
Abstract
The mRNA epitranscriptome imparts diversity to gene expression by installing chemical modifications. Advances in detection methods have identified chemical modifications in eukaryotic, bacterial, and viral messenger RNAs (mRNAs). The biological functions of modifications in mRNAs still remain to be understood. Chemical modifications are introduced in synthetic mRNAs meant for therapeutic applications to maximize expression from the synthetic mRNAs and to evade the host immune response. This overview provides a background of chemical modifications found in mRNAs, with an emphasis on pseudouridine and its known effects on the mRNA life cycle, its potential applications in synthetic mRNA, and the methods used to assess its effects on mRNA translation.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
The emerging field of epitranscriptomics has uncovered an additional layer of complexity in the regulation of gene expression that stems from the addition of RNA modifications post-transcriptionally. More than 150 modifications have been identified in highly abundant noncoding RNAs (ncRNAs, such as ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), and small nuclear RNAs (snRNAs)) [1, 2]. It was thought for a long time that post-transcriptional chemical modifications of RNA that affected the RNA’s biogenesis, function, and stability were limited to ncRNAs. Additionally, the only modifications that were reported for protein-coding messenger RNAs (mRNAs) were those in the mRNA 5′ cap (N7-methylguanosine (m7G)) and 3′ poly(A) tail, internal inosine (I) modifications, and modifications of internal adenosines to N6-methyladenosine (m6A) [1]. With advances in next-generation sequencing technologies and in analytical chemistry, a wide range of RNA modifications have now been identified in mRNAs, albeit at lower levels. Among the modifications that have been identified in mRNAs, m6A and pseudouridine (Ψ) occur most frequently. Although the first chemical modifications on mRNA were identified more than 40 years ago, their functional significance has only just begun to be uncovered. The localization of modifications throughout the body of the mRNAs suggests that modifications could potentially alter protein production by multiple mechanisms, and it is becoming increasingly clear that these modifications play diverse roles in a variety of physiological and disease settings.
Here, the effects of mRNA modifications on the life cycle of an mRNA, with a specific emphasis on the process of translation, are discussed. The discovery of mRNA modifications, and the pros and cons of the various detection methods, has been covered extensively in the literature. Because m6A has been discussed in detail in other chapters in this issue, I will focus on Ψ, the second most abundant mRNA modification. I also summarize the current state of knowledge of mRNA modifications, highlighting their known effects on protein expression, and also discuss the applications of mRNA modifications in the synthesis of functional mRNAs. Finally, I provide an overview of the methods used to assess the biological functions of mRNA modifications.
2 Overview of mRNA Life Cycle
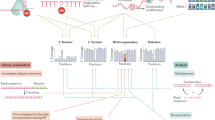
Gene expression in eukaryotes is a complex and highly regulated process, and mRNAs are subjected to multiple regulatory mechanisms throughout the course of their life cycle (Fig. 1) [3]. Eukaryotic pre-mRNAs are processed by the capping, splicing, and polyadenylation machineries in the nucleus. The mature mRNA, which is now ready for export, travels to the nuclear envelope and is translocated into the cytoplasm, where it is accessed by either the translation machinery to begin the process of protein synthesis or the RNA degradation machinery. Under certain circumstances, some mRNA species also undergo an additional localization process that enables the migration of translationally silenced mRNAs to specific cellular destinations before subsequent translation. In the cytosol , the number of protein molecules produced from each mRNA molecule is a combinatorial effect of the mRNA’s translational efficiency and its half-life. Early notions of the interplay between translation and decay were simplistic and based on the idea that mRNAs undergoing translation were protected from decay [3, 4]. It is now well understood that the decay of individual mRNAs can be either accelerated or antagonized by loss of translation efficiency, and changes in mRNA half-life can further alter translational fidelity [4]. A complex set of protein–mRNA and RNA–RNA interactions determine the fate of the mRNA, and perturbation to this set of interactions can have consequences on the mRNA’s life cycle. The presence of chemical tags on the mRNA is one such factor.

A simplified illustration showing the different steps in the life cycle of an mRNA. The pre-mRNA undergoes 5′- and 3′-processing, splicing, and polyadenylation, and the mature mRNA is then exported to the cytosol where it undergoes either translation or decay. Chemical modifications can influence all of the steps in mRNA metabolism, including splicing, maturation, translation, and decay
2.1 Chemical Modifications of mRNA
It is well documented that chemical modifications in ncRNAs can alter the charge, sterics, and conformation of the target RNA molecule, thus modulating the RNA’s biogenesis, stability, and function. Therefore, it is not surprising that the presence of chemical modifications in the mRNA can have similar effects in altering gene expression (Fig. 1). Previous studies have shown that the levels of different mRNA modifications vary widely and that there can be >1000-fold differences in the levels of different modifications [2]. Across organisms, the m6A modification has been found to be the most prevalent, and the abundances of Ψ, N4-acetylcytidine (ac4C), Cm, and Gm have been observed to be approaching those of m6A [2]. On the other hand, 3-methylcytidine (m3C), 1-methylguanine (m1G), and 5-hydroxymethylcytidine (hm5C) are rare, with levels at least 500-fold lower than that of m6A, making accurate detection of these modifications more difficult [2]. In general, the absence of high-quality quantitative mRNA modification maps makes it difficult to correlate the effects of modifications with biological phenotypes, and thus, to understand their physiological relevance, especially with respect to translation and mRNA stability. Furthermore, another critical factor to consider when dissecting the functional consequences of a specific modification is the relative position of the modified nucleotide in the mRNA. On the one hand, the presence of a modification in the coding sequence (CDS) of an mRNA has the potential to alter translation fidelity, decoding, and ribosome transit (Fig. 2). On the other hand, the presence of a modification in the untranslated regions (UTRs) can result in changes to the stability of the RNA structure, thereby modulating its ability to form RNA–protein interactions, which can consequently impact mRNA maturation, translation, and decay through pathways dependent on these interactions. Understanding the effects of modified nucleotides in mRNAs is further complicated by the fact that some of these modifications are reversible, whereas others are irreversible, suggesting that modifications have the potential to dynamically control gene expression. This form of dynamic control of gene expression can be viewed as a coordinated effort between writer proteins (RNA-modifying enzymes), reader (RNA-binding) proteins, and eraser proteins (which remove the modification from the RNA) (Table 1). The interactions of the reader and eraser proteins with the m6A-containing transcripts have been critical in dissecting the effects of m6A on the mRNA life cycle [5, 6]. In addition, positional information of m6A in the mRNA has greatly contributed to a functional understanding of m6A in biological contexts; depending on its location along the mRNA, m6A can affect fundamental processes of cell metabolism—such as splicing, transport, stability, and translation of the target mRNA—by providing interaction platforms for specific reader proteins [7]. Transcriptome-wide studies in conjunction with genetic and biochemical investigations have helped to elucidate how the presence of m6A in an mRNA affects the processes of mRNA degradation, transcript maturation, and translation [5, 8, 9]. Mounting evidence has now shown the involvement of m6A methylation status in embryonic development, stem cell regulation, adipogenesis, development of obesity and pathogenesis of type 2 diabetes, immunological processes, and carcinogenesis [10,11,12,13].

A simplified illustration showing the consequences of mRNA chemical modifications on translation. In contrast to an mRNA without any modification (a), the presence of a modification can result in aberrant translation termination, resulting in a protein pool with truncated peptides as observed for N1-m-pseudouridine (b). The presence of pseudouridine can also result in translation recoding and misincorporation of amino acids in the growing peptide (c). Modification-induced changes in secondary structure or RNA-protein interactions can alter ribosome recycling and ribosome transit time (as observed for N1-m-pseudouridine), subsequently affecting mRNA stability or even protein folding (d). All of these can lead to a change in the contents of the protein pool
In the subsequent sections, chemical modifications (other than m6A) that have been identified in mRNAs and their potential effects on mRNA stability and translation are discussed.
