
Abstract
Plant cell-free lysates contain all the cellular components of the protein biosynthesis machinery, providing an alternative to intact plant cells, tissues, and whole plants for the production of recombinant proteins. Cell-free lysates achieve rapid protein production (within hours or days) and allow the synthesis of proteins that are cytotoxic or unstable in living cells. The open nature of cell-free lysates and their homogeneous and reproducible performance is ideal for protein production, especially for screening applications, allowing the direct addition of nucleic acid templates encoding proteins of interest, as well as other components such as enzyme substrates, chaperones, artificial amino acids, or labeling molecules. Here we describe procedures for the production of recombinant proteins in the ALiCE (Almost Living Cell-free Expression) system, a lysate derived from tobacco cell suspension cultures that can be used to manufacture protein products for molecular and biochemical analysis as well as applications in the pharmaceutical industry.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- ALiCE system
- BYL
- Cell-free biosynthesis
- High-throughput screening
- In vitro transcription-translation
- Posttranslational modifications
- Protein expression
- Tobacco cell-free lysate
1 Introduction
Cell-free protein biosynthesis based on crude lysates offers several advantages over living cells, including shorter process times and the ability to prepare lysates in advance so that they can be stored for as long as necessary before starting protein production. Crude cell lysates contain the basic components required for translation, protein folding, and energy metabolism. Almost any protein encoded by an RNA template can be synthesized by adding amino acids, energy substrates, nucleotides, and salts. In coupled transcription-translation systems, recombinant proteins can be synthesized in a single batch process following the addition of the appropriate RNA polymerase (e.g., T7 RNA polymerase) and a DNA template (PCR product or vector ) containing an expression cassette with the corresponding promoter. In contrast to cell-based expression methods, cell-free expression results in limited protein hydrolysis and confers the ability to express difficult-to-produce or even toxic proteins or proteins containing specific chemical groups or unnatural amino acids at defined positions. Furthermore, the open nature of the system allows the reaction to be controlled and monitored directly.
The most widely used cell-free systems are based on Escherichia coli and are advantageous because of their low cost, scalability, and high productivity (up to 2.7 mg/mL) [1, 2]. However, because the lysates originate from bacteria, they are generally unsuitable for the production of complex proteins with multiple subdomains or posttranslational modifications due to inefficient oxidative folding and the absence of chaperones and glycosylation machinery. More recent bacterial lysates have been developed to facilitate the production of complex proteins [3,4,5] and to perform specific posttranslational modifications such as glycosylation [6] but they do not have the universal capabilities of eukaryotic cell-free systems. Several eukaryotic cell-free lysates have been described, including rabbit reticulocyte lysate, yeast cell extract, Chinese hamster ovary cell lysate, HeLa cell lysate, and wheat germ extract [7]. However, eukaryotic cell-free systems are generally expensive, extract preparation is laborious, and protein yields are low. Wheat germ extract is regarded as the gold standard for recombinant protein expression in a cell-free eukaryotic system, and commercial lysates achieve yields of up to 0.1 mg/mL [8]. Higher yields of 1 mg/mL have been achieved in a dialysis bag continuously fed with substrates and with small inhibitory byproducts continually removed [9, 10], but this approach is inconvenient, expensive, and difficult to scale up, particularly when running dozens or hundreds of parallel reactions in microtiter plates for screening studies.
We recently developed a new cell-free expression platform based on tobacco BY-2 cells [11], and achieved yields of up to 0.27 mg/mL when using the enhanced yellow fluorescent protein (eYFP) as a model in a coupled transcription-translation process for 18 h in a simple batch reaction [12]. Tobacco BY-2 cell suspension cultures are easy to prepare in large-scale batches, providing ample cell material for lysate preparation [13]. The resulting BY-2 lysates (BYL) can be prepared within 5 h by isolating protoplasts, followed by density centrifugation to remove the vacuole (containing most of the nucleases and proteases that reduce protein yields), and finally the mechanical disruption of evacuolated protoplasts [12].
