
Abstract
Miniaturized head-mounted microscopes for in vivo recording of neural activity have gained much recognition within the past decade of neuroscience research. In combination with fluorescent reporters, these miniature microscopes allow researchers to record the neural activity that underlies behavior, cognition, and perception in freely moving animals. Single-photon miniature microscopes are convenient for widefield recording but lack the increased penetration depth and optical sectioning capabilities of multiphoton imaging. Here we discuss the current state of head-mounted multiphoton miniature microscopes and introduce a miniature head-mounted two-photon fiber-coupled microscope (2P-FCM) for neuronal imaging with active axial focusing enabled using a miniature electrowetting lens. The 2P-FCM enables three-dimensional two-photon optical recording of structure and activity at multiple focal planes in a freely moving mouse. Detailed methods are provided in this chapter on the 2P-FCM design, operation, and software for data analysis.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Two-photon microscopy
- Fluorescence microscopy
- Lens system design
- Fiber optics
- Ultrafast pulse propagation
- Neural imaging
- Image processing
1 Introduction
Two-photon laser scanning microscopy (2P-LSM) has opened up opportunities for in vivo biological imaging at the sub-cellular level [1, 2]. In the neuroscience field, 2P-LSM combined with fluorescent indicators can now capture brain activity at high resolution and in real time. Although ingenious methods to study behavior and perception in a head-fixed animal have been developed, there are some behaviors that simply cannot be studied. Examples include navigation and social behaviors, while additionally there is evidence that head fixation can alter behavior and underlying neural processes [3]. Due to these constraints, head-mounted microscopes are now being developed that can allow the animal to be unrestricted with more naturalistic behavior. The first miniature microscopes to be developed for this purpose were one-photon widefield miniscopes [4,5,6,7]. These systems rely on innovations in compact CMOS imaging sensors and efficient visible wavelength light emitting diodes for the excitation source. Despite the success of the miniscopes with thousands deployed in laboratories internationally, there are limitations. One-photon widefield excitation in tissue results in high levels of light scattering, reducing signal to noise as the out-of-focus fluorescence is also collected on the imaging detector. Computational methods are typically used to select out transient fluorescent signals from the background from the image data. Detailed structural information of the brain region being imaged is sometimes not possible to obtain. Recently, modified miniscopes have been developed that place a phase plate [8] or microlens array [9] in the optical detection path which provides additional information for computational reconstruction in three dimensions (3D). However, light scattering, particularly in densely labelled samples, ultimately limits the imaging depth in these 3D miniscopes.
In contrast to one-photon widefield, two-photon microscopy removes the out-of-focus fluorescence and provides cross-sectional imaging only at the focus. 2P imaging provides excellent signal to noise and detailed structural information allowing high-resolution images of cells and processes. Previous work demonstrated miniature laser-scanning multiphoton fiber-coupled microscopes for head-mounted neuronal imaging in freely moving rodents using different methods of laser scanning including miniaturized microelectromechanical systems (MEMS) actuated mirror [10, 11] or piezoelectric scanner [12] within the head mount. However, none of these designs leverages the optical sectioning capabilities for full volumetric 3D imaging without introducing cumbersome complexity and weight. Recently, Zong et al. integrated an optical design using a MEMS mirror for lateral scanning and a tunable lens for axial scanning in a head-attached miniature microscope [13, 14]. However, this miniature microscope is complex to align and involves custom lenses and tunable optics. In contrast, the design reported here incorporates a miniature, compact electrowetting tunable lens (EWTL) for increasing the versatility of two-photon capabilities by adding active axial focusing, while using a coherent imaging fiber bundle for lateral scanning. The design uses only commercially available components and can be implemented on any standard bench-top laser scanning microscope.
2 Background
2.1 Optical Design Considerations for Miniature 2P Microscopes
The objectives used for two-photon excitation microscopy in deep tissues typically have a numerical aperture (NA) greater than 0.8 [2]. Imaging with high NA objectives reduces the spot size of the laser, thereby increasing the two-photon fluorescence signal for a given excitation power. However, large NA objectives are a problem for the manufacture of small imaging systems, because the system aperture is constrained by the diameter of the physical optics, which in turn forces the optical design to compromise on other important parameters, such as working distance and field-of-view [15]. Assuming a fixed aperture size, larger NA lenses require a reduction in the working distance. Additionally, the complexity of a lens design can be viewed as a function of its optical invariant, i.e., ability to collect light over a large angle and field-of-view (FOV). Increasing the NA has the effect of making it more challenging to design for a large FOV as well. When considering the fluorescence signal detection path, the fluorescent photons are only emitted at the focus and detected on a photo-multiplier tube (PMT) such that the background noise of the image is not degraded by scattered photon trajectories. Therefore, the excitation NA and the collection NA, more commonly referred to as Etendue, must be considered separately. The time-averaged intensity of fluorescence collected geometrically is:
where Ωf is the fractional collection efficiency of an imaging system, summarized as:
When imaging into scattering tissue, the excitation NA is not as important as the collection NA. This is because high-angle rays have longer optical paths, with higher probability of scattering, and are affected by aberrations. Additionally, there are diminishing returns as the focal volume decreases with higher NA. Therefore, a good efficiency two-photon imaging system can be designed that has a relatively low NA (~0.4–0.5) excitation path if the collection NA path can be increased independently. Such systems are often employed by using light collection paths that involve large-area detectors, which can be replicated with large effective-area optical fibers in miniaturized microscopes [10, 16].
2.2 Optical Design Considerations for Axial Scanning with a Tunable Lens
Unlike methods of axial scanning that physically move the sample or the objective lens, the use of a tunable lens to adjust the axial focus changes the way light propagates through the optical system. More specifically, the curvature at the back focal plane of the objective lens is modified. When propagated through a lens, this curvature of the wavefront gets transformed into an axial displacement of the paraxial focus spot. This is analogous to how a tilted wavefront becomes transformed by a lens into a transverse displacement of the focus.
One important consideration when designing such a system is the magnitude of curvature that is needed to achieve a desired axial scan range. An EWTL placed in the back focal plane of a lens directly shapes the wavefront entering the lens.
The wavefront phase term ϕ can be written as a function of the pupil radius ρ [17]:
where M is the objective magnification and Zc is the distance at which the focus is formed with a given quadratic phase. Rearranging this in terms of the amount of axial change for a given change in wavefront phase curvature gives:
This last expression shows that the ability of a tunable lens to change the axial focus of a lens system is dependent inversely on the square of the objective magnification. This means that designing for high magnification with the purpose of increasing resolution or NA will result in a decrease in axial scan range for a given EWTL.
Another consideration is that any imaging system that produces magnification cannot perfectly represent both the axial and transverse focus transformations simultaneously. This effect is illustrated in Fig. 1. Botcherby et al. describe the consequences of this on imaging properties [18]. In lens design, optical engineers typically optimize for either transverse or axial focusing. Most commonly transverse imaging is preserved because imaging lenses are expected to have uniform performance throughout the field-of-view (FOV). The consequence is an accumulation of spherical aberrations along the axial range of the lens, shown in Fig. 1c. One can also design for the Herschel condition, which instead forms perfect focus spots along the optical axis but accumulates aberrations in the transverse dimension.

Axial focusing by wavefront curvature shaping at the objective back focal plane. (a) An input converging or diverging wavefront is transformed into an axial translation away from the designed focal length of the lens. A lens producing an ideal diffraction limited focus at the design working distance (b) will not produce a diffraction limited focus when the input wavefront is divergent (c) due to spherical aberrations as a result of the Abbe sine condition
The FCM imaging system designs presented here are optimized primarily for the Herschel condition, by constraining the optimization parameters in Zemax optical design software to maintain consistent imaging properties throughout the full focal range of the imaging system. One important characteristic that is maintained is telecentricity, which ensures that there is nearly no magnification change as the focal length is tuned. However, this choice sacrifices some imaging performance in the transverse dimension, and so imaging performance is maintained only up to the expected FOV. Further, it is necessary to balance high magnification, which is required to achieve high resolution, with axial scanning range. In the end, the relationships between aperture size, focal length, transverse and axial consistency, and magnification become a careful interplay of compromises to reach a useful solution for miniaturized imaging. In this work, the actual optical designs are sought empirically based on initial designs and guiding principles. The designs are restricted by the parameters for size, resolution, FOV, and axial scan range and additionally restricting the designs to commercially available optics to allow for easier replication and affordability.
2.3 Cranial Windows
Cranial windows provide optical access to surface-level brain structures with traditional microscope objectives. Typically, two-photon laser scanning microscopy (2P-LSM) is performed on a head-fixed mouse with a coverslip implanted in a small craniotomy above the brain region of interest, normally 3 mm2 or less in size. There are several variations on windows that show the flexibility of this technique [19,20,21]. A similar procedure is skull-thinning and polishing, which can be healthier and stable for the brain tissue, but is time-consuming and may not be as optically clear [22]. Recently, Wang et al. demonstrated that application of index-matching epoxy to the bone can reduce scattering and enhance optical access [23]. Removable windows and permeable windows have also been described [24].
Some examples of common regions accessed by cranial windows are olfactory bulb [25], barrel cortex [26], and visual cortex [27]. These regions display high levels of behavior-dependent neuronal activity relatively close to the surface. The extended depth-limit of 2P-LSM, compared with widefield microscopy or confocal laser scanning microscopy, allows access to neurons about 500 μm below the brain surface. In terms of cortical layers in mouse, 500 μm is approximately the depth of the cell bodies found in layers 4/5 of structures such as the motor and somatosensory cortex. Three-photon excitation microscopy has been recently demonstrated to reach structures such as the hippocampus, located around 1 mm below the surface in the mouse brain. However, for deeper structures, it is necessary to excavate the tissue above the target structure or to implant relay optics, such as GRIN lenses [28].
2.4 Gradient-Refractive Index (GRIN) Lenses
GRIN lenses have been classically used for simple and space-efficient collimation of the divergent light exiting a single fiber core. In the past decade, GRIN lenses have been re-purposed for miniaturized imaging applications [29, 30]. The radial profile of the refractive index, ng, in a GRIN lens varies according to [31]:
where g is the gradient constant, r is the radial distance from the optical axis, and ng.0 is the index of refraction at r = 0. Rays launched into one end of the GRIN lens will be focused based on the axial length of the lens, defined by the pitch length, pL:
A pitch of 1 is a full sinusoidal period at a given wavelength. GRIN lenses are also available in a variety of NA values, which are defined by the focus angle in the external medium with index n1. This value can be approximated as [31]:
where d is the GRIN lens diameter. These expressions show that GRIN lenses with smaller diameter, d, need a shorter pitch length to achieve the same NA. To achieve a long and thin GRIN lens for deep-brain implants at high NA, it may require a very high pitch. To get around this constraint, a relay lens of lower NA is used in front of a high NA lens. Figure 2 shows an illustration of two kinds of GRIN lens assemblies used for imaging. A singlet lens is shown compared to a two-element lens with a low NA relay. By increasing the pitch number of the low NA relay, GRIN lenses of various lengths can be constructed for reaching deep-brain structures.