3 Modifications at the Ends of the mRNA
3.1 Modifications of mRNA 5′ Termini
The mRNA 5′ cap is a well-studied and well-documented structure that is post-transcriptionally added to the pre-mRNA. It consists of an untemplated guanosine that is then methylated to form 7-methylguanosine (m7G) via three enzymatic steps involving an RNA triphosphatase (TPase), an RNA guanylyltransferase (GTase), and a guanine-N7 methyltransferase (guanine-N7 MTase) [14]. m7G is linked to the first transcribed nucleotide via a 5′–5′ triphosphate bond to form the minimal Cap 0 structure (m7GpppRNA) in lower eukaryotes. mRNAs in higher eukaryotes can further have an additional methyl group on the ribose 2′-O position of the first transcribed nucleotide (2′-O-methylated versions of A, U, C, G) to form Cap 1 RNA, and the adjoining nucleotide can also be subsequently methylated to yield the Cap 2 structure [15]. Furthermore, certain vertebrate mRNAs have either an m6A or a 2′-O-dimethyladenosine (m6Am) as the first nucleotide [5, 8, 16, 17]. The modifications at the 5′ end of the mRNA act as a platform for multiple mRNA–protein interactions. The m7G cap is first recognized by the nuclear cap-binding complex (CBC), which facilitates mRNA export from the nucleus and is later replaced by the translation initiation factor eIF4E. Furthermore, the Cap 1 and Cap 2 structures (presence of the 2′-O methylation) have been shown to be pivotal for distinction between self and foreign RNA via altered recognition by RIG-I, MDA5, and IFIT-1 (cellular sensors that induce type I interferon (IFN) signaling) [18,19,20,21]. Binding of IFIT-1 to the mRNAs at the extended cap structure can also result in suppression of translation by competing for binding by eIF4E [18]. As for a direct effect on translation and mRNA stability, a recent study has shown that presence of the 2′-O-methylation of the first transcribed nucleotide blocks the decapping and 5′–3′-exoribonuclease activity of DXO (a noncanonical decapping enzyme) on RNA substrates [22].
The m6Am modification is formed by methylation of an adenosine nucleotide that is adjacent to the m7G cap (denoted as “Am”) by a unique writer protein, phosphorylated CTD-interacting factor 1 (renamed as CAPAM—cap-specific adenosine N6-methyltransferase) [16, 23]. The reversibility of m6Am has been observed only very recently, and FTO (originally identified as a demethylase for internal m6A) has been identified as the demethylase that acts on m6Am in snRNAs [24]. Whether or not the m6Am modification is reversible in mRNAs is still unknown. Some studies have shown an increased half-life for an m6Am-containing mRNA versus mRNAs that do not contain this modification, which can be attributed to an increased resistance of the modified mRNA to the action of the DCP2 decapping enzyme [17]. The molecular mechanisms of these altered interactions of DCP2 with m6Am-containing mRNAs are not well understood and need to be explored further. A direct correlation between the presence of an m6Am cap and translation of the m6Am-containing mRNA is missing, even though knockout of CAPAM has shown decreased translation for a subset of mRNAs [16]. It has been reported that the presence of a methylated nucleotide adjacent to the 5′ cap alters the binding of eIF4E to the mRNA cap [25]. Whether or not a methylated nucleotide adjacent to the mRNA cap affects the binding of other components of the translation initiation complex or the cytoplasmic CBC to the mRNA is unclear; this information is needed to further our understanding of how this particular modification might differentially affect translation of a subset of mRNAs. Furthermore, it remains a possibility that understanding the interaction of the cytosolic CBC and its interaction with the m6Am cap could help us understand more about eraser proteins.
In addition to the classical m7G cap found at the 5′ ends of the majority of eukaryotic mRNAs, nicotinamide adenine dinucleotide (NAD+), NADH, and dpCoA caps have been identified in eukaryotic mRNAs [26, 27]. In contrast to the m7G cap, the NAD+ cap cannot support translation in human cells and destabilizes mRNAs through the deNADding activity of the DXO proteins [28]. The discovery of alternative types of cap structures and the elucidation of their functions are certainly exciting areas of research, and the development of tools for cap metabolism will be pivotal for our understanding of the biological consequences of modifications at the 5′ termini of mRNAs.
3.2 Modifications of mRNA 3′ Termini
Because of the impact of an RNA’s ends on the stability of the RNA , post-transcriptional modifications at the ends of mRNA are tightly controlled. 3′-end processing generates a mature 3′ terminus, which is typically polyadenylated by canonical poly(A) polymerases. The poly(A) tail is bound by poly(A)-binding proteins (PABPs), which facilitates mRNA export from the nucleus, enhances protein synthesis through interactions with translation initiation factors, and prevents exoribonucleolytic decay of the mRNA [3]. Shortening a poly(A) tail in the cytoplasm to fewer than 15–20 nt destabilizes its interaction with the PABP, and release of the last PABP makes the mRNA translationally inactive and susceptible to degradation either through the decapping and 5′-to-3′ decay pathway or by the 3′-to-5′ decay pathway [3]. Deadenylated mRNAs can also be uridylated with terminal uridylyltransferases (TUTases) or noncanonical poly(A) polymerases [29]. Uridylation of the 3′ end is diverse and has physiological consequences, illustrating functional diversity of mRNA 3′ modifications in controlling RNA stability and degradation. Our understanding of the complex regulatory networks of protein–protein and protein–RNA interactions involved in capping/decapping, polyadenylation/deadenylation, and uridylation is expanding and so is the knowledge of the modifications decorating the 5′ and 3′ extremities of an RNA.
4 Modifications in the Body of the mRNA
4.1 Inosine
Inosine is a widespread modification found in RNA and DNA, and it is formed by the hydrolytic deamination at the C6 position of adenosine. Originally discovered as a precursor of the purine synthesis pathway, inosine is introduced in RNA either nonenzymatically by spontaneous hydrolysis or enzymatically by deaminases [30]. Adenosine-to-inosine (A-to-I) editing sites have been mapped extensively, and most of the sites have been mapped to noncoding regions of transcripts such as introns or UTRs [31, 32]. However, more than 1000 sites have been located in the CDSs of transcripts. When present in RNA, inosine can base pair to A, C, or U, thus allowing inosine to function as a wobble base [32]. The presence of inosine in the RNA has the potential to affect multiple stages in the RNA life cycle because the cellular machinery (splicing and translation factors) recognizes inosines as a G. Not surprisingly, inosine has been demonstrated to alter mRNA stability, splicing, and translational recoding [33, 34]. A-to-I editing in the coding region of the mRNA results in translation recoding and amino acid misincorporation mediated in part by the destabilization of the codon-anticodon interactions [34]. It has been reported that even though inosine is primarily interpreted as guanosine, it can also be decoded, in a context-dependent way, as either adenosine or uracil. Ribosome profiling data have demonstrated inosine-dependent ribosome stalling (up to nine ribosomes) in vivo and premature peptide termination upstream of the inosine-containing codon. Recoding is rare, most likely because the consequences of recoding are dire, and therefore, it is not surprising that predominant A-to-I editing events occur in the UTRs of mRNAs.
4.2 N1-Methyladenosine (m1A)
Recent studies implementing transcriptome-wide analyses have suggested the widespread presence of m1A at internal sites of mRNAs with frequencies of 0.007–0.054% (modified/main base) in mammals [35,36,37]. m1A in the 5′ UTR of cytosolic mRNAs has been associated with increased translation efficiency. Additionally, m1A has been reported to be prevalent in the CDSs and 3′ UTRs of mitochondrion-encoded transcripts [35, 37]. Manipulation of m1A levels using the mitochondrion-localized m1A methyltransferase TRMT61B was found to result in alteration of translation, thus highlighting a link between m1A and translation regulation [38]. The presence of m1A can disrupt the U-A Watson–Crick base pairing, and the increased efficiency of translation initiation, early elongation, and enhanced translation rates could be a consequence of the altered base-pair interactions. A tRNA T-loop-like structure has been identified to have an increased number of m1A marks, which is dependent on the TRMT6/TRMT61A methyltransferase complex [37]. Even though the altered activity of the writer, reader, and eraser proteins of m1A has been implicated in certain diseases [39], the exact molecular function of m1A is not known, and whether or not there is any direct pathological outcome related to m1A status is unclear.
4.3 N7-Methylguanosine (m7G)
m7G is one of the most abundant modifications present in the mRNA 5′ cap, and it is also abundant in tRNAs and rRNAs. m7G sites have now been observed in internal sequences in mRNAs and ncRNAs, and mass spectrometry (MS)-based quantification analyses have revealed the presence of internal m7Gs in decapped mRNAs with a frequency of 0.02–0.05% (modified/main base) [40]. Internal m7G in mRNA has been mapped to single-nucleotide resolution, which has revealed enrichment of this modification in CDSs, 3′ UTRs, some 5′ UTRs, and AG-rich contexts (AGm7GA, GGm7GAA, AUCGm7GA, and UUm7GAU), a feature that has been observed to be well conserved across different cell lines and tissues [41, 42]. Knockdown of METTL1 (methyltransferase complex for m7G) expression has shown a decrease in the translation efficiency of a subset of transcripts with hypomethylated sites [41, 42]. The exact mechanism of how methylation affects translation is not understood, and future mechanistic studies are required to understand whether internal m7Gs also act as a platform for the binding of translation factors, as has been described for the m7G at the 5′ end of the mRNA. Methylation can affect the binding of protein partners and can also alter the stability, or structure, of its target RNAs via steric effects. Furthermore, dynamic regulation of the internal m7G modification has been reported with increased methylation of the m7G sites and subsequent increased translation from the m7G-containing mRNAs in response to hydrogen peroxide and heat-shock treatments. Internal METLL1-mediated m7G marks have been identified in miRNAs as well and N7 methylation of the guanosine can regulate the biogenesis and structure of the miRNA [41]. It will be interesting to test if any of the effects of m7G on translation is mediated via miRNAs.