Most proteins are expressed and accumulate in the BYL cytosolic fraction. However, the BYL system contains actively-translocating microsomes generated by the disruption of the endoplasmic reticulum during lysate preparation. Therefore, protein products can be targeted to the microsomal vesicles by including N-terminal signal peptides, enabling the formation of disulfide bonds and the efficient folding and assembly of complex and multimeric proteins such as enzymes, full-size antibodies, and even membrane proteins. The BYL system also supports glycosylation, which is only possible in wheat germ extracts following the provision of additional microsomes [12, 14,15,16].
The productivity of the BYL system has been increased from 0.27 to 3 mg/mL by optimizing the lysate preparation and reaction conditions, and by extending the transcription-translation process to up to 48 h [7, 17]. This outperforms any other eukaryotic cell-free lysate. In addition, BYL reactions can be scaled up to 10 mL without loss of productivity, indicating the potential for even greater scalability (unpublished data), although special care is required with large reaction volumes to ensure a sufficient supply of oxygen for energy generation in the mitochondria. The BYL system has been commercialized by the company LenioBio GmbH and is marketed under the brand name ALiCE® [18].
In the following protocol, we describe how to produce recombinant proteins in the ALiCE system. We explain the preparation of the expression vector, the assembly and initiation of the cell-free reaction, and finally the purification of Strep-II-tagged target proteins by affinity chromatography.
2 Materials
RNase contamination destroys mRNA generated by the BYL system and therefore reduces or abolishes protein synthesis. Only use RNase-free filter tips and RNase-free water, and wear gloves at all times.
2.1 Preparation of the Expression Vector
-
1.
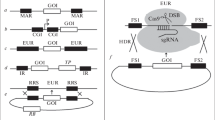
ALiCE expression vectors pALiCE01 and pALiCE02 (Figs. 1 and 2) (see Note 1).
-
2.
Restriction enzymes NcoI and KpnI for the insertion of cDNAs downstream from the T7 promoter and 5′ untranslated region (UTR) of pALiCE01.
-
3.
Restriction enzymes NcoI and NotI (or KpnI) for insertion of cDNAs downstream from the T7 promoter and 5′ UTR of pALiCE02.
-
4.
Competent E. coli DH5α cells.
-
5.
Plasmid DNA purification kit (see Note 2).
-
6.
5 mM Tris–HCl buffer (pH 8.5).

Map of the pALiCE01 and pALiCE02 vectors and sequence reference points. T7 = T7 promoter; 5′ UTR = 5′ untranslated region; MSP = melittin signal peptide sequence; 3′ UTR = 3′ untranslated region; bla = β-lactamase gene (ampicillin resistance); ori = plasmid origin of replication

Restriction sites of interest in pALiCE01 (a) and pALiCE02 (b). (a) Upper part: upstream cloning region of the gene of interest (here, eYFP). Lower part: downstream cloning region of the gene of interest. Please note that the start codon is a part of the NcoI restriction site (underlined). (b) Upper part: upstream cloning region of the gene of interest (here, eYFP). Lower part: downstream cloning region of the gene of interest. The choice between NotI and KpnI allows the insertion of an optional His6 tag. If using the KpnI restriction site, a stop codon should be added to the gene of interest
2.2 Coupled Transcription-Translation Reaction
-
1.
The ALiCE kit is commercially available from several companies (www.ambsbio.com; www.leniobio.com; www.sigmaaldrich.com) (see Note 3).
-
2.
RNase-free 96-well half-area tissue culture microplates with lids.
-
3.
RNase-free and DNase-free 50-mL bioreactor tubes with filter screw caps and 5–10 openings.
-
4.
Tabletop centrifuge.
-
5.
Orbital shaker (see Note 4).
2.3 Protein Recovery and Affinity Purification
-
1.
5% (w/v) n-dodecyl-β-maltoside (DDM) in 10× buffer W.
-
2.
Microtube shaking incubator.
-
3.
Tabletop centrifuge.
-
4.