GRIN lens illustrations. Top: Single GRIN lens with pitch ~0.5 with a magnification of 1. Bottom: Two-element GRIN lens with low NA relay at pitch ~0.75 and high NA objective with pitch ~0.25
GRIN lenses are engineered to be highly space-efficient, achieving relatively high NA (up to 0.5) for the small diameters (0.35–1.8 mm). The disadvantages are that they are highly wavelength-dependent and have rapidly deteriorating off-axis performance for imaging. This is mainly due to the small aperture, which vignettes the off-axis rays, effectively reducing the NA toward the edges of the lens. Longer GRIN lenses also suffer from accumulated geometric aberrations. Methods to extend the imaging field and resolution include using active adaptive optics [32] and custom fabricated microlenses [33].
Even with the disadvantages, GRIN lenses offer a tool for providing optical access to otherwise deep-brain structures. Deep-brain implants of GRIN lenses have been used with both widefield fluorescence microscopy and 2P-LSM in structures such as the hippocampus [34]. Animals may be head-restrained with an appropriate objective used to image near the top surface of the GRIN lens. Usually a baseplate is implanted on the skull to provide stability to the fragile GRIN lens assembly.
2.5 Fiber-Coupling of Excitation Laser
For 2P-FCM imaging a laser-source and point-scanning system are required. For the laser source, high-intensity laser light must be brought into the miniature microscope enclosure using an optical fiber. Fibers can be selected for the most efficient propagation of the laser light, generally such that the fiber-core supports only the fundamental transverse electromagnetic (TEM00) mode, in which case it is known as a single-mode fiber. In the special case of ultrashort laser pulses required for 2P-LSM, the maintenance of the peak pulse power is dependent on chromatic dispersion, modal dispersion, polarization dispersion, and non-linear effects [35]. Pre-conditioning the pulses can help to cancel these different effects of propagation through the fiber to maintain high peak powers [36,37,38]. Alternatively, it is possible to use hollow-core photonic bandgap fibers [39], in which the electric field exists mostly in an air-filled core, minimizing chromatic dispersion and nonlinearities. The choice of fiber should also depend on its weight, flexibility, and cost for the practical purposes of mouse imaging. Regardless of the choice, a single fiber core must be focused onto the sample and actively scanned to form an image.
Fiber-coupled scanning microscopy is a relatively well-developed field, owed to the progress in endoscopic surgical and diagnostic clinical tools that use miniaturized imaging heads coupled with fiber-optics to a detection system [40]. There are many techniques for accomplishing this, two of the most common being piezoelectric resonance scanning of the fiber-tip [12, 41,42,43,44] and integrated microelectromechanical systems (MEMS) actuated mirror scanning [10, 11, 45].
An extra consideration for these single-fiber delivery systems is that single-mode fiber-cores are inefficient for the collection of fluorescence emission. Solutions include the use of large mode-area fibers flanking the excitation core [16], miniature single-element detectors mounted on the microscope head [46], and a separate emission path for large-area collection fibers [10, 11, 47, 48].
Overall, these single-fiber-delivery techniques allow access to high-efficiency laser-transmission, which is especially useful for 2P-LSM because of the difficulty required in maintaining ultrashort pulse integrity at the focus. The cost of these techniques is the added mechanical complexity required to implement miniaturized laser-scanning. These may result in challenges in the optical design that limit imaging performance. Frequently, it is difficult to achieve large scan ranges or maintain large beam apertures using small mirrors. So far, these systems have been discussed only in the context of transverse scanning. Miniature fiber-scanning microscopes that implement a third axial scanning dimension can become overly cumbersome in their complexity.
2.6 Coherent Imaging Fiber Bundles (CIFB)
A CIFB is made from a longitudinally ordered bundle of optical fiber preforms that are drawn into dense canes with only a small amount of cladding between the cores [49]. The resulting CIFB is spatially coherent on both ends, allowing a pattern of light on one end to be represented by the cores on the other end, illustrated in Fig. 3.

Properties of coherent imaging fiber bundles. Left: Illustration of proximal-to-distal image coherence of CIFB, as well as the pixelation of an image formed through the bundle because of the discrete fiber cores. Right: Fujikura CIFB fluorescence image of a uniform target showing core distribution. Image was acquired using a laser scanning microscope focused on the proximal surface of the CIFB and placing a uniform fluorescent sample at the distal end
CIFBs can be coupled with a variety of simple imaging lenses to allow for full-field imaging. One of the simplest methods is to use optical epoxy to adhere an imaging GRIN lens (pitch ~0.5) to the distal end of the fiber bundle for use as a handheld imaging probe [50]. An image of the Fujikura CIFB is shown in Fig. 3 with one region expanded to better visualize the core distribution. Lateral-laser scanning microscopy using a CIFB can be accomplished by simply scanning the excitation laser focus across the proximal surface of the fiber. The light is sequentially coupled into each core and transmitted to the distal surface. The distal surface can then be imaged onto a sample to create targeted excitation of fluorophores. The emission light can be collected through the same optical path. This setup is convenient because all the filters, detectors, and scanning optics can be located proximal to the fiber bundle, and can potentially be those of a standard bench-top microscope.
Even with the gained simplicity, there are some issues with using a CIFB for miniature laser-scanning microscopy:
-
The CIFB introduces pixelation to the image due to the necessary cladding between the fiber-cores. Some techniques can be used to process the images to reduce this artifact, which can be summarized by different forms of low-pass spatial filtering [51,52,53]. However, the fundamental lateral resolution limit of the imaging system is determined by the core-to-core spacing and magnification of the imaging system.
-
To achieve small mode-field area sizes for single-mode propagation, it is necessary that the core size is small. Commercial small-core CIFBs have cores of different shapes, sizes, and propagation properties [54] and may result in random signal heterogeneity that shows up in the captured images.
-
Core-to-core coupling is a phenomenon that occurs because of the close inter-core spacing in fiber bundles [55]. This artifactual spreading of light intensity is most critical with broadband light in the NIR wavelengths, as used in multiphoton microscopy. This also limits the density of fiber-cores that can be used, and consequently the imaging resolution and field-of-view.
-
CIFBs are much stiffer than a thin, single-core fiber, with the mechanical stiffness depending primarily on the diameter of the CIFB (and thus the number and density of the cores). This poses a challenge for developing a system for freely behaving rodents, as it limits their mobility in their behaving environment. Leached fiber-bundles, in which the cores are only fused at the ends of the bundle, may be a promising solution to this problem.
While appreciating these challenges, CIFBs greatly simplify the distal optical setup, which is desirable when implementing the distal axial focusing mechanism. Additionally, the use of a CIFB may greatly reduce the cost of adoption for fiber-coupled microscopes because it may allow researchers to use an existing bench-top laser scanning microscope for fiber-coupled imaging with few or no modifications.
2.7 Ultrafast Laser-Pulse Propagation Through Fiber
The time-averaged two-photon fluorescence intensity generated by a pulsed laser source with average power Pavg, pulse width τP, and pulse repetition rate fP is given by the following equation:
where δ2 is the two-photon cross-section, η is the quantum efficiency of the fluorophore, h is Planck’s constant, and c is the speed of light [56, 57]. This relationship shows that two-photon fluorescence signal is inversely proportional to the laser pulse duration. Chromatic dispersion through an individual core of the CIFB results in temporal broadening of the pulse, significantly reducing the fluorescence signal from two-photon excitation [58]. Figure 4 illustrates the principle of chromatic dispersion. Due to the spectral bandwidth of the pulse, the different frequencies travel with different velocities so that, at the exit of the fiber, the pulse is spread out in time. In normal material dispersion, the lower frequencies (red wavelengths) travel faster than the higher frequencies (blue wavelengths) and the resulting pulse is positively chirped. Dispersive optics can be used to counter-act the material dispersion. One method is a Treacy grating-pair compressor, which uses two parallel gratings to spectrally disperse the light and recombine it in a geometry where the blue light travels faster than the red [59]. Addition of the grating-pair compressor before the fiber can effectively cancel out material dispersion, resulting in a short pulse at the fiber exit.

Illustration of material dispersion. The initial pulse out of the laser is short with all spectral frequencies in phase. Inside the 1 m glass fiber, the different frequencies travel with different velocities, resulting in a pulse that is stretched out in time. The resulting pulse is positively chirped, where the lower frequencies (red) travel faster than the higher frequencies (blue)
In addition, the high peak powers required for efficient two-photon imaging result in non-linear effects in the fiber. In the nonlinear regime, there is an intensity-dependent change in refractive index of the material, called the optical Kerr effect:
where n0 is the linear refractive index and n2 is the second-order nonlinear refractive index of the material.
As a pulse propagates in a material, the Kerr effect results in a time varying refractive index, and therefore a modulation in the phase of the electric field, called self-phase modulation (SPM), resulting in a frequency shift that varies across the pulse, according to the following equation:
where Δω is the shift in the instantaneous frequency across the pulse in time, L is the length of the fiber, and n is the refractive index. Typically, SPM results in spectral broadening when propagating a high-peak power short pulse in fiber; however, in the special case where the pulse starts with a negative chirp, the frequency shift from SPM can cause spectral narrowing [60], resulting in a longer pulse duration, due to the time-bandwidth product. High-peak power pulse propagation through optical fibers for multiphoton imaging has been explored extensively in previous works [61,62,63,64].One solution to obtain short pulses at the fiber exit is to first spectrally broaden the pulse by propagating in a polarization maintaining fiber (PMF) before applying negative chirp from the Treacy grating-pair compressor. As the pulse propagates through the CIFB, the spectrum narrows but is balanced out to obtain the initial spectral width from the laser, resulting in the bandwidth to support a short pulse. In certain cases, it is possible to obtain a pulse that is shorter than the original pulse duration by generating more bandwidth.
In addition, the high-peak power pulses incident on the proximal glass surface of the CIFB also produce unwanted background from auto fluorescence and inelastic Raman scattering [65]. We find that the background signal from the fiber surface is greatly reduced when the pulse is pre-chirped with lower peak intensity, thus resulting in fewer non-linear interactions at the proximal fiber surface.
Furthermore, the variability in diameter, shape, NA, and amount of cladding between adjacent cores warrants an investigation into the feasibility of propagating ultrashort pulses in a CIFB for two-photon imaging. Figure 5 shows a comparison of an image of a uniform fluorescent target with one-photon versus two-photon excitation through a CIFB. As can be noted, there is more heterogeneity in signal for the two-photon case. The issue of core-to-core variability in transmission of short laser pulses through commercial CIFBs was further explored by Garofalakis et al. by measuring the transmitted power, polarization, mode, and pulse duration from different fiber cores. The authors found that variation in pulse durations was the predominate cause of this heterogeneity and concluded that it resulted from a variation in the amount of chromatic dispersion in different cores [66]. Further work to understand and correct for these effects could improve multiphoton imaging through CIFBs.