4.4 5-Methylcytosine (m5C)
m5C is a prevalent base modification that is present in tRNAs and rRNA; it has also been mapped broadly in the transcriptome, including in mRNAs [43, 44]. Although the presence of m5C in mRNA was first observed in the 1970s, its complete transcriptome-wide mapping has been performed only recently. Initial MS-based transcriptome-wide m5C mapping has suggested high levels of m5C in mRNAs; however, recent work has suggested that the m5C frequency in the transcriptome might be overrepresented due to contamination with m5C from rRNA and tRNAs [45]. Bisulfite sequencing has mapped m5C sites in mRNAs to the 5′ UTR; however, a caveat to these findings is that incomplete conversion of cytidine and m5C during bisulfite treatment could have potentially affected the precise mapping of m5C [44, 46, 47]. Overall, 1000 m5C sites have been identified in the 5′ UTRs of mRNAs from human cell lines such as HeLa cells. Polysome profiling of HeLa cell lysates followed by bisulfite conversion and RNA sequencing has demonstrated the prevalence of m5C sites near the start codons in mRNAs and has also identified sequence contexts, similar to the tRNA variable loop, where m5C sites are present [48]. Furthermore, the levels of modification were inversely related to ribosome association, suggesting that the presence of m5C results in translation repression. When present in the CDS, m5C has been observed to be enriched in the first and second positions of a few specific codons, suggesting site-specific functions in translation regulation, which could be mediated by the reader proteins that might either perturb ribosome loading during translation initiation or represent an obstacle in the transit of elongating ribosomes [48]. m5C may also have the potential to affect decoding. The presence of m5C in the second position of the codon altered decoding in a bacterial in vitro translation system; however, whether or not m5C can have a same effect in vivo is not understood and will require more in-depth studies [49]. m5C regulation in mRNAs can be mediated by a specific set of writer, reader, and eraser proteins. An indirect effect of m5C on transcript stability can occur via the m5C reader protein YBX1 and its interaction with PABP [50]. Identification of hydroxymethylcytosine (hm5C), 5-formylcytosine, and 5-carboxylcytosine in RNA further highlights potential mechanisms to “erase” m5C from RNA.
4.5 3-Methylcytosine (m3C)
m3C was originally identified in tRNAs and plant mRNAs, but stringent purification of mRNA and quantification by LC-MS/MS have now shown that it also occurs in mammalian mRNA. Quantification has demonstrated that the abundance of m3C (0.0006–0.0038%, modified/main base) is similar to that of m5C and m1A but is much lower than that of m6A [2, 51]. How m3C affects an mRNA’s fate is still unclear. m3C has been found to be installed on mRNAs via METTL8 in murine and human cell lines [51]. Mettl8 KO mice had lower m3C levels in mRNAs, although considerable changes in the m3C levels in tRNAs were also observed [51]. Mettl8 knockout in mice also decreased the ratio of polysomes to monosomes, suggesting that Mettl8 might affect translation. Whether or not the reduced polysome status is a direct effect of the m3C levels in mRNAs needs to be investigated.
4.6 N4-Acetylcytidine (ac4C)
In HeLa cells, ac4C has been detected (0.11%, modified/main base) in more than 4000 regions of the human transcriptome, and these marks have been mapped predominantly in the CDSs, with ac4C content gradually decreasing from the 5′ to 3′ end of the gene transcript [52]. mRNAs enriched in ac4C have been observed to have a longer half-life, and the effect of ac4C on mRNA life cycle is further highlighted by the observation that ac4C was enriched in a wobble site C codon and that it significantly promoted translation efficiency in a luciferase reporter assay [52]. It has been hypothesized that the presence of ac4C in the wobble position promotes translation by facilitating tRNA decoding and that ac4C increases translation efficiency by enhancing the thermal stability of base-associated guanosine, thus altering the tRNA–mRNA interaction during translation. The cytidine acetyltransferase NAT10 has been determined to catalyze the formation of ac4C in a variety of mRNAs, because knocking out NAT10 reduces the level of ac4C on the RNA [52]. Analysis of ac4C position on the acetylated transcripts indicated that transcripts with ac4C sites in the CDS or 3′ UTR are preferentially downregulated by loss of NAT10. Furthermore, Ribo-seq analyses have demonstrated that acetylated mRNAs have increased translation efficiency compared to non-acetylated mRNAs, and loss of NAT10 decreases the translation efficiency of acetylated mRNAs [52]. It remains to be seen whether the ac4C mRNA modifications exist in other eukaryotes and/or whether this modification is seen in bacteria or archaea. No known reader proteins or a dedicated deacetylation process has been identified for ac4C mRNAs.
4.7 5-Methyluridine (m5U)
m5U is a common modification in the tRNAs of bacteria and eukaryotes, and its formation is catalyzed by TrmA in E. coli. A recent high-throughput sequencing approach with increased sensitivity led to the identification of m5U in mRNAs of various mammalian cells and tissues [53]. The measured contents of m5U in the mRNAs of various types of human cells and mouse tissues range from 0.001% to 0.0059% (m5U/U), comparable to that of m6Am and m3C in mRNAs of mammals. The potential role of m5U in mRNA remains elusive, but it is plausible that m5U in mRNA might affect interaction of the modified codon with the ribosome or lead to the alteration of RNA secondary structures and recruitment of m5U-specific binding proteins. Future work on transcriptome-wide mapping of m5U will further uncover the functional roles of m5U in the mRNAs of mammals.
4.8 2’-O-Methylation
While the existence of 2′-O-methyl modifications has been documented since the 1960s, much of the function of 2′-O-methyl modifications still remains unknown. 2′-O-methyl methylation in CDSs induces ribosome stalling by disrupting codon reading [54]. Moreover, knockdown of the methyltransferase fibrillarin was found to result in a decrease in ribosome 2′-O-methylation and a corresponding increase in translational infidelity [55]. 2′-O-methylation also modulates translation of internal ribosome entry site-containing mRNAs [55]. Whether or not this is mediated by the rRNA modification alterations or alteration of distinct IRES-containing mRNA modification is unclear. Apart from a direct function in translation , self- and non-self-identification of mRNAs is dependent on the presence or absence, respectively, of 2′-O-methyl marks in humans [21].
4.9 8-Oxo-7, 8-Dihydroguanosine (8-Oxo-G)
Modifications are introduced into RNAs not just by nucleotide-specific modifying enzymes but also in response to environmental stress conditions (e.g., oxidative stress, heat shock, and UV irradiation), which can cause chemical damage [56]. Reactive oxygen species can oxidize RNA bases and generate 8-oxo-G, 8-dihydroadenosine, 5-hydroxyuridine, 5-hydroxycytidine, and cytosine glycol. 8-Oxo-G, the most abundant form of the oxidized base, has been detected in purified mRNA. Incorporation of 8-oxo-G in luciferase mRNAs leads to the presence of truncated proteins, suggesting that 8-oxo-G can alter decoding [57]. The presence of 8-oxo-G in the CDS has also been reported to alter peptide bond formation and subsequent triggering of an mRNA surveillance pathway, no-go decay (NGD), that targets stalled ribosomes during translation elongation [57]. 8-Oxo-G-containing mRNAs have been reported to be degraded with the involvement of the reader proteins YBX1, AUF1 (also known as hnRNP D), and PCBP1 [58, 59]. The exact mechanism by which the reader proteins trigger the degradation of 8-oxo-G-containing mRNAs, and the interactions that lead to decay factor recruitment, is not well understood. Whether or not 8-oxo-G is reversible or whether this modification preferentially forms in specific mRNAs is not known.