Strep-Tactin XT Superflow high-capacity 50% suspension (IBA Lifesciences).
-
5.
10× buffer W: 1 M Tris–HCl pH 8.0, 1.5 M NaCl, 10 mM EDTA (see Note 5).
-
6.
Buffer BXT: 100 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 50 mM biotin (see Note 5).
-
7.
Gravity-flow column.
3 Methods
3.1 Preparation of the Expression Vector
-
1.
For standard cytosolic expression, insert the cDNA encoding the desired protein into vector pALiCE01 (Fig. 1) via the restriction sites NcoI and KpnI (Fig. 2). If microsomal targeting is required, insert the cDNA into vector pALiCE02 (Fig. 1) using the restriction sites NcoI and NotI, or NcoI and KpnI (see Notes 6–8).
-
2.
Introduce the recombinant vectors into competent E. coli DH5α cells (see Notes 9 and 10).
-
3.
Create a master plate of 4–8 transformants and isolate plasmids from the transformed bacteria by anion exchange chromatography for purification (see Note 11).
-
4.
Verify the presence of the cDNA insert by DNA sequencing using standard protocols [19].
-
5.
If larger amounts of plasmid DNA are required, a positive clone from the master plate can be used to inoculate liquid cultures for additional plasmid preparation steps (see Note 2). For long-term storage, prepare a glycerol stock from a positive clone.
-
6.
Adjust the final plasmid DNA to the desired concentration depending on the size of the insert (e.g., 125 nM) by adding an appropriate volume of 5 mM Tris buffer (pH 8.5). The final plasmid concentration in the cell-free reaction mix should be 5 nM (see Note 12).
-
7.
Store the plasmid in aliquots at −20 °C.
3.2 Coupled Transcription-Translation Reaction
3.2.1 General Procedures
-
1.
The typical reaction volume for a coupled cell-free transcription-translation reaction is 52 μL in 2-mL tubes or 50 μL in microtiter plates, with larger volumes prepared in 50-mL tubes. Accordingly, choose the reaction vessel as follows: (a) a 2-mL tube provided in the ALiCE kit, already containing 50 μL of reaction mix; (b) an empty 96-well half-area plate; or (c) an empty 50-mL tube.
-
2.
Thaw the plasmid DNA template at room temperature (20–25 °C). Mix gently by flipping and centrifuge briefly to collect all liquid at the bottom of the tube, then place on ice.
-
3.
Remove the ALiCE reaction mix from storage and thaw at room temperature (20–25 °C) in a water bath. Mix gently by inverting the tube (see Note 13).
-
4.
Place the ALiCE reaction mix on ice immediately after thawing. Ensure that the reactions are initiated within 30 min after thawing. Freeze any remaining lysate immediately at −80 °C (see Note 14).
-
5.
Assemble and initiate the cell-free coupled transcription-translation reaction as described in Subheadings 3.2.2–3.2.4 depending on the reaction volume and reaction vessel (see Note 15).
-
6.
The protein can be detected by SDS-PAGE and Coomassie staining or immunoblot using antibodies specific for the target protein or Strep-II tag [18] (see Notes 16 and 17).
3.2.2 Coupled Transcription-Translation Reaction: 50-μL Scale in Microtiter Plates
-
1.
Add 48 μL of the reaction mix to each well of an RNase-free 96-well half-area microtiter plate.
-
2.
Add 2 μL of plasmid DNA to each well, resulting in a final concentration of 5 nM in the total reaction volume of 50 μL (see Notes 18 and 19).
-
3.
Pipette water into the spaces between the reaction compartments of the plate (75 μL each) to maintain high humidity inside the plate during incubation (Fig. 3) (see Note 20).
-
4.
Incubate the plates in an orbital shaker at 500 rpm and 25 °C for up to 48 h. A 12.5 mm or 25 mm shaking diameter and controlled humidity of 80% is recommended for optimal results. If no humidity control is available, open containers filled with water should be placed in the shaker to provide a higher than ambient relative humidity (see Note 21).