Imaging heterogeneity in CIFB. Images taken using the 2P-FCM showing the raw image of the proximal end of the CIFB focused on a NIR phosphorescent detector card to demonstrate one-photon excitation and a uniform green fluorescent test slide to demonstrate two-photon excitation
2.8 Tunable Focus by Liquid Lens Technology
Liquid lenses are a common variety of electrically tunable lens (ETL) that use one or more liquids as a shape-changing refractive interface to allow for focus adjustment with minimal mechanical motion [67]. There are several common liquid ETL types that may be used for performing high-speed optical focusing for laser-scanning microscopy [68]. Large-aperture liquid ETLs are particularly suited for bench-top systems because of their large focal range, high speed, and repeatability. An example of a commercially available 10-mm aperture, 30-mm diameter ETL (EL-10-30-C-MV, Optotune AG, Switzerland) functions by rapidly changing the volume of liquid in a container with a flexible polymer surface, which results in a focal length shift. Large aperture liquid ETLs have enabled rapid axial focusing, up to 100 Hz, in compact laser scanning confocal microscopes (LSCM), 2P-LSM, and selective plane illumination microscopy (SPIM) systems without mechanical movement of the objective or sample [69,70,71,72]. However, shape-changing polymer ETLs are not suitable for miniaturized head-attached microscopes because of their large size and susceptibility to orientation and vibration.
Electrowetting tunable lenses (EWTLs) are another type of liquid ETL that has applications in microscopy [73]. An example is the Arctic 316, made by Varioptic, France, with outer diameter 7.8-mm and 2.5-mm aperture. Electrowetting is a method for changing the wettability of a liquid on a dielectric surface by applying a voltage across the interface, effectively changing the contact angle of the liquid with the surface. To form a lens, two fluids with dissimilar refractive-indices and hydrophobicity are placed in a container with dielectric sidewalls. The hydrophilic polar liquid has dissolved impurities to allow it to react to an external electric field. The contact angle of the liquid interface to the sidewalls of the lens can be changed by applying an electric field across the lens. Eventually an equilibrium is reached between the electrostatic forces acting on the polar liquid and the surface tension of the system to create a lens surface with a stable curvature.
Carefully matching the density of the liquids at these scales makes EWTLs very resistant to the effects of gravity due to orientation and vibrations, which makes it ideally suited for a miniature head-mounted system. Commercial EWTLs have also demonstrated very large tuning ranges, on the order of 50 diopters. Further, the responses of EWTLs to input voltages are well described by simple oscillators. Although EWTL focusing speed is not as rapid as some other ETLs, by engineering the voltage input function, the lens response time can be brought to less than 20 ms, which is within the range of what is required for an active scanning system [74]. EWTLs also have the potential to perform extended optical functions, such as beam steering and wavefront shaping [75, 76]. Because of the small aperture size, these lenses are not frequently used in bench-top microscopy applications. However, EWTLs are good candidates for robust axial focusing solution for miniature microscopes.
3 Methods
This section describes in detail the design, construction, and testing of the two-photon fiber-coupled microscope (2P-FCM), with axial scanning enabled by an integrated EWTL and lateral scanning achieved with the use of a CIFB. The use of an EWTL is ideal for this application as it is lightweight, is compact, has low-power requirements, and is immune to motion and orientation [68, 77]. The optics are packaged in a light-weight 3D-printed enclosure. Pulse propagation through the CIFB is controlled by careful pre-compensation of the dispersion of glass and verified by spectrally resolved auto-correlation measurements. Testing and verification of the 2P-FCM performance is done using imaging resolution test targets and fluorescent beads in thick agarose preparations. Finally, we describe how to image in vivo neuronal activity by 3D-imaging of neurons in the motor cortex of a freely behaving mouse and in the piriform cortex using a GRIN relay lens combined with the head-attached 2P-FCM. A baseplate is permanently affixed to the mouse skull for attachment and alignment for repeated imaging over the same brain region.
3.1 Overall System Design
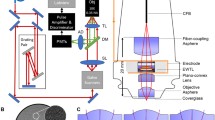
The experimental setup for imaging with the 2P-FCM is shown in Fig. 6. The distal optics of the 2P-FCM are housed in a two-part 3D-printed enclosure, which can be repeatedly attached to a baseplate affixed to the animal’s head. A CIFB couples the distal imaging optics to a custom 2P-LSM system to relay the excitation laser and collect emitted fluorescence from the sample. Laser-scanning over the CIFB forms an image of the sample while the fluorescence collected back through the fiber cores is detected by photodetectors housed in the 2P-LSM after spectral filtering. The individual components of the system are discussed in detail below.

2P-FCM imaging system. Pulses from a Ti:Sapphire laser source are spectrally broadened through polarization maintaining fiber (PMF) and chirped using a grating-pair pulse stretcher. Output pulses are focused and scanned onto the surface of the CIFB through scanning mirrors, scan/tube lens relay, and 10x Objective. Fluorescence emission is collected by the CIFB and directed to a photon counting PMT through a dichroic filter. The collected signal is amplified and transformed to logic levels to be detected by the DAQ counter
3.1.1 Laser-Source
The excitation source is a Spectra-Physics MaiTai HP Ti:Sapphire pulsed laser, with ~80-fs duration pulses tuned to a center wavelength of 910 nm and operating at 80-MHz repetition rate. The beam power is controlled by a half-wave plate on a rotation mount (Newport Conex-AG-PR100P) followed by a Glan-Taylor polarizer (Thorlabs GT10-B).
3.1.2 Spectral and Temporal Pulse Pre-compensation
In order to obtain the shortest pulses at the sample, the laser is first sent through a pre-compensation setup optimized for propagating through a 1.0 m length CIFB (FIGH-15-600 N, Fujikura). The output from the laser is focused into a 0.5 m length polarization maintaining (PM) fiber (PM780-HP, Thorlabs) that causes spectral broadening of the pulse. The output of the PMF is collimated with a fixed fiber-collimation lens (F220APC-780, Thorlabs). The 1.0 mm beam is then expanded through a 3.75× beam expander to reduce the irradiance on the gratings. The beam is reflected off of the two gratings at near Littrow angle separated by 265 mm. It is double passed through the grating pair using a retroreflector with the beam exiting at a different height. The gratings are reflective ruled gold with a density of 300 grooves/mm (49-572, Edmund Optics). The grating-pair stretcher applies ~66,000 fs2 of negative GDD to the laser pulse to compensate for the positive dispersion from the 0.5 m length PMF, 1.0 m length CIFB, and additional non-linear dispersion at the power levels used in the experiments. The precise grating separation distance was determined empirically by maximizing the fluorescence signal while imaging a fluorescent test slide (Chroma Technology) through the 2P-FCM. After traveling through the grating pair, the beam size is reduced by 2.5× with a reverse beam expander to optimize the beam diameter for coupling into the CIFB.
3.1.3 2P-LSM Bench-Top System
The beam is scanned through a galvanometric mirror scanning system (6215H, Cambridge Technologies), relayed through a 50 mm FL scan lens and 180 mm FL tube lens in an Olympus IX71 microscope. The beam is focused and laterally scanned on the surface of the CIFB using a 10×/0.4 NA Olympus UPLANSApo objective lens. A XYZ-translation stage (CXYZ05, Thorlabs) is used to accurately align the fiber to the focus of the objective lens. The beam propagates through the CIFB and then through the distal miniature optics and focused onto the sample. The fluorescence emission is collected by the same optics, back through the CIFB. Because the miniature optics are not chromatically corrected, green emission light is projected onto a ~50 μm diameter area on the CIFB surface. Power transmission through the CIFB was measured using a 532 nm light source and was found to have a ~ 67% efficiency through the CIFB when collected through multiple fiber-cores. The 10× objective collects the fluorescence emission from the CIFB and is separated from the excitation laser path by a primary dichroic mirror (T670LPXR, Chroma Technology). A second dichroic mirror (FF562-Di02, Semrock) splits the red and green fluorescence that is focused using an achromatic doublet lens (LB1761-A, Thorlabs) on separate large-area photon counting photo-multiplier tubes (PMT) (H7422P-40, Hamamatsu). The output pulses from the PMTs are amplified by a high-bandwidth amplifier (ACA-4-35db, Becker & Hickl GmbH) and converted to logic-level pulses by a timing discriminator (6915, Phillips Scientific). The pulses are counted by a data-acquisition card (PCIe-6259, National Instruments) at a rate of 20 MHz. The counts are sampled and binned by pixels and converted into an image in custom software in Labview (National Instruments) that also controls the EWTL driver.
3.1.4 2P-FCM Miniature Optical System Design
The imaging system for the 2P-FCM was designed in Zemax optical design software. Models for the stock lenses and for the EWTL were obtained from the manufacturers (Edmund Optics, Thorlabs, and Varioptic). The CIFB (Fujikura Ltd. FIGH-15-600 N) has an outer diameter of 700 μm, an active image diameter of 550 μm, and a length of 1.0 m. There are ~15,000 cores with core-to-core spacing of 4.5 μm and core diameter of 3.2 μm, as previously reported by Chen et al. measured with scanning electron microscopy [55].
The miniature optics contained in the head-mounted 2P-FCM imaging system are shown in Fig. 7. The fiber-coupling lens is an asphere (FL: 6.2-mm, diameter: 4.7-mm, Edmund Optics 83-710) to collimate the light diverging from the fiber bundle. The electrowetting lens is placed in the collimated beam and the light is refocused onto the sample by an objective lens consisting of a plano-convex lens (FL: 7.5-mm, diameter: 3.0-mm, Edmund Optics 49-177), and an aspheric lens (FL: 2.0 mm, diameter: 3.0 mm, Thorlabs 355151-B). The nominal magnification of this imaging system is 0.4× and field-of-view (FOV) is ~220-μm, corresponding to the de-magnified CIFB active imaging diameter. Similarly, the lateral sampling resolution is the de-magnified core spacing, which is ~1.8 μm.