4.10 5-Hydroxymethylcytosine (hm5C)
hm5C can be generated in RNA by the oxidization of m5C via deoxygenases of the ten-eleven translocation (Tet) family. Transcriptome-wide approaches to map hm5C (hydroxymethylated RNA immunoprecipitation followed by sequencing; hMeRIP-seq) demonstrated enrichment of hm5C sites in the CDS [60]. It has been postulated that hm5C acts as an eraser of m5C and can restore mRNA translation in mRNAs whose translation is otherwise inhibited by the presence of m5C [61]. Furthermore, m5C and hm5C are thought to affect mRNA secondary structure and thereby regulate target mRNA recognition by RNA-binding proteins . The relative abundance of hm5C in mRNAs is debated because hm5C has not been detected in highly purified mRNA preparations.
5 Pseudouridine in mRNAs
Pseudouridine , one of the first post-transcriptional RNA modifications discovered, is now recognized as one of the most abundant types of RNA modifications. Although Ψ was first discovered in rRNAs, tRNAs, and snRNAs, recent transcriptome-wide analysis of Ψ profiles in humans and yeast has now revealed that hundreds of mRNAs contain this modification [2, 62,63,64,65]. The ratio of Ψ/uridine in mammalian mRNA, as measured by LCMS/MS, has been observed to be about 0.2–0.6%, a frequency comparable to that of m6A [66]. Pseudouridine is generated by the C–C glycosidic isomerization of a uridine base, and there are two distinct mechanisms that have been identified for conversion of uridine into Ψ in vivo [67, 68]. The first is an RNA-independent mechanism in which Ψ is deposited by various Ψ synthases (PUSs) that have different substrate specificities and subcellular localizations; the second involves an RNA-dependent mechanism that is guided and catalyzed by Box H/ACA small nucleolar RNAs (snoRNAs). The enzymatic cores of the PUS enzymes are conserved, and substrate RNA recognition is mediated via the various protein domains [69]. However, the Ψ synthase NAP57/DKC1, also known as dyskerin, uses H/ACA snoRNAs as guides to position a uridine at the active site of the enzyme, which then installs the Ψ marks in rRNAs and snRNAs [70]. Interestingly, studies have shown that knocking down the expression of individual PUSs does not drastically alter Ψ levels in mRNA, suggesting that there is functional redundancy among the PUS proteins. The mechanism by which PUS proteins regulate pseudouridylation in specific mRNAs is an intriguing and outstanding question in the field. Recent work combining in silico RNA-folding predictions with systematic substrate mutagenesis and quantitative kinetic analyses has provided insights into the structural features in mRNA that might help determine pseudouridylation by yeast PUS1 and has demonstrated that, in vitro, PUS1 can recognize substrates in a structure-dependent manner [71]. These studies suggest that PUS1-mediated pseudouridylation in vivo requires the substrates to be structured; however, because mRNA structures are likely to be dynamic in vivo, the presence of cellular RNA-binding proteins probably influences the pseudouridylation process. By extension, the findings also indicate that modulation of an mRNA’s structure might be a critical checkpoint for the Ψ-mediated regulation of gene expression; additional studies to understand mRNA structural dynamics in vivo and their role in the process of pseudouridylation will help shed more light on this aspect. In humans, DKC1 has been reported to catalyze the isomerization of a subset of uridines in mRNAs [63].
The chemical properties of Ψ differ from those of uridine, and Ψ has an altered strength of base-pairing with adenine because the phosphodiester backbone is more rigid. The presence of Ψ55 in tRNA has been proposed to stabilize the tertiary structure of tRNAs, particularly under extreme thermal stress. The Ψ modification has also been observed to be in locations crucial for intermolecular RNA–RNA and RNA–protein interactions such as the peptidyl transferase center (for peptide bond formation), in tRNAs and rRNAs (to provide structural stabilization), and in major and minor spliceosomal snRNA complexes. In mRNAs, Ψ has not been found to have any positional bias and has been detected in 5′ UTRs, CDSs, and 3′ UTRs [62]. Because Ψ can alter base-pairing interactions, it is plausible that Ψ could directly or indirectly influence mRNA stability and translation (Fig. 3). Early evidence for this hypothesis came from in vitro studies showing that targeted conversion of U to Ψ in translation termination codons (UAA, UGA, and UAG) converted these stop codons into missense codons, and U-to-Ψ conversion of premature termination codons inhibited the accelerated degradation of these mRNAs triggered by the process of nonsense-mediated mRNA decay (NMD) [72]. Multiple lines of evidence have demonstrated a role for Ψ in maintaining mRNA stability, although the effects have been dependent on the system in which this was evaluated. In vitro-transcribed mRNAs containing Ψ have been observed to have higher stability than unmodified mRNAs in cell-based assays. Deletion of Pus7 in yeast resulted in reduction of Ψ-containing mRNAs, suggesting that the presence of Ψ can stabilize mRNAs [63]. Contradictory to these observations, in Toxoplasma gondii it has been reported that PUS1-targeted mRNAs have an increased half-life in PUS1 mutant [73]. These examples highlight that Ψ can have a multifold effect on mRNA structure and function, and a better understanding of Ψ’s effect on mRNA stability will require a greater comprehension of how the other coupled mRNA processes, especially translation, are affected by the presence of Ψ. There is a growing body of evidence showing that Ψ can affect both the rate and fidelity of translation. Structural studies of the 70S ribosome with ΨUU in the A site and tRNAPhe bound to the ribosome have demonstrated that the 3′-CCA end of tRNAPhe is not properly positioned in the peptidyl-transferase center in the presence of Ψ despite correct mRNA: tRNA base-pairing interactions [74]. Kinetic data have further revealed that replacing a single uridine with Ψ impedes amino acid incorporation in vitro; this misincorporation of amino acids in presence of Ψ has been recapitulated in vivo using cell-based assays where complete substitution of U with Ψ in synthetic mRNA showed misincorporation of amino acids, albeit at low frequencies. Taken together, these studies indicate that Ψ-containing codons can slow the elongating ribosome. The magnitude by which the presence of Ψ in an mRNA can affect translation will likely depend on several parameters, including sequence context and mRNA secondary structures . The sequence context of the Ψ-containing codon can affect the interaction between the ribosome and the codon, as has been observed for inosine-containing codons. Translation of the Ψ-containing mRNAs can also be affected by the indirect effect of Ψ on mRNA secondary structure, and RNA secondary structure mapping studies (e.g., using DMS or SHAPE reagents) comparing mRNAs with modified or unmodified nucleotides have now provided additional insights into how mRNA structure affects ribosome transit time. Complete substitution of uridines in an mRNA with N1-methyl-pseudouridine (m1Ψ) resulted in m1Ψ-containing mRNAs that have a higher translation efficiency that could, in part, be attributed to a highly structured CDS that extended the half-life of the mRNA [75]. Additionally, increased ribosome occupancy on m1Ψ-containing mRNAs can further facilitate translation initiation or ribosome recycling, resulting in enhanced overall translation from the mRNA [76]. The effect of modifications on the translation can thus be imparted by multiple mechanisms.

Pseudouridine affects mRNA in multiple ways. (a) The presence of Ψ provides rigidity to the mRNA backbone and can alter RNA–protein interactions. (b) Secondary structures in the mRNA can be stabilized in the presence of Ψ. (c) Presence of Ψ in a sense codon can lead to translation recoding, and in a nonsense codon can lead to nonsense suppression
Identifying the consequences of Ψ in mRNAs is complicated in cells because the enzymes that incorporate Ψ into mRNAs also catalyze Ψ addition in ncRNA species. Furthermore, the impact of Ψ on mRNA stability can be difficult to deconvolute from its effects on translation in cells. It is also unknown whether readers for Ψ exist or whether Ψ can be reverted back to U in vivo; the identification of Ψ reader and eraser proteins is needed to further our functional understanding of Ψ marks in mRNA. Similar to the other modifications discussed in this review, Ψ has been implicated in various human diseases, although a direct association of the modification with the disease state has not been elucidated.
6 Applications of RNA Modifications
As has been discussed, chemical tags in RNAs impart an additional level of gene expression regulation, and modified nucleotides (or modified RNAs, per se) are being explored for a number of different applications, including extending their use to antisense oligonucleotides and miRNA derivatives to enhance the therapeutic potential of these RNA species. Because chemical modifications have the potential to alter RNA–RNA and RNA–protein interactions, chemically modified synthetic RNA-based biosensors are being evaluated for improved interactions between the modified RNA and the target molecule. Modified ribonucleosides or their chemical analogs (e.g., ribavirin, acyclovir, and valacyclovir) are being used as antiviral agents or as cancer therapies. Although the mechanism of action of these analogs varies, they typically mimic the canonical nucleosides in terms of uptake and incorporation into the host nucleic acids, which results in chain termination of the newly synthesized DNA, leading to incomplete replication and viral or cell death.