Orientation of spaces within a microtiter plate that should be filled with water to increase humidity, thus avoiding the evaporation of the reaction mix during in vitro transcription-translation
3.2.3 Coupled Transcription-Translation Reaction: 52-μL Scale in 2-mL Tubes
-
1.
Add 2 μL of plasmid DNA to the 50 μL reaction mix in the 2-mL tubes of the ALiCE kit resulting in a final plasmid concentration of 5 nM (see Notes 18 and 19).
-
2.
Only use the supplied punctured caps to close the 2-mL tubes (see Note 22).
-
3.
Incubate the 2-mL tubes in an orbital tabletop shaker at 700 rpm and 25 °C for up to 48 h (see Note 23).
3.2.4 Coupled Transcription-Translation Reaction at Volumes of 4–6 mL in 50-mL Bioreactor Tubes
-
1.
Add 40–60 μL of plasmid DNA to the 4–6 mL reaction mix in a 50-mL bioreactor tube, resulting in a final plasmid concentration of 5 nM (see Notes 18 and 19).
-
2.
Close the tube using a filter cap with openings to ensure a constant oxygen supply.
-
3.
Incubate the 50-mL tube on a tube roller or in a roller drum at 40 rpm and 25 °C for up to 48 h (see Note 4).
3.3 Protein Recovery and Affinity Purification
Recombinant proteins accumulating in the cytosolic fraction of the cell-free lysate (following expression using the pALiCE01 vector ) can be directly applied to affinity chromatography columns after centrifugation at 16,000 × g for 10 min at room temperature and the dilution of the supernatant with 10× buffer W at a 9:1 ratio (Subheading 3.3.2). Cell-free production using the pALiCE02 vector will result in the accumulation of recombinant proteins in the microsomes. The target protein can be released from the microsomes by following the steps described in Subheading 3.3.1.
3.3.1 Recovery of Recombinant Proteins from the Microsomes
-
1.
Add DDM from a 5% (w/v) stock solution in 10× buffer W to the reaction mix to achieve a final concentration of 0.5%, which will lyse the microsomes (see Note 24).
-
2.
Incubate for 10 min at room temperature, shaking at 700 rpm (see Note 25).
-
3.
Centrifuge the preparation at 16,000 × g for 10 min at room temperature. Transfer the supernatant containing the protein to a fresh reaction tube (see Note 26).
-
4.
The supernatant can be stored at 4 °C for a maximum of one day, or for longer time at −20 °C, or can be used directly for affinity chromatography as described in Subheading 3.3.2 (see Note 27).
3.3.2 Purification of Strep-II-Tagged Proteins by Affinity Chromatography
The presence of epitope tag sequences in the expression vector allows target protein purification using a universal affinity chromatography step. Here, we describe affinity purification via the Strep-II tag in a 5-mL reaction mix. Smaller or larger volumes will require different sizes of purification column and different amounts of purification resin.
-
1.
Add 2 mL of Strep-Tactin XT Superflow high-capacity 50% suspension (corresponding to 1 mL resin) to the gravity-flow column and remove the resin storage buffer by gravity flow (see Note 28).
-
2.
Equilibrate the column twice by adding 1 mL 1× buffer W (two column bed volumes). Remove buffer W by gravity flow.
-
3.
Apply the supernatant fraction from the untreated reaction mixture (for cytosolic proteins) or the supernatant fraction derived from the reaction mixture treated with DDM (for proteins recovered from microsomes). Allow the lysate to run through the column by gravity flow so that the Strep-II tag binds to the resin. Collect the flow-through fraction (FT).
-
4.
Wash the column five times with 1 mL of 1× buffer W (five column bed volumes) and let the buffer run through the column by gravity flow. Collect each wash fraction (W1–W5).
-
5.
Elute the bound protein by sequentially adding six 0.5-mL aliquots of buffer BXT (six half column bed volumes) and collect the eluate fractions (E1–E6).
-
6.
Analyze the proteins in the FT, W1–W5 and E1–E6 fractions by SDS-PAGE [18] (see Notes 16 and 17).