Optics of the 2P-FCM miniature microscope head that focuses excitation light from CIFB cores onto the tissue. The CIFB-coupling asphere collects the light from the cores of the CIFB, which are then passed through the aperture of the EWTL. The plano-convex lens and the objective asphere focus the light onto the tissue through a #1 coverglass with 0.15-mm thickness
A commercially available EWTL (Varioptic Arctic 316) is used to control the axial focusing of the 2P-FCM imaging system. The predicted working distance and other imaging properties from Zemax at these three different voltage settings are summarized in Table 1. The optical power range of the EWTL is specified as −16 to +36 diopters. The optical system is optimized through the full focal range of the EWTL to minimize magnification change, maximize the axial scan range, and maximize the working distance of the 2P-FCM.
The 2P-FCM has low excitation NA of 0.45; however, the large effective area CIFB results in a higher collection NA of ~0.6. This is illustrated in Fig. 8 showing the Zemax model for 910-nm forward excitation and 532-nm backward emission.

Numerical aperture comparison of forward excitation light at 910 nm (top) and backward emission light at 532 nm (bottom). Note that the blue lines indicate the optical rays entering the system on axis, while the green and red are off-axis rays. Largest emission and excitation field positions are matched at 220-μm FOV. Note that the backward emission is defocused on the fiber bundle end so that multiple fibers are used collect the fluorescent emission
3.1.5 3D-Printed Miniature Head-Mount Design
The enclosure for the 2P-FCM optics is designed in Solidworks 3D CAD software (Dassault Systemes). The packaging is split into three sections: top, bottom, and baseplate, shown in Fig. 9. The top-section contains the CIFB ferrule, held in place by two set-screws, and the fiber-collimating asphere. The bottom-section contains the objective lenses. The unmounted lenses are held in by friction in precisely sized openings. The top section has two curved tabs that interface with slots in the bottom section, which help to ensure reproducible alignment. The EWTL and the electrode are sandwiched between the bottom section and the top section with an O-ring that ensures good electrical contact. The flat-flex electrode cable exits the enclosure through a small slot between the sections. The top-section tabs have a single thread at the end, which interfaces with the baseplate as shown in Fig. 9a. In this way, the baseplate is pulled up against the bottom section by the top section. This greatly improves rigidity when attached to a moving animal. The baseplate is designed with ridges and holes to improve the adhesion of the cement for attachment to the animal skull. The entire enclosure is 3D-printed using a high-resolution projection-based resin printer (Kudo3D Titan 1), with a resolution of 50 μm. The material used is a photo-curable resin (3DM-XGreen) dyed with 0.5% molybdenum disulfide to decrease light scattering and thus increase feature resolution. A photo of the top and bottom sections immediately after printing is shown in Fig. 9b. 3D printing allows optimization of the prototype and easily enables design changes, such as the inclusion of GRIN lenses for deep-brain imaging.

3D-printed enclosure. (a) A two-part 2P-FCM snaps together to secure the EWTL and electrode. (b) Photo of 3D-printed parts, top: before any processing with supports still attached and bottom: assembled 2P-FCM
3.2 Test Sample Preparation
Resolution and axial scan range measurements were performed by imaging fluorescent beads embedded in agarose (Sigma-Aldrich A9414) and a USAF 1951 resolution target (Edmund Optics 38-257). 2-μm yellow-green fluorescent beads (Invitrogen F8853) were used to measure axial scanning extent as well as lateral and axial resolution.
Low-melting-point agarose was prepared at a concentration of 0.5% in water. The 2-μm diameter fluorescent yellow-green beads were diluted in the agarose to a concentration of ~2.0 × 107 beads/mL. Approximately 2.0 mL of solution was placed on a #1 coverglass and allowed to set at room temperature. The beads were imaged in sequential axial planes by a 20x 0.75 NA Olympus UPLANSApo objective with a motorized stage and separately by the 2P-FCM by changing the voltage applied to the EWTL.
3.3 Mouse Imaging Setup
All experiments were approved and conducted in accordance with the Institutional Animal Care and Use Committee of the University of Colorado Anschutz Medical Campus. Three-month-old male C57BL/6 mice (neocortex recordings, Jackson Laboratories stock No 000664) or Nst1-Cre (piriform recordings, MMRRC stock No 030648-UCD) were anesthetized by intraperitoneal ketamine-xylazine injection. The skin above the target site was numbed by lidocaine injection and retracted to expose the skull. For cranial window recordings in neocortex, the mouse was injected with an adeno-associated virus driving the expression of GCaMP6s under the synapsin promoter (AAV5.Syn.GCaMP6s), similar to procedures in [78]. The coordinates of the injection targeted the hindlimb somatosensory cortex, 0.2 mm posterior to bregma and 1.5 mm lateral to the midline, at a depth of 300 μm [79]. The injection volume was 0.66 microliters delivered with a glass micro-pipette through a 0.5-mm hole drilled at the target site. For GRIN lens recordings in piriform cortex, the mouse was injected with 1 microliter of AAV5-hsyn-DIO-GCaMP6s into anterior piriform cortex (AP:0.1 mm, ML:3 mm, DV:4.1 mm).
One month after AAV injection, the mice used for neocortex recordings were implanted with an optical cranial window near the injection site, using standard techniques as previously described. Briefly, mice were anesthetized by isoflurane inhalation and the skin under the scalp was numbed by subcutaneous lidocaine injection. The skin above the skull was removed to expose the injection site and skull surface. A 2-mm square window of skull was removed immediately anterior to the injection hole to expose the dura mater. The opening was covered with a 2-mm square #1 coverglass and secured in place with cyanoacrylate glue. Dental acrylic cement (C&B-Metabond) was used to cover the skull surface. The presence of fluorescence signal was confirmed with standard 2P-LSM using a 20× 1.0 NA Zeiss Plan-Apochromat water-immersion objective. For piriform recordings an Inscopix 1-mm diameter 9-mm length GRIN lens (Inscopix, 1050-004596) was implanted to the region targeted for AAV virus injection and a custom-made headplate was placed with dental acrylic cement (C&B-Metabond) 2 weeks after AAV injection.
The baseplate attachment procedure is similar to what has been described for other miniature head-attached microscopes. While the mouse was still anesthetized, the 2P-FCM was held and positioned above the window with a micromanipulator (Sutter MP-285) until fluorescence signal could be observed with widefield epifluorescence through the bundle. The target region was chosen with two-photon imaging and the 2P-FCM was positioned to the region with the baseplate attached. The baseplate was then secured to the existing acrylic with black acrylic cement (Lang Dental Jet Acrylic). After allowing to set for ~30 min, the 2P-FCM was removed, leaving the baseplate in place, and the mouse was allowed to recover.
The imaging setup is illustrated in Fig. 10. The mouse was lightly anesthetized with isoflurane inhalation. The baseplate was carefully gripped by thumb and forefinger and the 2P-FCM was inserted and secured with a quarter-turn. The EWTL electrode was connected to light-gauge wires that were draped, along with the CIFB, over a horizontal metal post above the behavior cage. The mouse was allowed to recover in the behavior cage for imaging. The cage was illuminated by red light to minimize coupling into the fluorescence detection path, and a camera (Logitech C615) was positioned above the cage to monitor behavior during imaging.

Mouse attachment. (a) The CIFB is attached to a coupling objective on the proximal end and the 2P-FCM on the distal end. (b) 2P-FCM is attached to the permanent baseplate on the mouse with a quarter-turn
3.4 Image Processing
The images from the 2P-FCM show a honeycomb pixelation pattern due to the packing of the cores of the CIFB. Several methods have been described to de-pixelate images from CIFBs [53, 80, 81]. The simplest methods involve low-pass filtering with either a blurring function [82] or masking the image in the frequency domain [83]. However, two-photon imaging through a CIFB has the additional complication of the non-uniformity of the fiber cores. Each core is assumed to have a unique sensitivity, due to the variability in diameter, shape, NA, and amount of cladding between adjacent cores. This manifests as discrete variations in image intensity across the FOV.
This was addressed by programmatically dividing out the sensitivity of each core and interpolating the core values to remove the pixelation pattern. A flat map of the full field CIFB fiber-cores was taken by imaging a fluorescent test slide (Chroma Technologies) with the 2P-FCM, with an example shown in Fig. 11a. The flat-map stores the centroid coordinates of the cores and their corresponding sensitivity. The processing was performed with custom software (Matlab, Mathworks). Each image to be analyzed was registered to the flat-map, which allowed the identification of the cores. An example of a raw pixelated image is shown in Fig. 11b. The relative sensitivity of each core was compensated by dividing by the flat map values. The honeycomb pattern was eliminated by using the nearest neighbor interpolation method [84]. A Savitsky-Golay filter was used to reduce the added single-pixel noise introduced by the core multiplication factor during flat normalization. During the interpolation, the fiber cores were registered to a square pixel grid for straightforward analysis. An example output frame is shown in Fig. 11c.

Image processing of two-photon imaging through CIFB. (a) Flat-field map showing non-uniformity due to differences in sensitivity in individual cores in the CIFB. (b) Unprocessed image of cells in mouse cortex with fiber-pixelation. (c) Post-processed image after fiber-cores were corrected with flat-field mask and re-gridded into a typical square pattern. Grid lines added to emphasize pixels
For the processing of temporal scans, each frame was processed with the same flat-map alignment so that the cores are static in the field. Once the honeycomb pattern and CIFB-induced intensity variation were removed, a clustering algorithm was used to identify regions of interest (ROIs) of high correlation [85]. Significant changes in cytosolic Ca2+ were identified as changes in fluorescence larger than three standard deviations above baseline within each ROI.
3.5 Testing and Calibration of Resolution, Magnification, and Axial Scan Range
The lateral and axial resolution and the axial focusing range of the 2P-FCM can be tested by imaging 2-μm diameter yellow-green fluorescent micro-beads embedded in clear agarose. The lateral resolution is fundamentally limited by the average spacing of the fiber cores in the CIFB. With the core-to-core spacing of ~4.5 μm and 2P-FCM magnification factor of 0.4×, the theoretical lateral sampling at the object is ~1.8 μm.
The beads are imaged with the 2P-FCM at sequential focal planes by tuning the EWTL focus in discrete steps to obtain a Z-stack. The images are processed to remove the fiber-pixelation pattern as described in the previous section. Figure 12 shows a comparison of processed images of the beads imaged with a 20× 0.75 NA objective and the 2P-FCM. The average lateral and axial line profiles of 5 beads measured from different focus positions are fit to a Gaussian function. The axial bead size measured by the 20× objective is 4.5-μm FWHM and with the 2P-FCM it is 9.9-μm FWHM. The lateral bead size measured by the 20× objective is 1.7-μm FWHM (dotted grey line) and with the 2P-FCM it is 2.6-μm. The lateral bead size is larger than the diffraction limit as it is limited by the fiber bundle spacing. With a bead size of ~2 μm, non-uniform sampling of the bead with multiple fiber cores causes a larger effective lateral profile. The axial profile measurements of both the 20x objective at 0.75 NA and the 2P-FCM at 0.45 NA are similar to what is expected from the diffraction-limited calculations.