6.1 Synthetic mRNA Vaccines
A prominent application of chemical modifications in RNA is in synthetic mRNA-based therapeutics. mRNA-based therapeutics is based on the idea that a synthetic mRNA can be delivered in vivo and the mRNA can utilize the cellular machinery to generate the protein of interest. The field of RNA-based therapeutics is emerging as a promising new alternative to conventional protein-based vaccine approaches [77, 78]. Use of mRNA as a therapeutic modality is advantageous because the molecule is noninfectious and the mRNA does not get integrated into the genome. Furthermore, the mRNA’s half-life can be regulated by the cellular machineries, thus making expression from the mRNA transient and amenable to regulation. In addition, the manufacturing process to produce an mRNA vaccine is rapid, inexpensive, and scalable, making it a lucrative platform. Even though the first report for the successful in vivo use of synthetic mRNA was published in the early 1990s, the idea failed to gain traction until more recently, mainly due to concerns associated with mRNA instability in vivo, low translation efficiency, immunogenicity concerns, and inefficient in vivo delivery. The pharmacokinetic (PK) and pharmacodynamic (PD) properties of mRNA-based therapeutics are affected by the half-life of the synthetic mRNA, which subsequently affects the efficacy of the drug. Several approaches have been used to increase the efficiency of protein synthesis from a synthetic mRNA, including optimization of the 5′ UTR sequences to minimize secondary structures that could impair ribosome recruitment or transit, removal of alternate initiation codons in the 5′ leader to maximize translation initiation from the main CDS, addition of stabilizer elements in the 3′ UTR, elimination of miRNA-binding sites from the 3′ UTR, and optimization of poly-A tail length and codon sequences (which affect translation efficiency and mRNA stability) [79,80,81,82]. Over the past decade, technological innovations together with an increased understanding of the interconnected mRNA processes have enabled advancement of synthetic mRNAs for therapeutics and vaccines—use of chemically modified mRNAs for therapeutics being at the forefront to address the issues of mRNA instability in vivo and immunogenicity of synthetic mRNAs [83].
6.2 Modifications of the mRNA Termini
As discussed in previous sections, the mRNA 5′ cap structure not only promotes translation via its interaction with the translation initiation factor 4E (EIF4E), but also modulates mRNA decay via interactions with the DCP1/DCP2 complex (Fig. 1) [3]. Synthetic mRNAs delivered in vivo interact with the same cytosolic proteins as an endogenous mRNA, and therefore, the interaction of the 5′ cap with either the translation or the decay machinery is pivotal in determining the therapeutic efficacy of a synthetic mRNA molecule. An added level of complication arises from the fact that a synthetic mRNA can also encounter cytosolic immune receptors that recognize viral RNAs [19,20,21]. Translation efficiency and immune evasion can be enhanced by the use of cap analogs during mRNA synthesis that have chemical modifications, and several mimics of the 5′ cap have been evaluated for these purposes [84,85,86,87,88]. The most common cap analogs used are the anti-reverse cap analogs (ARCAs), which have modifications within the ribose moiety of the m7G cap [87]. ARCA possesses a 3′-O-methyl group on the sugar adjacent to the m7G, which prevents incorrect cap incorporation during the mRNA synthesis process, resulting in a higher percentage of capped mRNA species and an increased population of mRNAs that can be translated in vivo. A single phosphorothioate (O-to-S) substitution in the triphosphate bridge of the ARCA cap analog has been reported to increase the affinity of this cap structure for EIF4E, thereby reducing an mRNA’s susceptibility to DCP1/DCP2-mediated decapping and subsequent decay [86]. Because the presence of a 2′-O-methylation modification in Cap 1 prevents IFIT-1-induced translation inhibition, cap analogs that confer such structures are also routinely used to reduce binding to IFIT-1 and to subsequently prevent activation of an immune response. Systematic analyses of methylation status of the first nucleotide using cap analogs have further demonstrated that the identity of the first nucleotide and its methylation status in an mRNA influences the expression in a cell-type-specific manner [25]. Whether or not this increased translation is due to a direct effect on the binding of eIF4E or an indirect effect of reduced IFIT-1 binding is not clear.
Modifications at the 3′ end of the mRNA also affect mRNA translation and stability (Fig. 1), and poly(A) tails of optimum length are added to synthetic mRNAs for efficient translation. For certain synthetic mRNAs, the combination of a cap modification with a poly(A) tail length of 100 residues has been shown to increase translation in dendritic cells. The correlation between poly(A) tail length and translation is not clear, but in naturally occurring mRNAs, regulation of poly(A) tail length is critical for maintaining specific biological behaviors. During eukaryotic mRNA degradation, deadenylation by 3′-exonucleases such as the poly(A)-specific ribonuclease (PARN) selectively deadenylates single-stranded mRNA via recognition of the 2′,3′-adenosine diol intermediate [89]. Chemical modification of the end 2′,3′-diol group in a synthetic Luciferase mRNA resulted in a threefold increase in protein synthesis due to an increase in transcript stability [90]. These findings suggest that chemical modification of the mRNA’s 3′ end might interfere with the cellular interactions that lead to decay. Even though the mRNA half-life can be extended, whether or not these modifications alter the stable RNP formation or affect translation indirectly needs to be investigated.
6.3 Internal Modifications of the Synthetic mRNA
In vivo studies of synthetic mRNAs modified with Ψ have demonstrated increased translational rates and protein expression compared with mRNAs lacking Ψ [82, 91,92,93,94,95]. For example, injection of Ψ-modified erythropoietin (EPO) mRNAs into mice led to 10- to 100-fold higher levels of expression from the modified mRNAs than from unmodified mRNAs, making this modification very attractive for the purposes of mRNA therapeutics. However, the effects of Ψ on protein expression have been quite varied, and translation enhancement was not observed in other systems. The discrepancies in the effects of Ψ on expression can be explained by differences in the sequences tested and differences in the innate immune responses in the target cells. In vitro, Ψ has been shown to repress translation when introduced at all three nucleotide positions in a codon. m1Ψ has been shown to enhance translation by increasing ribosome pausing, and consequently ribosome density, on synthetic mRNAs [76]. A correlation between protein expression and structure of the mRNA has also been demonstrated for synthetic mRNAs modified with m1Ψ, with the modified mRNAs having an increased functional half-life [75]. An mRNA with a combination of Ψ, m5U, and m1Ψ is currently being evaluated in preclinical research. The effect of Ψ on translation could also be indirect and mediated by interference with global protein synthesis shutdown during a cellular immune response. Uridine-containing mRNAs have been reported to activate protein kinase R (PKR)-mediated phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α), thus leading to global downregulation of protein synthesis . In contrast, reduced phosphorylation of eIF2α has been observed with Ψ-containing mRNAs, suggesting that the increased translation is imparted by reducing the activation of PKR [96]. Reduced activation of 2′–5′-oligoadenylate synthetase (OAS) by Ψ-containing mRNAs and reduced 2′–5′-oligoadenylate-mediated activation of RNase-L have also been observed. Increased resistance of the Ψ-containing mRNAs to RNase-L-mediated degradation further results in increased stability of the mRNA and, subsequently, in increased protein synthesis. Another modification that is currently being evaluated for its therapeutic potential is m5C, because earlier studies found that m5C incorporated in synthetic mRNAs stimulated expression from these transcripts in vivo.
Immunostimulatory effects of synthetic mRNAs are imparted by two different structural elements—the cap structure (as discussed in the previous section) as well as the nucleotide content of the synthetic RNA. Synthetic RNA is recognized in nonimmune cells through the RIG-I receptor and in immune cells through the Toll-like receptors (TLRs). It has been observed that the presence of U-rich sequences in the RNA is a trigger for TLRs, and thus reduction of the U-content of the mRNA, as well as the substitution of uridine with modified uridine in the mRNA, is a strategy that has been implemented to evade recognition by the TLRs. Replacement of uridine with ψ, m1Ψ, m5U, 2-thiouridine (s2U), or 5-methoxyuridine (5moU), and replacing the natural cytidine with m5C, has demonstrated a reduction in the immune responses to synthetic mRNAs. It has also been hypothesized that the effect of uridine can be counterbalanced by altering the poly(A) tail to generate mRNAs with low immunogenicity because the U content decreases or is shielded in the sequence.
In light of these findings, chemical modifications of mRNA are applied to not only modulate translation efficiency but also alter mRNA recognition by the innate immune system. Understanding these mechanisms underlying how modifications affect interrelated mRNA processes can aid in the rationalized design of efficacious synthetic mRNA molecules. Modifications can be introduced to affect the mRNA secondary structure without affecting the codon when required, and rationalized incorporation of the modifications can be done when minimal secondary structure changes are desirable. In addition, modifications can also be introduced site-specifically to affect immunogenicity without affecting decoding. In a cellular context, destabilization of the local secondary structure (as seen with m6A) alters the accessibility of RNA-binding proteins to specific sequence elements; the same can be achieved for synthetic mRNAs. Decoupling structural stability from primary sequence changes can be instrumental in altering the expression from the synthetic mRNA.