4 Notes
-
1.
Both expression vectors (pALiCE01 and pALiCE02) are provided in the pALiCE kit. The pALiCE01 vector causes proteins to accumulate in the cytosolic fraction resulting in high product yields for most target proteins but no posttranslational modifications. The pALiCE02 vector should be used when expressing more complex proteins (e.g., requiring disulfide bonds and/or glycosylation). This vector contains a sequence encoding the N-terminal melittin signal peptide enabling the translocation of the target protein to the microsomes (organelle-like structures that form parts of the endoplasmic reticulum and the Golgi-apparatus during the ALiCE manufacturing process). Even when using the melittin signal peptide sequence, some portion of the target protein may accumulate in the cytosolic fraction. Both vectors contain the T7 RNA polymerase promoter to enable efficient transcription, a 5′ UTR, a Strep-II tag sequence to purify the target protein by affinity chromatography, and the sequence encoding the fluorescent protein eYFP. Therefore, both vectors can also be used as a positive control for expression, accumulating eYFP in the cytosol or microsomes, respectively. In addition, pALiCE02 also carries a His6 tag for protein detection and purification. The vector maps of pALiCE01 and pALiCE02 are shown in Fig. 1. Expanded views of the cloning sites in the 5′ and 3′ directions are shown in Fig. 2. The complete plasmid sequences are provided in the instruction manual for the ALiCE kit [20].
-
2.
Highly-purified plasmid DNA is required to facilitate an efficient transcription-translation reaction. A plasmid preparation method based on anion exchange chromatography is therefore recommended (e.g., NucleoBond Xtra Midi from Macherey-Nagel). For plasmid DNA preparation kits based on silica matrices, we recommend the addition of RNase inhibitors or phenol-chloroform purification of the template before the transcription-translation reaction.
-
3.
The ALiCE kit is shipped on dry ice. Upon arrival, immediately store the individual components as indicated in the instruction manual. The cell-free reaction mix is stable for at least 12 months under these conditions.
-
4.
Reactions in microtiter plates require a shaker with controlled humidity (~80%), a plate holder, and a shaking diameter of 12.5 or 25 mm. Use RNase-free 96-well half-area tissue culture microplates. Reactions in tubes provided with the ALiCE kit require a tabletop shaker with 2-mL tube inserts and a shaking diameter of 3 mm. Larger reaction volumes (up to 6 mL) can be accommodated in 50-mL tubes using a tube roller or roller drum. If possible, use an incubator with controlled humidity, or place open vessels filled with water inside the incubator to increase humidity.
-
5.
EDTA should not be included in the buffer used for protein purification if the target is a metalloprotein.
-
6.
The cDNA encoding the target protein can be subcloned from an existing vector or the coding region can be amplified by PCR using primers that introduce appropriate restriction sites. Alternatively, the cDNA can be inserted into the expression vector by Gibson assembly [21]. If a Strep-II tag is used for purification, ensure that the cDNA includes the corresponding sequence.
-
7.
If using KpnI for cloning, make sure the open reading frame contains a stop codon.
-
8.
Standard cloning procedures are recommended to generate large quantities of plasmid DNA. However, to save the time and labor involved in cell-based cloning, various cell-free methods have been described, including rolling circle replication [22, 23].
-
9.
E. coli strains other than DH5α may have an adverse effect on target protein yields due to the lower quality of the plasmid DNA.
-
10.
The vectors encode a β-lactamase (bla) to allow selection on media containing ampicillin.
-
11.
Plasmid preparation and purification using a “miniprep” kit (e.g., NucleoBond from Macherey-Nagel) is sufficient, but RNAse inhibitors should be added to avoid transcript degradation. However, “midiprep” kits are recommended to maximize the yields of transcription-translation reactions (see Note 2).
-
12.
The DNA concentration significantly affects the protein yield.
-
13.
Do not mix by vortexing because this will disrupt the reaction components and significantly reduce yields.
-
14.