Axial and lateral resolution tested by imaging fluorescent beads with a 20x Olympus objective (dashed lines) or with the 2P-FCM (solid lines). Reprinted from [92]
In order to calibrate the electrowetting lens voltage, the sample is imaged by a 20× 0.75 NA objective and 2P-FCM. Figure 13a shows side projections of beads overlaid in green and red, respectively. The same region of the agarose-bead sample was imaged in both cases, such that most of the same beads appear in both Z-stacks. This made it possible to compare directly their apparent size and axial location to determine the 2P-FCM scan range. The predicated axial focus plane through the EWTL focusing range from Zemax and actual bead positions are shown in Fig. 13b (gray line and black dots, respectively). The full focusing range did not span the range predicted (240-μm predicted vs. 180-μm measured), likely due to under-performance by the EWTL at the high end of the optical power range.

Measurement of axial scan range. (a) Side-projection of ~2-μm diameter fluorescent beads suspended in clear agarose and imaged with a 20x 0.8 NA Olympus objective (green) using 910 nm excitation light and a motorized stage or with the 2P-FCM while varying the EWTL power (red). (b) Predicted scan range as the EWTL optical power is changed modeled in Zemax (grey line) and Z-positions of measured beads (black circles)
The optical magnification from the CIFB to the target was evaluated by imaging the group 6, element 2 square on the USAF 1951 resolution target with the 2P-FCM as shown in Fig. 14. The imaging diameter of the CIFB is 550 μm. The magnification at three different optical power settings for the EWTL is measured by comparing the scaled size of the resolution target through the CIFB with the actual size. The results are summarized in Table 2. The magnification is measured to be ~0.4×, varying by less than 5% through the focusing range, agreeing closely with the predictions from the Zemax model.

Measurements of magnification of 2P-FCM. Elements of a USAF 1951 resolution target were imaged through the CIFB at three different focus settings. The known size of the elements is indicated
4 Discussion
4.1 2P-FCM Imaging Capabilities
The following experiments on three-dimensional, tilted-field scanning, and two-color imaging demonstrate the potential features of the 2P-FCM as an extremely versatile device.
4.1.1 3D Imaging
To demonstrate volumetric imaging, a 1 mm thick sample of fixed mouse cortical tissue was imaged with the 2P-FCM. The sample was fixed with 4% paraformaldehyde and has cells that express enhanced green fluorescent protein (eGFP) driven by proteolipid protein promoter. The eGFP labels oligodendrocyte cell bodies in the cortex. Axial scanning was accomplished by sequentially tuning the EWTL across the full focal range in 36 discrete steps, acquiring 36 image planes to form a Z-stack. The images were processed to remove the fiber-core pattern as described in the Methods section. A 3D-volume image was created using ImageJ/Fiji software and is shown in Fig. 15. Over 200 cells were observed in this volume.

3D imaging with 2P-FCM of fixed tissue with oligodendrocytes expressing eGFP. (a) 3D volume acquired by the 2P-FCM (220 μm dia. Lateral × 180 μm axial) with over 200 cells in the image. (b) Processed image of a single slice in the stack after filtering to remove pixelation pattern. (Reprinted from [92])
4.1.2 Tilted-Field Imaging
A simple but effective way of increasing the functionality of a microscope that has a rapid axial focusing mechanism is tilted-field imaging. Further, because there are no moving parts, there is no concern for the stability of the specimen during the scan. The tilted-field scan is accomplished by driving the EWTL with a control voltage that changes linearly with the slow axis voltage during the scan. Figure 16 shows the results of performing this test on fixed, thick brain tissue with sparse fluorescence signal from neurons expressing GCaMP6s. Twenty-five total angles were acquired, 12 in each direction, varying the voltage on the EWTL from 40 to 50 VRMS across the slow axis of the raster scan.

Tilted-field imaging enabled by rapid focusing of the EWTL. (a) Maximum intensity projection of fixed mouse brain tissue expressing GCaMP6s in neurons, acquired by the 2P-FCM. Arrows indicate cell bodies retained in fields. (b) Side projection of the volume. The same cell bodies are indicated by the arrows. The planes for the horizontal and angled scan limits are indicated. (c) Tilted field images taken from selected planes at angles (−30,0,30) degrees as indicated in (b). (Reprinted from [92])
4.1.3 Multi-Color Imaging
Separating fluorescence colors is difficult in many miniaturized microscopes because the detection path is exclusive and often also needs to be miniaturized. Multi-color imaging can be an important feature for researchers who want to investigate the co-localization or relative abundance of certain physiological markers. The 2P-FCM offers an advantage in that two-photon excitation at a single wavelength can excite dyes that have separated emission spectra. A common example is GFP and tdTomato, which are both efficiently excited at ~900 nm with pulsed light, but have spectrally separate emission bands. Another feature of a fiber-coupled system is that the emission signal can be spectrally de-mixed after returning through the fiber-bundle. The same filters and detectors used in a benchtop multiphoton microscope for multi-channel imaging can be readily employed for 2P-FCM imaging.
Figure 17 shows two-color imaging with 900 nm two-photon excitation of tdTomato and GFP simultaneously with both a 20x Olympus objective and the 2P-FCM. The cells are GFP-expressing mature oligodendrocytes and tdTomato-expressing oligodendrocyte lineage cells and sparsely labeled astrocytes (Mobp-EGFP; Olig2-Cre; R26-lsl-tdTomato triple transgenic mice). The two experiments were performed using the same detectors and filters. This provides an example of how the 2P-FCM can act as a swappable objective lens for mouse imaging, without needing to change the filters or detection path to acquire images on different channels.

Multi-color imaging. (a) Maximum intensity projection of a region of brain tissue acquired with a 20x 0.75 NA objective. Yellow cells are oligodendrocytes (Green and Red), while red-only cells are astrocytes and oligodendrocytes. Two astrocytes are marked with arrows and are easily identified by the characteristic bushy morphology. (b) Same tissue imaged with the 2P-FCM, using the same detectors, filters, and excitation wavelength. Green and red cells are visible in the field, with likely astrocytes marked by arrows. (Modified from [92])
4.2 2P-FCM Imaging In Vivo
For in vivo 2P-FCM imaging a 3D-printed baseplate is permanently attached to the mouse. During each imaging session, the 2P-FCM is attached to the baseplate with little force on the skull. A photo of a baseplate attached to a mouse with a cranial window is shown in Fig. 18a. Figure 18b shows a photo of a mouse with the 2P-FCM attached. The CIFB and electrode wire for the EWTL are draped passively over a horizontal post to reduce the weight on the mouse. When attached, the mouse is able to move around freely in a small area (about 12″ square). The movement is somewhat restricted by the length of the CIFB (1.0 m) and torsional resistance of the CIFB, which prevents the rotation of more than about 180°. It was found that, after short acclimation (~30 min), mice could traverse the entire behavioral area and habituated to the restrictions. Future implementation may include a commutator with rotation encoder for realignment, which has been shown previously [86].

Mouse 2P-FCM attachment photos. (a) Baseplate implanted on mouse with cranial window. (b) Mouse behaving with 2P-FCM attached
Figure 19 shows a widefield epifluorescence image of the region for an implanted mouse through the 2P-FCM. The left image shows the background fluorescence and vasculature on the day of implantation. The right image was taken 17 days later showing that the baseplate is stable and only a slight lateral shift in alignment is observed.

Implant stability over 17 days imaged with widefield epi-fluorescence. Scale bar 50 μm
The expression level in the neurons virally transfected with GCaMP6s in the cortex was verified by histology shown in Fig. 20. Neuronal cell bodies are seen in layers 4/5, as well as layers 2/3 in lower density.

Histological coronal section of mouse injected with GCaMP6s virus, 12 weeks after injection, showing good expression in layers 2–5 of motor cortex
4.2.1 2P-FCM In Vivo Mouse Imaging Through Cranial Window
In vivo two-photon imaging of neuronal activity through the 2P-FCM was performed in an awake and mobile mouse expressing GCaMP6s in cortical neurons. The mouse was allowed to wander freely in a 7″ by 11″ plastic cage. The cage was filled with sawdust as well as food and various novel objects, such as raisins and tissue paper, to motivate the mouse to explore the environment while attached to the 2P-FCM. A 3D projection of a Z-stack taken by tuning the EWTL focus, showing the imaging volume was acquired in Fig. 21b, c. Bright cell bodies and processes were visible down to 160 μm, corresponding to cortical layers 2/3. Time-courses were acquired sequentially at three focal planes at z = 50, 95, and 140 μm below the cranial window. At the deepest focal plane (z = 140-μm), the active regions are round objects ~10–20 μm in diameter, which are likely cell bodies. At the middle depth of 95 μm, there is a mixture of processes and cell bodies, while closer to the brain surface, activity appears predominately from processes as shown in Fig. 21d. Figure 21f shows the time courses of the ΔF/F signal for the five indicated regions of interest at the three different depths.