7 Approaches to Understand the Effects of mRNA Modifications on Translation
As we continue to identify modifications in mRNAs, the next question that needs to be addressed is how these modifications affect the mRNA life cycle. The effects of modifications on the mRNA life cycle are complicated to tease out in a biological context, i.e., in vivo, because the modifications are installed not only in the mRNA but also in other noncoding mRNAs by the same set of writer proteins. Therefore, cell-based assays using synthetic modified mRNAs are routinely implemented to evaluate the effect of mRNA modifications. Protein expression from the transfected mRNAs in cell-based assays is measured a few hours after transfection, making it difficult to study the direct effects of the modifications; also, the kinetics of protein synthesis are difficult to analyze. Transfection of synthetic mRNAs also results in degradation of a significant proportion of the mRNAs in endosomes, and the final readout does not reflect the actual efficiency of protein expression. Differences in delivery modes, cellular uptake, and endosomal targeting can affect the mRNA pool for translation. Furthermore, deconvoluting the effects of mRNA modifications on translation versus their effects on mRNA stability is not trivial. Because chemical modifications of mRNAs have been shown to affect mRNA translation and stability, it is critical that cell-based assays be combined with in vitro assays that can help tease out the function of the mRNA modification.
7.1 mRNA Termini Modifications
The discovery of diverse modifications in endogenous mRNA caps has provided versatility to mRNA function, and because the cap structure helps distinguish endogenous mRNAs from viral RNAs, the 5′ termini of synthetic mRNAs are routinely modified to recapitulate these endogenous structures [14, 15]. The biological significance of the 5′ cap variations is routinely evaluated using in vitro-synthesized RNA in either cell-based or biochemical assays to understand the effects of the modifications in enhancing translation and evading immune responses [25, 88, 97, 98]. There are some considerations that should be taken into account when utilizing cell-based assays in combination with synthetic modified mRNAs. Current methods involve using cap analogs during in vitro transcription to introduce the cap modifications at the 5′ end of the mRNA. However, the incorporation of the cap analog is not always efficient, and the resulting RNA population can have a mix of capped and uncapped RNAs [86, 87]. Furthermore, the purity of the in vitro-transcribed RNA can also affect the immune response generated against the mRNA preparation, which can subsequently affect its translation efficiency [99, 100]. There are multiple parameters that can play a role in expression from an exogenously delivered mRNA. Interaction with cellular immune receptors (RIG-I, MDA5, IFIT-1) is dependent on the nature and the modification status of the 5′ cap and the presence of impurities in the synthetic mRNA can also activate PKR and 2′-5′ OAS [96]. PKR-mediated phosphorylation of eIF2α represses translation initiation and 2′-5′ OAS-mediated activation RNase-L can lead to degradation of cellular RNAs and a subsequent shutdown of global protein biosynthesis [96]. Teasing out the direct effects of the 5′ cap modification in translation (via binding to the cytoplasmic CBC, specifically, eIF4E) versus the indirect effects due to global protein synthesis shutdown is critical. Not choosing the right cell type for cell-based assays can also bias the results; in a recent study evaluating the effect of modifications in the first transcribed nucleotide in the RNA, it was observed that the effect of the methylation status of the nucleotide after the cap is strongly dependent on the type of cultured cells [25].
Biochemical assays to determine the binding kinetics allow determination of apparent KD; however, they do not give insights into the kinetics of IFIT1–RNA interactions. Biophysical assays allowing direct mRNA–protein binding have also been used in combination with cell-based assays. Binding of the 5′ ends of mRNA with either the cap-binding protein eIF4E, the cytosolic receptors (IFIT1), or the decapping enzyme (DcpS) has been evaluated to determine the kinetics of these interactions and deduce a role of the 5′ cap modification [25, 97].
7.2 Internal Modifications
Cell-free in vitro translation systems , such as those derived from Krebs cells, HeLa cells, and rabbit reticulocyte lysates, have been used to understand the effects of RNA modifications on translation and to recapitulate the increase in protein expression seen using the modified mRNAs in cell-based assays [34, 74, 76, 83, 93,94,95,96]. There are discrepancies in the studies using synthetic modified mRNAs, and an emerging body of evidence is making clear that the translation systems and the cell types used for evaluating the effects of modifications can have an impact on the final outcome. The effect of m5C on translation yield has been found to be quite different when tested in rabbit reticulocyte extracts, wheat germ extracts, or bacterial translation systems [49, 76, 101]. Discrepancies may also arise because of differences in sequence contexts of the modifications in a synthetic mRNA. A concerted evaluation of multiple mRNAs—including mRNAs of varied length, structure, and sequence—would be required to get a comprehensive idea of the effects of modifications in vitro. Current cell-based or in vitro assays have utilized approaches where the entire body of the mRNA has been modified with a specific analog, which does not reflect the modification status of a naturally occurring mRNA, and it should not be surprising that the consequences of modifying the entire mRNA will be quite distinct from the consequences of introducing a modification at a single position in a codon. In order to circumvent the unnatural representation of modifications in these synthetic mRNAs, systematic modification of the first, second, or third position of the codon has been performed, and the peptide products of the corresponding mRNAs have been analyzed [101]. Site-specific introduction of modifications in the CDS of the mRNA followed by translation in HEK293 cells revealed a strong position-dependent effect of modifications on translation efficiency and accuracy. Whereas single m5C or Ψ modifications did not have any effect on product formation, m1A impeded translation, and a codon-position-dependent effect on translation was observed for 2′-O-methylated nucleotides and m6A. Miscoding in the presence of modification can be measured either in vitro or using cell-based assays. Eukaryotic translation is highly accurate, and miscoding in the presence of modifications seems unlikely. However, in a recent study, in vitro biochemical and structural studies were combined together with cell-based assays to demonstrate miscoding in the presence of Ψ (Fig. 4, [101]). Misincorporation in the presence of m5C has also been reported in bacterial systems [49]. However, in vivo experiments in human cells have yielded conflicting results for miscoding that can be attributed to the sensitivity of the methods implemented, as well as to the sequence contexts of the codons.

Experimental procedure to test translation recoding from modified mRNAs (containing pseudouridine) in vivo. The modification is incorporated during in vitro transcription, and the mRNA is transfected into HEK293 cells. After optimal expression, the protein is purified using an N-terminal hemagglutinin (HA) and a C-terminal FLAG tag, ensuring enrichment of full-length protein (normal and recoded) and not prematurely terminated products. The purified protein is then subjected to digestion and mass spectrometry (MS) analyses to identify amino acid misincorporation
Understanding how mRNA modifications impact translation is critical, but it is also important to gain quantitative insights into how mRNA–protein interactions affect the mRNA life cycle. Alteration of protein–mRNA interactions has been investigated for m6A using a combination of immunoprecipitation and pull-down assays, mass spectrometry, and RNA-seq approaches. m6A can both weaken and enhance RNA–protein interactions. The YTH family of m6A reader proteins differentially interact with methylated and unmethylated transcripts, a discrimination more prominent than for other RNA-binding proteins [102]. Unlike m6A, reader proteins for other mRNA modifications are not known yet, and the plausible interactions between the translation and mRNA decay machineries have not yet been mapped.
8 Future Perspectives
The identification of modifications in mRNA has unveiled an uncharted layer of regulation of RNA stability and translation. The study of mRNA modifications is bound to grow rapidly as researchers seek to understand the influence of these modifications on human health and biology. Advancements in analytical chemistry and deep sequencing-based methods have furthered our understanding of RNA modifications. Future work combining the quantitative transcriptome-wide mapping of modifications with in-depth characterization of the writer, reader, and eraser proteins using structural biology and in vitro biochemistry will be pivotal in getting a comprehensive landscape of mRNA modifications and their function in the regulation of gene expression. Structural and biochemical analyses to unveil modification-induced changes in mRNAs, and the factors driving the selection of modification sites and the stoichiometry of the modifications, are all outstanding questions in the field, and their answers will enable a better understanding of how cellular machineries encounter the modifications and the consequences of such an event. Most of the current studies have been performed using single types of modifications, and the combinatorial effect of different types of modifications when present in the same mRNA will be of interest. How mRNA modifications contribute to a specific disease state can only be addressed when a comprehensive understanding of the molecular mechanisms is available. A better understanding of the correlation between mRNA modifications and the regulation of factors involved in the dynamic control of these modifications will help screening of inhibitors of mRNA modification regulators that will open up new therapeutic avenues. It is not surprising that new mechanisms of mRNA modification will be discovered as the analyses of modifications are refined and technological advances are made.
