of the imaging chamber. To ensure that the cells in focus are indeed located within the matrix, an entire layer of cells above, as well as below the chosen focus range, should be visible.
-
17.
A z-step size of 1 μm is advised as nuclear protrusions and chromatin herniations as small as 1–2 μm in diameter can occur during confining migration [6]. However, during live imaging, bigger z-step sizes of around 2 μm together with a more open pinhole should be chosen to not compromise cell viability during imaging.
-
18.
Experiments with immune cells, for example, are usually monitored for 0.5–4 h, while tumor cells may be monitored for 2–48 h or even longer periods.
-
19.
A macro in Fiji can be installed by opening the tab “Plugins -> Macros -> Install”. Installed macros can be opened by clicking on “Plugins -> Macros -> Run” followed by selecting the appropriate file, or the macro file can be dragged directly onto the Fiji window.
-
20.
The “Nuclear shape analysis.ijm” macro assumes a z-stack image. Before running the script, the numerical value in line 10 needs to correspond to the nuclear stain (DAPI) channel.
-
21.
The macro collapses the z-stack into one image. To the DAPI channel, the Li’s Minimum Cross Entropy thresholding method [18] is applied in order to generate a binary image and creates size-defined ROIs that segment the boundaries of the nuclei. It is advisable to test several thresholding methods and select the one that provides the most reliable segmentation.
-
22.
Proper thresholding of the nuclear signal is required to achieve a correct morphometric analysis. The thresholding can be done with “Nuclear shape change analysis.ijm” or manually. Further details, as well as an example TIFF file with nuclear signal only, can be found in “Nuclear Shape Change Analysis Manual” available at https://github.com/RTC-Microscopy/Nuclear-Shape-Analysis.
-
23.
The free TrackMate plugin available for Fiji is used to generate a stack of labeled tracks. A description on how to use TrackMate is beyond the scope of this paper and can be found on the internet. Moreover, settings required for correct cell tracking depend on many factors and have to be optimized for respective data sets. Of note, after successful tracking the stack with labeled tracks can be exported from TrackMate via “Export label image -> Execute -> Export spots as single pixels -> OK”.
-
24.
Due to automatic labeling of segments in TrackMate, cell nuclei are annotated as “Tracks”. Note that as a result of filtering strategies in TrackMate, the final data set may lack particular tracks. In Fig. 6c, for instance, tracks 1, 5, 6, etc. are missing.
References
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3(6):453–458. https://doi.org/10.1038/nrc1098
Frantz C, Stewart KM, Weaver VM (2010) The extracellular matrix at a glance. J Cell Sci 123(Pt 24):4195–4200. https://doi.org/10.1242/jcs.023820
Thiam HR, Vargas P, Carpi N, Crespo CL, Raab M, Terriac E, King MC, Jacobelli J, Alberts AS, Stradal T, Lennon-Dumenil AM, Piel M (2016) Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat Commun 7:10997. https://doi.org/10.1038/ncomms10997
Keeling MC, Flores LR, Dodhy AH, Murray ER, Gavara N (2017) Actomyosin and vimentin cytoskeletal networks regulate nuclear shape, mechanics and chromatin organization. Sci Rep 7(1):5219. https://doi.org/10.1038/s41598-017-05467-x
Stephens AD, Banigan EJ, Marko JF (2018) Separate roles for chromatin and lamins in nuclear mechanics. Nucleus 9(1):119–124. https://doi.org/10.1080/19491034.2017.1414118
Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P (2013) Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 201(7):1069–1084. https://doi.org/10.1083/jcb.201210152
Davidson PM, Denais C, Bakshi MC, Lammerding J (2014) Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell Mol Bioeng 7(3):293–306. https://doi.org/10.1007/s12195-014-0342-y
Patteson AE, Pogoda K, Byfield FJ, Mandal K, Ostrowska-Podhorodecka Z, Charrier EE, Galie PA, Deptula P, Bucki R, McCulloch CA, Janmey PA (2019) Loss of vimentin enhances cell motility through small confining spaces. Small 15(50):e1903180. https://doi.org/10.1002/smll.201903180
Micalet A, Moeendarbary E, Cheema U (2021) 3D in vitro models for investigating the role of stiffness in cancer invasion. ACS Biomater Sci Eng. https://doi.org/10.1021/acsbiomaterials.0c01530
Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, Deryugina E, Friedl P (2009) Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol 20(8):931–941. https://doi.org/10.1016/j.semcdb.2009.08.005
Weigelin B, Bakker GJ, Friedl P (2012) Intravital third harmonic generation microscopy of collective melanoma cell invasion: Principles of interface guidance and microvesicle dynamics. Intravital 1(1):32–43. https://doi.org/10.4161/intv.21223
Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P (2007) Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 9(8):893–904. https://doi.org/10.1038/ncb1616
Chen B, Co C, Ho CC (2015) Cell shape dependent regulation of nuclear morphology. Biomaterials 67:129–136. https://doi.org/10.1016/j.biomaterials.2015.07.017
Ankam S, Teo BKK, Pohan G, Ho SWL, Lim CK, Yim EKF (2018) Temporal changes in nucleus morphology, Lamin a/C and histone methylation during Nanotopography-induced neuronal differentiation of stem cells. Front Bioeng Biotechnol 6:69. https://doi.org/10.3389/fbioe.2018.00069
Filippi-Chiela EC, Oliveira MM, Jurkovski B, Callegari-Jacques SM, da Silva VD, Lenz G (2012) Nuclear morphometric analysis (NMA): screening of senescence, apoptosis and nuclear irregularities. PLoS One 7(8):e42522. https://doi.org/10.1371/journal.pone.0042522
Krause M, Yang FW, Te Lindert M, Isermann P, Schepens J, Maas RJA, Venkataraman C, Lammerding J, Madzvamuse A, Hendriks W, Te Riet J, Wolf K (2019) Cell migration through three-dimensional confining pores: speed accelerations by deformation and recoil of the nucleus. Philos Trans R Soc Lond Ser B Biol Sci 374(1779):20180225. https://doi.org/10.1098/rstb.2018.0225
Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW (2017) TrackMate: An open and extensible platform for single-particle tracking. Methods 115:80–90. https://doi.org/10.1016/j.ymeth.2016.09.016
Li CH, Tam PKS (1998) An iterative algorithm for minimum cross entropy thresholding. Pattern Recogn Lett 19(8):771–776. https://doi.org/10.1016/S0167-8655(98)00057-9
Acknowledgments
We would like to acknowledge the Microscopic Imaging Centre core support at Radboudumc for the use of facilities. This work was supported by a grant from the Dutch Cancer Foundation KWF to K.W. [grant number 11199].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
1 Electronic Supplementary Materials
Cell movement in 3D collagen. Maximal projections of nuclear signal are merged with reflection signal to visualize collagen (AVI 884 kb)
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Authors
About this protocol

Cite this protocol
Svoren, M., Camerini, E., van Erp, M., Yang, F.W., Bakker, GJ., Wolf, K. (2023). Approaches to Determine Nuclear Shape in Cells During Migration Through Collagen Matrices. In: Margadant, C. (eds) Cell Migration in Three Dimensions. Methods in Molecular Biology, vol 2608. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2887-4_7
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2887-4_7
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2886-7
Online ISBN: 978-1-0716-2887-4
eBook Packages: Springer Protocols