Two-photon Ca2+-imaging in an awake and freely moving mouse using the 2P-FCM. (a) Widefield camera image of the epifluorescence taken through the FCM showing vasculature in the FOV. A black rectangle indicates the acquisition region for the following images. (b) 3D projection of a Z-stack acquired by sequentially imaging and focusing through the tissue using the EWTL. (c) Side projection of the same Z-stack indicating the three depths at which time series were acquired: at 50 μm, 95 μm, and 140 μm depth, recorded at frame rates of 2.5, 1.3, and 2.0 Hz, respectively. (d) Maximum intensity projections of image frames that coincide with Ca2 + −transients, showing structural changes through the focal depths. Scale bar is 50 μm. (e) Selected ROIs that contain fluorescence transients that exceed the 6 SD threshold. Time traces for five representative ROIs are selected for each depth. (f) Detailed time-courses of the ΔF/F signal for the five indicated regions. (g) All identified transients, aligned by the peak ΔF/F signal, shown in gray with averaged intensity signal in black. (Modified from [92])
The mouse was recorded with a camera during the imaging session to correlate the imaging results to the motion of the mouse. Lateral motion artifacts were present during some of the recordings. A motion correction algorithm was used to correct for the laterally shifting field [87]. The results of the motion correction indicate a time-averaged motion artifact magnitude of <2 μm, but had peaks up to 10 μm in some cases. The intensity of bright cells that did not exhibit fluorescence activity did not vary with the motion artifacts, so we conclude that the extent of the motion in the axial-dimension is lower than the depth-of-focus for the 2P-FCM (<10 μm). Overall, these motion artifacts were similar to those reported in head-fixed 2PE imaging studies [88, 89] and widefield imaging, which benefits from a much larger depth of field [4]. The 2P-FCM did not become loose or dislodged during the imaging sessions, which lasted between 1 and 4 h.
4.2.2 2P-FCM In Vivo Mouse Imaging in Deep Brain Regions Through GRIN Lens
The 2P-FCM can additionally be used for imaging in deep brain regions by coupling to a GRIN lens. The baseplate is attached such that the 2P-FCM is aligned at the center of the GRIN lens and with the focus of the 2P-FCM positioned at the image working distance of the GRIN lens. The 1:1 GRIN relay lens used did not change the magnification of the 2P-FCM image, although aberrations from imaging through the GRIN lens reduced the field–of-view (FOV) in comparison to the cranial window imaging. Additionally, actuating the EWTL changed the axial focal plane imaged by the 2P-FCM through the GRIN lens as shown in Fig. 22. As an example of behaviorally relevant imaging of neuronal activity in deep brain regions, the 2P-FCM was used to image activity in anterior piriform cortex in a freely moving female Ntsr1-cre mouse exploring a novel environment with male bedding. GCaMP6s was expressed virally using AAV_CWSL.hSyn.DIO.Synaptophysin-GCaMP6s.P2A.mRuby. Imaging data was processed to remove the fiber bundle artifact using the custom Matlab software and then analyzed by manually selecting ROIs where ΔF/F transients exceeded 6 standard deviations threshold. Behavioral video was analyzed with the DeepLabCut [90] software package which provides markerless tracking of body parts. The mouse was allowed to explore the environment for 5 min while recording behavioral video and imaging neural activity with the 2P-FCM. Piriform cortex is the largest recipient of projections from olfactory bulb, and it is expected to show an increase in neural activity when the mouse smells novel odors. DeepLabCut was used to track the snout of the mouse during the image sequence. The measured distance of the mouse to the bedding was compared with activity for manually selected ROIs. Distinct increases in activity can be observed when the mouse is in close proximity to the novel male bedding.

Behaviorally dependent responses from 2P-FCM recording in piriform cortex in a mouse implanted with a GRIN lens and GCaMP6s expressing neurons. (a) Still frame of video of mouse behavior environment containing familiar bedding and novel unfamiliar bedding during freely moving behavior with attached 2P-FCM. (b) Example maximum projections at two separate focal planes, shown with relative distance of focus from GRIN lens. (c) Distance between the female mouse’s snout and the foreign male mouse’s bedding versus time. Manual correction was made to marker placement when DeepLabCut misplaced or lost markers. (d) An example frame of the behavioral video labeled by DeepLabCut. The green marker tracks the mouse’s body, the blue marker tracks the mouse’s snout, and the red marker is placed on the male bedding. (e) Summary of activity, showing greater activity in some ROIs as the mouse approaches the novel odor. (f) Max projection showing six manually selected regions used in the analysis
5 Materials
Pulse Pre-compensator
-
1x Polarization Maintaining Fiber – Thorlabs Custom Fiber Patch Cable: PM780-HP, FC/APC connectors both ends, keyed to slow axis, FT800SM-Blue Tubing, 0.5 m in length
-
1x Input Coupler – Thorlabs ZC618APC-B – Variable Magnification and Focal Length
-
1x Output Coupler – Thorlabs F810APC-850 – 7.8 mm 1/e2 beam diameter
-
2x Gratings – Edmund 37–127 – 300 line per mm blazed at 760 nm
-
1x D-mirror – Thorlabs PFD10-03-M01 – 1” Protected Gold D-Shaped Mirror
-
1x Retro reflector – Thorlabs HRS1015-AG – Hollow Roof Mirror, Ultrafast Enhanced Silver Coating
-
1x linear translator – OptoSigma TSD-65171SUU (Imperial) +/− 25 mm travel
-
1x dovetail optical rail
-
Additional optomechanics – kinematic mirror mounts, grating holders.
Objective Adapter to Hold CIFB
-
1x - SMA Fiber Adapter – Thorlabs SM1SMA
-
1x - Base Cage Plate – Newport OC1-BT
-
1x - Z Stage – Newport OC1-TZ
-
1x - XY Mount – Newport OC1-LH-XY
-
1x - Holder for Objective – Newport OC1-LH1-TZ
-
4x - Cage Rod – Thorlabs ER4
-
1x - RMS to SM1 Thread Adapter for Microscope – Thorlabs SM1A4
-
1x - SMA Fiber Adapter – Thorlabs SM05SMA
-
1x - Coupling Objective – Olympus 10x/0.4 NA.
2P-FCM Fiber and Head Mount
-
1x – Coherent Imaging Fiber Bundle – Fujikura FIGH-15-600 N – 1.0 m SMA connector one end and plastic ferrule on other (Myriad)
-
1x Electrowetting lens – Varioptic Artic 360 and controller
-
1x - Fiber-coupling asphere (FL: 6.2 mm, diameter: 4.7 mm, Edmund Optics 83-710)
-
1x - Plano-convex lens (FL: 7.5 mm, diameter: 3.0 mm, Edmund Optics 49-177)
-
1x - Aspheric lens (FL: 2.0-mm, diameter: 3.0-mm, Thorlabs 355151-B)
-
1x – 3D printed FCM head mount and baseplate
Imaging Test Targets
-
1x – Green fluorescent flat (TedPella 2273)
-
1x – Green fluorescent resolution target II-IV (Max Levy DA113)
6 Notes
The following includes detailed notes on how to align the pre-compensation optics before the microscope and align the 2P-FCM for freely moving mouse imaging. Details including Zemax optical design, solidworks files, and software can be found at https://github.com/CUNeurophotonics/2PFCM.
6.1 Optimizing Alignment into Single Mode Fiber
One of the challenges is setting up the single-mode fiber and grating pair compressor in front of the microscope. In particular, aligning the beam through the single-mode fiber can be challenging. Make sure to terminate the fiber with an APC (angled physical contact) on both fiber ends. APC termination minimizes back reflection because the fiber face is cut at an angle as opposed to a flat surface which can reflect back along the same optical path and cause the ultrafast laser to stop modelocking. For input coupling into the single mode fiber, we use a fiber collimator (Thorlabs ZC618APC-B) that allows for the adjustment of beam diameter and divergence. Before the input coupler, set up two steering mirrors in x, y kinematic mounts to optimize for the beam input position and angle. It is ideal for the last mirror before the input coupler to be as close to the input coupler as possible so that it mostly controls the input angle while the first mirror controls the position. To perform the initial alignment of the laser to the fiber input coupler, set the laser to 780 nm with 10–20 mW of power. Higher power will damage the fiber if misaligned and 780 nm is used to be able to see the laser.
One method to get alignment started is to use a visual fault locator connected to the fiber end face. One can then spatially overlap the incoming femtosecond laser beam with the back propagating light from the locator using the input alignment mirrors, shown in Fig. 23.

Method for alignment of single mode fiber. Visualization of red back-propagating beam from visual fault locator with forward propagating beam from femtosecond laser on IR card. Femtosecond laser is incident on back of IR card and seen as a green spot on the front side
After performing the course alignment, remove the fault locator and replace with a power meter. Proceed to “walk-in” the laser with the mirrors: Turn the horizontal knob of the first mirror and use the other mirror’s horizontal knob to maximize the power and repeat. If this keeps lowering the power from the initial amount you had, try turning the first knob in the opposite direction and repeat. After the horizontal alignment is optimized, do this for the vertical as well. If this does not work, you are likely in a side lobe of the Airy disk. In that case walk it out of the local peak by moving steadily in each direction. It should go down and then up in one of the directions. Then start the walk in again!
At the end of the alignment, the output power from the fiber should be ~70–80% of the power measured before the input collimator.
6.2 Setup and Alignment of the Grating Pair Compressor
Alignment steps for the grating pair compressor (shown in Fig. 24) are as follows:
-
1.
Adjust the output beam collimator (A) until the output beam from the fiber is traveling parallel to one of the hole lines on the optical table and at a constant height.
-
2.
Insert a D-shaped mirror mount (B) and adjust until the beam is traveling approximately 90 degrees to the fiber output, again parallel to a hole line. Finely adjust the position of the mirror until clipping is minimized on the D-shaped mirror and beam is again traveling at a constant height and parallel to the hole line.
-
3.
Turn the linear stage knob holding the retro-reflector (C) forward until it intercepts the incoming beam. Adjust the position until the beam is reflected at a lower height so it passes below the D-shaped mirror. Adjust the tilt of the retro-reflector to move the reflected beam laterally until it is again parallel to the hole line, ensuring that the reflection is close to perfect in the lateral dimension with only a change in height.
-
4.
Insert a mirror (D) to intercept the reflected beam after passing beneath the D-shaped mirror to send to the microscope input. Again, adjust the mount until the beam is perfectly perpendicular.
-
5.
Using a final mirror mount, re-align the output beam with the beam path into the microscope.
-
6.
Fit an adjustable iris into a threaded kinematic mount (E) that usually holds the beam-expander tube. Adjust the height and position of the threaded kinematic mount and its post holder until the beam is traveling directly through the center of the iris. Two irises may be used to ensure that the tip and tilt of the output beam is correctly aligned.
-
7.
Attach the gratings with posts onto the linear dovetail rail.
-
8.
Move the linear stage back such that the beam fully avoids the retro-reflector and lands entirely on the first grating surface (F). The grating should be adjusted primarily in the tilt axis (lateral movement of beam) until it lands on the surface of the second grating (G) with no clipping. The second grating may be moved laterally on the compound rail carrier assembly to minimize the clipping of the input beam if necessary.
-
9.
Adjust the tilt of the second grating until the beam once again aligns well onto the retro-reflector and back through the grating system, exit mirror (D), and iris.
-
10.
Move the linear stage to again push the retro-reflector into the beam path, such that the gratings no longer interact with the beam. Check that the alignment is still good. Put a card to monitor the beam position some distance after the iris (E). Make a mark on the card where the beam intersects. Move the linear stage out of the beam path again and check that the grating alignment is maintained, and clipping is minimal to none. Finely adjust the gratings until the beam is realigned perfectly with the mark on the card. At this point, the gratings should be nearly perfectly parallel to each other.
-
11.
Adjust the distance between the two gratings to desired length, performing adjustments of the rail carriers to avoid clipping when possible. For 1.0 m fiber bundle and 1.0 m polarization maintaining fiber the best separation between 300 groove/mm grating surfaces is ~265 mm. When properly aligned the pre-compensator should yield 25% of the incoming power from the laser.