References
McCown PJ, Ruszkowska A, Kunkler CN, Breger K, Hulewicz JP, Wang MC, Springer NA, Brown JA (2020) Naturally occurring modified ribonucleosides. Wiley Interdiscip Rev RNA:e1595. https://doi.org/10.1002/wrna.1595
Jones JD, Monroe J, Koutmou KS (2020) A molecular-level perspective on the frequency, distribution, and consequences of messenger RNA modifications. Wiley Interdiscip Rev RNA 11(4):e1586. https://doi.org/10.1002/wrna.1586
Roy B, Jacobson A (2013) The intimate relationships of mRNA decay and translation. Trends Genet 29(12):691–699. https://doi.org/10.1016/j.tig.2013.09.002
Bicknell AA, Ricci EP (2017) When mRNA translation meets decay. Biochem Soc Trans 45(2):339–351. https://doi.org/10.1042/BST20160243
Hawley BR, Jaffrey SR (2019) Transcriptome-wide mapping of m(6) A and m(6) am at single-nucleotide resolution using miCLIP. Curr Protoc Mol Biol 126(1):e88. https://doi.org/10.1002/cpmb.88
Zaccara S, Ries RJ, Jaffrey SR (2019) Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20(10):608–624. https://doi.org/10.1038/s41580-019-0168-5
Shi H, Wei J, He C (2019) Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 74(4):640–650. https://doi.org/10.1016/j.molcel.2019.04.025
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR (2015) Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12(8):767–772. https://doi.org/10.1038/nmeth.3453
McIntyre ABR, Gokhale NS, Cerchietti L, Jaffrey SR, Horner SM, Mason CE (2020) Limits in the detection of m(6)A changes using MeRIP/m(6)A-seq. Sci Rep 10(1):6590. https://doi.org/10.1038/s41598-020-63355-3
Wu J, Frazier K, Zhang J, Gan Z, Wang T, Zhong X (2020) Emerging role of m(6) A RNA methylation in nutritional physiology and metabolism. Obes Rev 21(1):e12942. https://doi.org/10.1111/obr.12942
Paramasivam A, Vijayashree Priyadharsini J (2020) Novel insights into m6A modification in circular RNA and implications for immunity. Cell Mol Immunol 17(6):668–669. https://doi.org/10.1038/s41423-020-0387-x
Chen M, Wong CM (2020) The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis. Mol Cancer 19(1):44. https://doi.org/10.1186/s12943-020-01172-y
Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, Schurer A, Gourkanti S, Bruning JC, Vu LP, Jaffrey SR, Landau DA, Kharas MG (2019) m(6)A RNA methylation maintains hematopoietic stem cell identity and symmetric commitment. Cell Rep 28(7):1703–1716. e1706. https://doi.org/10.1016/j.celrep.2019.07.032
Ramanathan A, Robb GB, Chan SH (2016) mRNA capping: biological functions and applications. Nucleic Acids Res 44(16):7511–7526. https://doi.org/10.1093/nar/gkw551
Padgett RA (2019) New roles for old RNA cap flavors. Nat Chem Biol 15(4):317–318. https://doi.org/10.1038/s41589-019-0252-3
Akichika S, Hirano S, Shichino Y, Suzuki T, Nishimasu H, Ishitani R, Sugita A, Hirose Y, Iwasaki S, Nureki O, Suzuki T (2019) Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science 363(6423):eaav0080. https://doi.org/10.1126/science.aav0080
Sun H, Zhang M, Li K, Bai D, Yi C (2019) Cap-specific, terminal N(6)-methylation by a mammalian m(6)Am methyltransferase. Cell Res 29(1):80–82. https://doi.org/10.1038/s41422-018-0117-4
Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, Mann A, Kindler E, Gil-Cruz C, Ziebuhr J, Thiel V, Pichlmair A (2013) Sequestration by IFIT1 impairs translation of 2’O-unmethylated capped RNA. PLoS Pathog 9(10):e1003663. https://doi.org/10.1371/journal.ppat.1003663
Xu XX, Wan H, Nie L, Shao T, Xiang LX, Shao JZ (2018) RIG-I: a multifunctional protein beyond a pattern recognition receptor. Protein Cell 9(3):246–253. https://doi.org/10.1007/s13238-017-0431-5
Lassig C, Hopfner KP (2017) Discrimination of cytosolic self and non-self RNA by RIG-I-like receptors. J Biol Chem 292(22):9000–9009. https://doi.org/10.1074/jbc.R117.788398
Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F, Khan AG, Patel SS, Marcotrigiano J (2016) Structural basis for m7G recognition and 2’-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc Natl Acad Sci U S A 113(3):596–601. https://doi.org/10.1073/pnas.1515152113
Picard-Jean F, Brand C, Tremblay-Letourneau M, Allaire A, Beaudoin MC, Boudreault S, Duval C, Rainville-Sirois J, Robert F, Pelletier J, Geiss BJ, Bisaillon M (2018) 2’-O-methylation of the mRNA cap protects RNAs from decapping and degradation by DXO. PLoS One 13(3):e0193804. https://doi.org/10.1371/journal.pone.0193804
Cowling VH (2019) CAPAM: the mRNA cap adenosine N6-methyltransferase. Trends Biochem Sci 44(3):183–185. https://doi.org/10.1016/j.tibs.2019.01.002
Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, Rentmeister A, Gross SS, Pellizzoni L, Debart F, Goodarzi H, Jaffrey SR (2019) FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol 15(4):340–347. https://doi.org/10.1038/s41589-019-0231-8
Sikorski PJ, Warminski M, Kubacka D, Ratajczak T, Nowis D, Kowalska J, Jemielity J (2020) The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5′ cap modulates protein expression in living cells. Nucleic Acids Res 48(4):1607–1626. https://doi.org/10.1093/nar/gkaa032
Grudzien-Nogalska E, Bird JG, Nickels BE, Kiledjian M (2018) “NAD-capQ” detection and quantitation of NAD caps. RNA 24(10):1418–1425. https://doi.org/10.1261/rna.067686.118
Wang J, Alvin Chew BL, Lai Y, Dong H, Xu L, Balamkundu S, Cai WM, Cui L, Liu CF, Fu XY, Lin Z, Shi PY, Lu TK, Luo D, Jaffrey SR, Dedon PC (2019) Quantifying the RNA cap epitranscriptome reveals novel caps in cellular and viral RNA. Nucleic Acids Res 47(20):e130. https://doi.org/10.1093/nar/gkz751
Jiao X, Doamekpor SK, Bird JG, Nickels BE, Tong L, Hart RP, Kiledjian M (2017) 5′ end nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO-mediated deNADding. Cell 168(6):1015–1027. e1010. https://doi.org/10.1016/j.cell.2017.02.019
Menezes MR, Balzeau J, Hagan JP (2018) 3’ RNA uridylation in epitranscriptomics, gene regulation, and disease. Front Mol Biosci 5:61. https://doi.org/10.3389/fmolb.2018.00061
Zinshteyn B, Nishikura K (2009) Adenosine-to-inosine RNA editing. Wiley Interdiscip Rev Syst Biol Med 1(2):202–209. https://doi.org/10.1002/wsbm.10
Roth SH, Levanon EY, Eisenberg E (2019) Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat Methods 16(11):1131–1138. https://doi.org/10.1038/s41592-019-0610-9
Brachova P, Alvarez NS, Hong X, Gunewardena S, Vincent KA, Latham KE, Christenson LK (2019) Inosine RNA modifications are enriched at the codon wobble position in mouse oocytes and eggs. Biol Reprod 101(5):938–949. https://doi.org/10.1093/biolre/ioz130
Licht K, Hartl M, Amman F, Anrather D, Janisiw MP, Jantsch MF (2019) Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res 47(1):3–14. https://doi.org/10.1093/nar/gky1163
Hoernes TP, Faserl K, Juen MA, Kremser J, Gasser C, Fuchs E, Shi X, Siewert A, Lindner H, Kreutz C, Micura R, Joseph S, Hobartner C, Westhof E, Huttenhofer A, Erlacher MD (2018) Translation of non-standard codon nucleotides reveals minimal requirements for codon-anticodon interactions. Nat Commun 9(1):4865. https://doi.org/10.1038/s41467-018-07321-8
Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar-Yaacov D, Erlacher M, Rossmanith W, Stern-Ginossar N, Schwartz S (2017) The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 551(7679):251–255. https://doi.org/10.1038/nature24456