Do not use liquid nitrogen to store the reaction mix and avoid more than one freeze-thaw cycle.
-
15.
The ALiCE reaction mix already contains all components required for in vitro transcription-translation, including T7 RNA polymerase, nucleotides, and amino acids. Only the DNA template should be added to the reaction mix.
-
16.
To ensure the target protein is efficiently separated from lysate-derived background proteins, high-resolution SDS-PAGE should be carried out with an appropriate gel concentration (e.g., 10% (w/v)). We recommend an SDS-PAGE gradient gel (e.g., 4–12% (w/v)) and a maximum of 0.5–1 μL of sample per lane. Heating the sample to 95 °C for 10 min before loading is required for denaturing SDS-PAGE . Insoluble compounds can be pelleted by centrifugation allowing the separate analysis of the soluble and insoluble fractions by SDS-PAGE .
-
17.
When using eYFP as positive control, a successful reaction is indicated by the yellow coloration of the lysate. However, precise protein yield can be determined by measuring the fluorescence emission at 528 nm (excitation wavelength = 485 nm) compared to known eYFP standards.
-
18.
The ideal concentration of the plasmid DNA is 5 nM, but an approximate mass of plasmid DNA required as the template may be calculated using the following formula: plasmid DNA mass = length of the complete vector [bp]/3000 bp × 0.5 μg. However, the optimal plasmid concentration may be dependent on the DNA template and therefore plasmid DNA concentrations in a range of 1–20 nM should be tested to identify the optimal concentration.
-
19.
If required, more than one plasmid can be added to a single reaction, but the total plasmid concentration should be 5 nM (see Note 18).
-
20.
Do not seal the plate because the cell-free lysate requires oxygen throughout the reaction for energy production in the mitochondria.
-
21.
When using a shaker with a shaking diameter of 3 mm, increase shaking speed to 700 rpm. Protein expression is still possible if the humidity falls below 80%, although evaporation may occur and this tends to reduce the yield.
-
22.
Because oxygen is needed throughout the reaction, the reaction volume in the tubes can be increased to 200 μL but this will reduce productivity. For reaction volumes above 200 μL, the sample should be divided into smaller volumes or transferred to larger reaction tubes (Subheading 3.2.4).
-
23.
For optimal results, we recommend a shaking diameter of 3 mm and the use of a holding block for 2-mL tubes.
-
24.
Different DDM concentrations (or alternative detergents) may be more suitable for the recovery of certain proteins, depending on the features of the target protein.
-
25.
Do not mix by vortexing because this will disrupt the reaction components.
-
26.
Keep in mind that any DDM in the supernatant may affect subsequent steps, including activity assays or purification by chromatography.
-
27.
Purification is most efficient if the purification step is initiated immediately after protein recovery.
-
28.
The volume of lysate should be 0.5–10 column bed volumes.