Photo of grating pair compressor. Labels indicate (a) fiber output coupler on kinematic mount, (b) D-mirror, (c) retro-reflector roof mirror on kinematic mount and linear stage, (d) output mirror, (e) kinematic mount with iris for alignment, (f) first grating, and (g) second grating
6.3 Optimizing Imaging Through Coherent Fiber Bundle
The FCM objective holder, used to align the proximal end of the CIFB to the microscope focus, is shown in Fig. 25.
-
1.
Screw in the FCM objective holder into the 2P-LSM using the appropriate adapter. Adjust the micrometer on the linear stage (OC1-TZ) to position the surface of the CIFB at the focus of the objective. Visualizing the fiber surface is easiest when using widefield epifluorescence. Center the fiber using the xy adjustments on the cage adapter (OC1-LH-XY).
-
2.
If there is dirt on the fiber repeat cleaning with methanol/air puffs.
-
3.
Hold the FCM with a Sutter micromanipulator perpendicular to the plane you will image. Image an IR card with the laser at its lowest setting (0.1%). Check centering and re-set the focus.
-
4.
If possible, use an iris after the scan lens to control the field-of-view. Close down the iris to exclude the edge of the fiber which has strong fluorescence and can saturate the detectors.
-
5.
Use a green fluorescent flat (Ted Pella 2273) to take an image to normalize the fluorescence of the different cores. Make sure that the pixels in the image are not saturated.
-
6.
If there are high intensity pixels on the fiber, one way to remove these is to set the PMT gain to zero, increase the laser power and scan for several seconds.
-
7.
Image a pollen slide as a first step before moving on to your sample.

Diagram of FCM objective holder to mount the FCM to any two-photon laser scanning microscope. The holder screws into the objective turret on the microscope with the appropriate adapter. A linear stage (OC1-TZ) adjusts the focus of objective to the end face of the coherent imaging fiber bundle (CIFB). A cage mount with xy translation (OC1-LH-XY) adjusts the lateral position of the fiber to center it on the objective field-of-view
6.4 Installing the Pedestal for the 2P-FCM on the Cranium
Optimize 2P-FCM imaging as detailed above. Then perform the following steps to install the FCM over the cranial window in an anesthetized mouse.
-
1.
Hold the 2P-FCM attached to the baseplate perpendicular to the cranial window. Bring the 2P-FCM close to the cranial window and zero the Sutter micromanipulator. Focus on the GCaMP-labeled neurons.
Important
During the cranial window surgery place the head plate as close to the cranium as possible. A thick metabond layer between the cranium and the metal plate will make it impossible to place the 2P-FCM in the position necessary to be able to focus at depth. If necessary, you can thin the bottom of the FCM baseplate.
-
2.
Perform a z stack with the electrowetting lens and do a time course recording. When you have found the right location, mix adhesive and place small amounts around the baseplate to bond it with the cranium. Take care not to bond the baseplate to the 2P-FCM.
7 Conclusions
We describe a head-mounted 2P-FCM that achieves 3D imaging of neural activity in a freely moving mouse. The imaging volume is 220-μm diameter by 190-μm depth. The device is optimized for resolving neuronal somata with a lateral resolution of 2.6 μm and axial resolution of 9 μm. The 2P-FCM is compact, is lightweight, and includes an electrowetting lens for active axial scanning. We demonstrate the use of this device for tilted plane imaging, multi-color imaging, and fast multiplane imaging. The 2P-FCM was demonstrated for the imaging of neuronal activity in different planes of cortical layer 2/3 of a freely moving mouse with minimal motion artifacts. The 2P-FCM differs from previous head-mounted microscopes [4, 12, 46, 47, 86, 91] because it allows live-focusing for a range of ~200-μm depth suitable for imaging neurons in layer 2/3 of cortex.
The goal of this work has been to create miniature fiber-coupled microscopes (FCMs) that have the capability to compete with head-fixed imaging setups for awake and behaving mouse brain imaging. There have been two key technologies that have driven the progress of this work. First, the coherent imaging fiber-bundle (CIFB), which is a tool that has large adoption in the field of clinical endoscopy. CIFBs allow passive imaging that functions remarkably well for both single- and multi-photon fluorescence fiber-coupled imaging. Second, the electrowetting tunable lens (EWTL), because of its light-weight and mechanical simplicity, is an ideal tool for miniaturized microscope applications.
The design outlined here can be easily assembled and attached onto a standard benchtop multiphoton microscope, commonly available for many labs, making the technology inexpensive to implement. Future work can further this technology for multi-site brain recording or potentially combined with two-photon holography for imaging and photostimulation in the freely moving animal with 3D access.
References
Denk W, Strickler J, Webb W (1990) Two-photon laser scanning fluorescence microscopy. Science 248(4951):73–76
Helmchen F, Denk W (2005) Deep tissue two-photon microscopy. Nat Methods 2(12):932–940
Bjerre A-S, Palmer LM (2020) Probing cortical activity during head-fixed behavior. Front Mol Neurosci 13(30)
Ghosh KK, Burns LD, Cocker ED et al (2011) Miniaturized integration of a fluorescence microscope. Nat Methods 8(10):871–878
Cai DJ, Aharoni D, Shuman T et al (2016) A shared neural ensemble links distinct contextual memories encoded close in time. Nature 534(7605):115–118
Aharoni D, Khakh BS, Silva AJ et al (2019) All the light that we can see: a new era in miniaturized microscopy. Nat Methods 16(1):11–13
de Groot A, van den Boom BJ, van Genderen RM et al (2020) NINscope, a versatile miniscope for multi-region circuit investigations. elife 9:e49987
Yanny K, Antipa N, Liberti W et al (2020) Miniscope3D: optimized single-shot miniature 3D fluorescence microscopy. Light Sci Appl 9:171
Skocek O, Nobauer T, Weilguny L et al (2018) High-speed volumetric imaging of neuronal activity in freely moving rodents. Nat Methods 15(6):429–432
Zong W, Wu R, Li M et al (2017) Fast high-resolution miniature two-photon microscopy for brain imaging in freely behaving mice. Nat Methods 14(7):713–719
Klioutchnikov A, Wallace DJ, Frosz MH et al (2020) Three-photon head-mounted microscope for imaging deep cortical layers in freely moving rats. Nat Methods 17(5):509–513
Sawinski J, Wallace DJ, Greenberg DS et al (2009) Visually evoked activity in cortical cells imaged in freely moving animals. Proc Natl Acad Sci 106(46):19557–19562
Zong W, Wu R, Chen S et al (2021) Miniature two-photon microscopy for enlarged field-of-view, multi-plane and long-term brain imaging. Nat Methods 18(1):46–49
Zong W, Obenhaus H, Skytøen E et al (2021) Large-scale two-photon calcium imaging in freely moving mice. bioRxiv. https://doi.org/10.1101/2021.09.20.461015
Liang C, Sung K-B, Richards-Kortum RR et al (2002) Design of a high-numerical-aperture miniature microscope objective for an endoscopic fiber confocal reflectance microscope. Appl Opt 41(22):4603–4610
Wu Y, Xi J, Cobb MJ et al (2009) Scanning fiber-optic nonlinear endomicroscopy with miniature aspherical compound lens and multimode fiber collector. Opt Lett 34(7):953–955
Botcherby EJ, Juskaitis R, Booth MJ et al (2007) Aberration-free optical refocusing in high numerical aperture microscopy. Opt Lett 32(14):2007–2009
Botcherby EJ, Juškaitis R, Booth MJ et al (2008) An optical technique for remote focusing in microscopy. Opt Commun 281(4):880–887
Grutzendler J, Yang G, Pan F et al (2011, 2011) Transcranial two-photon imaging of the living mouse brain. Cold Spring Harb Protoc (9):pdb.prot065474
Isshiki M, Okabe S (2014) Evaluation of cranial window types for in vivo two-photon imaging of brain microstructures. Microscopy (Oxf) 63(1):53–63
Holtmaat A, Bonhoeffer T, Chow DK et al (2009) Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4(8):1128–1144
Shih AY, Mateo C, Drew PJ et al (2012) A polished and reinforced thinned-skull window for long-term imaging of the mouse brain. JoVE 61:3742
Wang T, Ouzounov DG, Wu C et al (2018) Three-photon imaging of mouse brain structure and function through the intact skull. Nat Methods 15(10):789–792
Goldey GJ, Roumis DK, Glickfeld LL et al (2014) Removable cranial windows for long-term imaging in awake mice. Nat Protoc 9(11):2515–2538
Wachowiak M, Economo MN, Diaz-Quesada M et al (2013) Optical dissection of odor information processing in vivo using GCaMPs expressed in specified cell types of the olfactory bulb. J Neurosci 33(12):5285–5300
Peron Simon P, Freeman J, Iyer V et al (2015) A cellular resolution map of barrel cortex activity during tactile behavior. Neuron 86(3):783–799
Andermann ML, Kerlin AM, Reid C (2010) Chronic cellular imaging of mouse visual cortex during operant behavior and passive viewing. Front Cell Neurosci 4(3)
Murray TA, Levene MJ (2012) Singlet gradient index lens for deep in vivo multiphoton microscopy. J Biomed Opt 17(2):021106
Jung JC, Schnitzer MJ (2003) Multiphoton endoscopy. Opt Lett 28(11):902–904
Knittel J, Schnieder L, Buess G et al (2001) Endoscope-compatible confocal microscope using a gradient index-lens system. Opt Commun 188(5–6):267–273
Jung JC, Mehta AD, Aksay E et al (2004) In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy. J Neurophysiol 92(5):3121–3133
Wang C, Ji N (2013) Characterization and improvement of three-dimensional imaging performance of GRIN-lens-based two-photon fluorescence endomicroscopes with adaptive optics. Opt Express 21(22):27142–27154
Antonini A, Sattin A, Moroni M et al (2020) Extended field-of-view ultrathin microendoscopes for high-resolution two-photon imaging with minimal invasiveness. elife 9:e58882
Barretto RPJ, Schnitzer MJ (2012, 2012) In vivo microendoscopy of the hippocampus. Cold Spring Harb Protoc (10):pdb.prot071472
Agrawal GP (2000) Nonlinear fiber optics. In: Christiansen PL, Sørensen MP, Scott AC (eds) Nonlinear science at the Dawn of the 21st century, Springer, vol 542. Berlin Heidelberg, Berlin, Heidelberg, pp 195–211
Agrawal GP, Potasek MJ (1986) Effect of frequency chirping on the performance of optical communication systems. Opt Lett 11(5):318–320
Shen X, Kahn JM, Horowitz MA (2005) Compensation for multimode fiber dispersion by adaptive optics. Opt Lett 30(22):2985–2987
Clark SW, Ilday FÖ, Wise FW (2001) Fiber delivery of femtosecond pulses from a Ti:sapphire laser. Opt Lett 26(17):1320–1322
Bouwmans G, Luan F, Knight JC et al (2003) Properties of a hollow-core photonic bandgap fiber at 850 nm wavelength. Opt Express 11(14):1613–1620
Jabbour JM, Saldua MA, Bixler JN et al (2012) Confocal Endomicroscopy: instrumentation and medical applications. Ann Biomed Eng 40(2):378–397
Engelbrecht CJ, Johnston RS, Seibel EJ et al (2008) Ultra-compact fiber-optic two-photon microscope for functional fluorescence imaging in vivo. Opt Express 16(8):5556–5564
Myaing MT, MacDonald DJ, Li X (2006) Fiber-optic scanning two-photon fluorescence endoscope. Opt Lett 31(8):1076–1078
Flusberg BA, Jung JC, Cocker ED et al (2005) In vivo brain imaging using a portable 3.9 gram two-photon fluorescence microendoscope. Opt Lett 30(17):2272–2274
Rivera DR, Brown CM, Ouzounov DG et al (2011) Compact and flexible raster scanning multiphoton endoscope capable of imaging unstained tissue. Proc Natl Acad Sci 108(43):17598–17603
Qiu Z, Piyawattanamatha W (2017) New endoscopic imaging technology based on MEMS sensors and actuators. Micromachines 8(7):210
Helmchen F, Fee MS, Tank DW et al (2001) A miniature head-mounted two-photon microscope. Neuron 31(6):903–912
Helmchen F, Denk W, Kerr JN (2013) Miniaturization of two-photon microscopy for imaging in freely moving animals. Cold Spring Harb Protoc 2013(10):904–913
Zhao Y, Nakamura H, Gordon RJ (2010) Development of a versatile two-photon endoscope for biological imaging. Biomed Opt Express 1(4):1159–1172
Wood HAC, Harrington K, Stone JM et al (2017) Quantitative characterization of endoscopic imaging fibers. Opt Express 25(3):1985–1992
Pierce M, Yu D, Richards-Kortum R (2011) High-resolution fiber-optic microendoscopy for in situ cellular imaging. JoVE 47:2306
Cheon GW, Cha J, Kang JU (2014) Spatial compound imaging for fiber-bundle optic microscopy. In: SPIE BiOS (ed) Optical fibers and sensors for medical diagnostics and treatment applications, vol XIV, p 893811. https://doi.org/10.1117/12.2038690
Han J-H, Yoon SM (2011) Depixelation of coherent fiber bundle endoscopy based on learning patterns of image prior. Opt Lett 36(16):3212–3214
Shinde A, Matham MV (2014) Pixelate removal in an image fiber probe endoscope incorporating comb structure removal methods. J Med Imaging Hlth Inform 4(2):203–211
Steelman ZA, Kim S, Jelly ET et al (2018) Comparison of imaging fiber bundles for coherence-domain imaging. Appl Opt 57(6):1455–1462
Chen X, Reichenbach KL, Xu C (2008) Experimental and theoretical analysis of core-to-core coupling on fiber bundle imaging. Opt Express 16(26):21598–21607
Xu C, Webb WW (1996) Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J Opt Soc Am B 13(3):481–491
Diaspro A, Chirico G, Collini M (2005) Two-photon fluorescence excitation and related techniques in biological microscopy. Q Rev Biophys 38(2):97–166
Lefort C, Mansuryan T, Louradour F et al (2011) Pulse compression and fiber delivery of 45 fs Fourier transform limited pulses at 830 nm. Opt Lett 36(2):292
Treacy E (1969) Optical pulse compression with diffraction gratings. IEEE J Quantum Electron 5(9):454–458
Oberthaler M, Höpfel RA (1993) Special narrowing of ultrashort laser pulses by self-phase modulation in optical fibers. Appl Phys Lett 63(8):1017–1019
Lelek M, Suran E, Louradour F et al (2007) Coherent femtosecond pulse shaping for the optimization of a non-linear micro-endoscope. Opt Express 15(16):10154–10162
Lefort C, Kalashyan M, Ducourthial G et al (2014) Sub-30-fs pulse compression and pulse shaping at the output of a 2-m-long optical fiber in the near-infrared range. J Opt Soc Am B 31(10):2317–2324
Kim D, Choi H, Yazdanfar S et al (2008) Ultrafast optical pulse delivery with fibers for nonlinear microscopy. Microsc Res Tech 71(12):887–896
Kalashyan M, Lefort C, Martínez-León L et al (2012) Ultrashort pulse fiber delivery with optimized dispersion control by reflection grisms at 800 nm. Opt Express 20(23):25624–25635
Udovich JA, Kirkpatrick ND, Kano A et al (2008) Spectral background and transmission characteristics of fiber optic imaging bundles. Appl Opt 47(25):4560–4568
Garofalakis A, Kruglik SG, Mansuryan T et al (2019) Characterization of a multicore fiber image guide for nonlinear endoscopic imaging using two-photon fluorescence and second-harmonic generation. J Biomed Opt 24(10):1–12
Chiu C-P, Chiang T-J, Chen J-K et al (2012) Liquid lenses and driving mechanisms: a review. J Adhes Sci Technol 26(12–17):1773–1788
Blum M, Büeler M, Grätzel C et al (2011) Compact optical design solutions using focus tunable lenses. In: SPIE optical systems design, Marseille, France, p 81670W. https://doi.org/10.1117/12.897608
Jabbour JM, Malik BH, Olsovsky C et al (2014) Optical axial scanning in confocal microscopy using an electrically tunable lens. Biomed Opt Express 5(2):645–652
Jiang J, Zhang D, Walker S et al (2015) Fast 3-D temporal focusing microscopy using an electrically tunable lens. Opt Express 23(19):24362–24368
Yang W, Carrillo-Reid L, Bando Y et al (2018) Simultaneous two-photon imaging and two-photon optogenetics of cortical circuits in three dimensions. elife 7:e32671
Ryan DP, Gould EA, Seedorf GJ et al (2017) Automatic and adaptive heterogeneous refractive index compensation for light-sheet microscopy. Nat Commun 8(1):612
Zhao Y-P, Wang Y (2013) Fundamentals and applications of Electrowetting. Rev Adhes Adhesives 1(1):114–174
Supekar OD, Zohrabi M, Gopinath JT et al (2017) Enhanced response time of Electrowetting lenses with shaped input voltage functions. Langmuir 33(19):4863–4869
Smith NR, Abeysinghe DC, Haus JW et al (2006) Agile wide-angle beam steering with electrowetting microprisms. Opt Express 14(14):6557–6563
Supekar OD, Ozbay BN, Zohrabi M et al (2017) Two-photon laser scanning microscopy with electrowetting-based prism scanning. Biomed Opt Express 8(12):5412–5426
Berge B, Peseux J (2000) Variable focal lens controlled by an external voltage: an application of electrowetting. Eur Phys J E 3(2):159–163
Chen T-W, Wardill TJ, Sun Y et al (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499(7458):295–300
Franklin KBJ, Paxinos G (2013) Paxinos and Franklin’s the mouse brain in stereotaxic coordinates, 4th edn. Academic Press, an imprint of Elsevier, Amsterdam
Lee C-Y, Han J-H (2013) Elimination of honeycomb patterns in fiber bundle imaging by a superimposition method. Opt Lett 38(12):2023–2025
Lee C-Y, Han J-H (2013) Integrated spatio-spectral method for efficiently suppressing honeycomb pattern artifact in imaging fiber bundle microscopy. Opt Commun 306:67–73
Göbel W, Kerr JND, Nimmerjahn A et al (2004) Miniaturized two-photon microscope based on a flexible coherent fiber bundle and a gradient-index lens objective. Opt Lett 29(21):2521–2523
Winter C, Rupp S, Elter M et al (2006) Automatic adaptive enhancement for images obtained with fiberscopic endoscopes. IEEE Trans Biomed Eng 53(10):2035–2046
Rupp S, Winter C, Elter M Evaluation of spatial interpolation strategies for the removal of comb-structure in fiber-optic images. In: 2009 annual international conference of the IEEE Engineering in Medicine and Biology Society, 3–6 Sept. 2009, pp 3677–3680. https://doi.org/10.1109/IEMBS.2009.5334719
Ozden I, Lee HM, Sullivan MR et al (2008) Identification and clustering of event patterns from in vivo multiphoton optical recordings of neuronal ensembles. J Neurophysiol 100(1):495–503
Flusberg BA, Nimmerjahn A, Cocker ED et al (2008) High-speed, miniaturized fluorescence microscopy in freely moving mice. Nat Methods 5(11):935–938
Dubbs A, Guevara J, Yuste R (2016) Moco: fast motion correction for calcium imaging. Front Neuroinform 10:6
Dombeck D, Tank D (2014) Two-photon imaging of neural activity in awake mobile mice. Cold Spring Harb Protoc 2014(7):pdb.top081810
Dombeck DA, Khabbaz AN, Collman F et al (2007) Imaging large-scale neural activity with cellular resolution in awake, Mobile mice. Neuron 56(1):43–57
Mathis A, Mamidanna P, Cury KM et al (2018) DeepLabCut: markerless pose estimation of user-defined body parts with deep learning. Nat Neurosci 21(9):1281–1289
Ziv Y, Burns LD, Cocker ED et al (2013) Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci 16(3):264–266
Ozbay BN, Futia GL, Ma M et al (2018) Three dimensional two-photon brain imaging in freely moving mice using a miniature fiber coupled microscope with active axial-scanning. Sci Rep 8(1):8108
Acknowledgments
We would like to thank Ethan Hughes for assistance with in vivo animal imaging work, Juliet Gopinath, Victor Bright, and Robert Cormack for helpful discussions. We acknowledge Nicole Arevalo for assistance with animal care, and Elizabeth Gould and Wendy Macklin for providing brain tissue from Plp-EGFP mice. Funding for this work was provided by the National Science Foundation DBI-1353757, CBET-1631912, and IIP-1602128 and the National Institutes of Health BRAIN Initiative NS099577.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this protocol

Cite this protocol
Ozbay, B.N. et al. (2023). Miniature Multiphoton Microscopes for Recording Neural Activity in Freely Moving Animals. In: Papagiakoumou, E. (eds) All-Optical Methods to Study Neuronal Function. Neuromethods, vol 191. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2764-8_7
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2764-8_7
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2763-1
Online ISBN: 978-1-0716-2764-8
eBook Packages: Springer Protocols