References
Caschera F, Noireaux V (2014) Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie 99:162–168
Des Soye BJ, Gerbasi VR, Thomas PM, Kelleher NL, Jewett MC (2019) A highly productive, one-pot cell-free protein synthesis platform based on genomically recoded Escherichia coli. Cell Chem Biol 26:1743–1754.e9
Goerke AR, Swartz JR (2008) Development of cell-free protein synthesis platforms for disulfide bonded proteins. Biotechnol Bioeng 99:351–367
Smith MT, Hawes AK, Shrestha P, Rainsdon JM, Wu JC, Bundy BC (2014) Alternative fermentation conditions for improved Escherichia coli-based cell-free protein synthesis for proteins requiring supplemental components for proper synthesis. Process Biochem 49:217–222
Higuchi K, Yabuki T, Ito M, Kigawa T (2020) Cold shock proteins improve E. coli cell-free synthesis in terms of soluble yields of aggregation-prone proteins. Biotechnol Bioeng 117:1628–1639
Jaroentomeechai T, Stark JC, Natarajan A, Glasscock CJ, Yates LE, Hsu KJ, Mrksich M, Jewett MC, DeLisa MP (2018) Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat Commun 9:2686
Schillberg S, Raven N, Spiegel S, Rasche S, Buntru M (2019) Critical analysis of the commercial potential of plants for the production of recombinant proteins. Front Plant Sci 10:720
Makino S, Beebe ET, Markley JL, Fox BG (2014) Cell-free protein synthesis for functional and structural studies. Methods Mol Biol 1091:161–178
Harbers M (2014) Wheat germ systems for cell-free protein expression. FEBS Lett 588:2762–2773
Buntru M, Vogel S, Spiegel H, Schillberg S (2014) Tobacco BY-2 cell-free lysate: an alternative and highly-productive plant-based in vitro translation system. BMC Biotechnol 14:37
Buntru M, Vogel S, Stoff K, Spiegel H, Schillberg S (2015) A versatile coupled cell-free transcription-translation system based on tobacco BY-2 cell lysates. Biotechnol Bioeng 112:867–878
Raven N, Rasche S, Kuehn C, Anderlei T, Klöckner W, Schuster F, Henquet M, Bosch D, Büchs J, Fischer R, Schillberg S (2015) Scaled-up manufacturing of recombinant antibodies produced by plant cells in a 200 L orbitally shaken disposable bioreactor. Biotechnol Bioeng 112:308–321
Havenith H, Kern C, Rautenberger P, Spiegel H, Szardenings M, Ueberham E, Lehmann J, Buntru M, Vogel S, Treudler R, Fischer R, Schillberg S (2017) Combination of two epitope identification techniques enables the rational design of soy allergen Gly m 4 mutants. Biotechnol J 12:1600441
Huck NV, Leissing F, Majovsky P, Buntru M, Aretz C, Flecken M, Müller JP, Vogel S, Schillberg S, Hoehenwarter W, Conrath U, Beckers GJM (2017) Combined 15N-labeling and tandemMOAC quantifies phosphorylation of MAP kinase substrates downstream of MKK7 in Arabidopsis. Front Plant Sci 8:2050
Wu D, von Roepenack-Lahaye E, Buntru M, de Lange O, Schandry N, Pérez-Quintero AL, Weinberg Z, Lowe-Power TM, Szurek B, Michael AJ, Allen C, Schillberg S, Lahaye T (2019) A pathogen effector subverts translational regulation to boost host polyamine levels. Cell Host Microbe 26:638–649.e5
Schillberg S, Finnern R (2021) Plant molecular farming for the production of valuable proteins – critical evaluation of achievements and future challenges. J Plant Physiol 258-259:153359. https://doi.org/10.1016/j.jplph.2020.153359
Green MR, Sambrook J (2012) Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
https://irp.cdn-website.com/f5971158/files/uploaded/ALiCE-Instruction-Manual-Rev-1.1-1.pdf
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345
Kumar G, Chernaya G (2009) Cell-free protein synthesis using multiply-primed rolling circle amplification products. BioTechniques 47:637–639
Wang K, Ma Q, Jiang L, Shujuan L, Lu X, Hou Y, Wu C, Ruan J (2016) Ultra-precise detection of mutations by droplet-based amplification of cirucularized DNA. BMC Genomics 17:214
Acknowledgments
We thank Dr. Richard M Twyman for editorial assistance. The protocols described herein are partly derived from the projects “Cell-Free Biosynthesis” (FKZ 0315942) and “ThinkBig” (FKZ 031B0830A/FKZ 031B0830A), both funded by the German Federal Ministry of Education and Research (BMBF), as well as ‘PEPPER’ (881025) funded by the EIC Horizon 2020.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Buntru, M., Vogel, S., Finnern, R., Schillberg, S. (2022). Plant-Based Cell-Free Transcription and Translation of Recombinant Proteins. In: Schillberg, S., Spiegel, H. (eds) Recombinant Proteins in Plants. Methods in Molecular Biology, vol 2480. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2241-4_8
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2241-4_8
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2240-7
Online ISBN: 978-1-0716-2241-4
eBook Packages: Springer Protocols