Khoddami V, Yerra A, Mosbruger TL, Fleming AM, Burrows CJ, Cairns BR (2019) Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc Natl Acad Sci U S A 116(14):6784–6789. https://doi.org/10.1073/pnas.1817334116
Schwartz S (2018) m(1)A within cytoplasmic mRNAs at single nucleotide resolution: a reconciled transcriptome-wide map. RNA 24(11):1427–1436. https://doi.org/10.1261/rna.067348.118
Li X, Xiong X, Zhang M, Wang K, Chen Y, Zhou J, Mao Y, Lv J, Yi D, Chen XW, Wang C, Qian SB, Yi C (2017) Base-resolution mapping reveals distinct m(1)A methylome in nuclear- and mitochondrial-encoded transcripts. Mol Cell 68(5):993–1005. e1009. https://doi.org/10.1016/j.molcel.2017.10.019
Zhao Y, Zhao Q, Kaboli PJ, Shen J, Li M, Wu X, Yin J, Zhang H, Wu Y, Lin L, Zhang L, Wan L, Wen Q, Li X, Cho CH, Yi T, Li J, Xiao Z (2019) m1A regulated genes modulate PI3K/AKT/mTOR and ErbB pathways in gastrointestinal cancer. Transl Oncol 12(10):1323–1333. https://doi.org/10.1016/j.tranon.2019.06.007
Enroth C, Poulsen LD, Iversen S, Kirpekar F, Albrechtsen A, Vinther J (2019) Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res 47(20):e126. https://doi.org/10.1093/nar/gkz736
Pandolfini L, Barbieri I, Bannister AJ, Hendrick A, Andrews B, Webster N, Murat P, Mach P, Brandi R, Robson SC, Migliori V, Alendar A, d'Onofrio M, Balasubramanian S, Kouzarides T (2019) METTL1 promotes let-7 MicroRNA processing via m7G methylation. Mol Cell 74(6):1278–1290. e1279. https://doi.org/10.1016/j.molcel.2019.03.040
Zhang LS, Liu C, Ma H, Dai Q, Sun HL, Luo G, Zhang Z, Zhang L, Hu L, Dong X, He C (2019) Transcriptome-wide mapping of internal N(7)-methylguanosine methylome in mammalian mRNA. Mol Cell 74(6):1304–1316. e1308. https://doi.org/10.1016/j.molcel.2019.03.036
Edelheit S, Schwartz S, Mumbach MR, Wurtzel O, Sorek R (2013) Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet 9(6):e1003602. https://doi.org/10.1371/journal.pgen.1003602
Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T (2012) Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 40(11):5023–5033. https://doi.org/10.1093/nar/gks144
Trixl L, Lusser A (2019) The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip Rev RNA 10(1):e1510. https://doi.org/10.1002/wrna.1510
Huang T, Chen W, Liu J, Gu N, Zhang R (2019) Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat Struct Mol Biol 26(5):380–388. https://doi.org/10.1038/s41594-019-0218-x
Amort T, Rieder D, Wille A, Khokhlova-Cubberley D, Riml C, Trixl L, Jia XY, Micura R, Lusser A (2017) Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol 18(1):1. https://doi.org/10.1186/s13059-016-1139-1
Schumann U, Zhang HN, Sibbritt T, Pan A, Horvath A, Gross S, Clark SJ, Yang L, Preiss T (2020) Multiple links between 5-methylcytosine content of mRNA and translation. BMC Biol 18(1):40. https://doi.org/10.1186/s12915-020-00769-5
Hoernes TP, Clementi N, Faserl K, Glasner H, Breuker K, Lindner H, Huttenhofer A, Erlacher MD (2016) Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code. Nucleic Acids Res 44(2):852–862. https://doi.org/10.1093/nar/gkv1182
Lyabin DN, Eliseeva IA, Skabkina OV, Ovchinnikov LP (2011) Interplay between Y-box-binding protein 1 (YB-1) and poly(A) binding protein (PABP) in specific regulation of YB-1 mRNA translation. RNA Biol 8(5):883–892. https://doi.org/10.4161/rna.8.5.16022
Xu L, Liu X, Sheng N, Oo KS, Liang J, Chionh YH, Xu J, Ye F, Gao YG, Dedon PC, Fu XY (2017) Three distinct 3-methylcytidine (m(3)C) methyltransferases modify tRNA and mRNA in mice and humans. J Biol Chem 292(35):14695–14703. https://doi.org/10.1074/jbc.M117.798298
Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, Hosogane M, Sinclair WR, Nanan KK, Mandler MD, Fox SD, Zengeya TT, Andresson T, Meier JL, Coller J, Oberdoerffer S (2018) Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175(7):1872–1886. e1824. https://doi.org/10.1016/j.cell.2018.10.030
Cheng Q, Xiong J, Ma C, Jiang D, Yu-Qi F (2020) Chemical tagging for sensitive determination of uridine modifications in RNA. Chem Sci 11:1878–1891
Choi J, Indrisiunaite G, DeMirci H, Ieong KW, Wang J, Petrov A, Prabhakar A, Rechavi G, Dominissini D, He C, Ehrenberg M, Puglisi JD (2018) 2'-O-methylation in mRNA disrupts tRNA decoding during translation elongation. Nat Struct Mol Biol 25(3):208–216. https://doi.org/10.1038/s41594-018-0030-z
Erales J, Marchand V, Panthu B, Gillot S, Belin S, Ghayad SE, Garcia M, Laforets F, Marcel V, Baudin-Baillieu A, Bertin P, Coute Y, Adrait A, Meyer M, Therizols G, Yusupov M, Namy O, Ohlmann T, Motorin Y, Catez F, Diaz JJ (2017) Evidence for rRNA 2'-O-methylation plasticity: control of intrinsic translational capabilities of human ribosomes. Proc Natl Acad Sci U S A 114(49):12934–12939. https://doi.org/10.1073/pnas.1707674114
Cadet J, Wagner JR (2013) DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol 5(2). https://doi.org/10.1101/cshperspect.a012559
Simms CL, Hudson BH, Mosior JW, Rangwala AS, Zaher HS (2014) An active role for the ribosome in determining the fate of oxidized mRNA. Cell Rep 9(4):1256–1264. https://doi.org/10.1016/j.celrep.2014.10.042
Ishii T, Hayakawa H, Sekiguchi T, Adachi N, Sekiguchi M (2015) Role of Auf1 in elimination of oxidatively damaged messenger RNA in human cells. Free Radic Biol Med 79:109–116. https://doi.org/10.1016/j.freeradbiomed.2014.11.018
Hayakawa H, Uchiumi T, Fukuda T, Ashizuka M, Kohno K, Kuwano M, Sekiguchi M (2002) Binding capacity of human YB-1 protein for RNA containing 8-oxoguanine. Biochemistry 41(42):12739–12744. https://doi.org/10.1021/bi0201872
Huber SM, van Delft P, Mendil L, Bachman M, Smollett K, Werner F, Miska EA, Balasubramanian S (2015) Formation and abundance of 5-hydroxymethylcytosine in RNA. Chembiochem 16(5):752–755. https://doi.org/10.1002/cbic.201500013
Fu L, Guerrero CR, Zhong N, Amato NJ, Liu Y, Liu S, Cai Q, Ji D, Jin SG, Niedernhofer LJ, Pfeifer GP, Xu GL, Wang Y (2014) Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J Am Chem Soc 136(33):11582–11585. https://doi.org/10.1021/ja505305z
Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515(7525):143–146. https://doi.org/10.1038/nature13802
Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, Fink G, Regev A (2014) Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159(1):148–162. https://doi.org/10.1016/j.cell.2014.08.028
Charette M, Gray MW (2000) Pseudouridine in RNA: what, where, how, and why. IUBMB Life 49(5):341–351. https://doi.org/10.1080/152165400410182
Li X, Zhu P, Ma S, Song J, Bai J, Sun F, Yi C (2015) Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol 11(8):592–597. https://doi.org/10.1038/nchembio.1836
Tardu M, Jones JD, Kennedy RT, Lin Q, Koutmou KS (2019) Identification and quantification of modified nucleosides in Saccharomyces cerevisiae mRNAs. ACS Chem Biol 14(7):1403–1409. https://doi.org/10.1021/acschembio.9b00369
Penzo M, Guerrieri AN, Zacchini F, Trere D, Montanaro L (2017) RNA pseudouridylation in physiology and medicine: for better and for worse. Genes (Basel) 8(11). https://doi.org/10.3390/genes8110301
Adachi H, De Zoysa MD, Yu YT (2019) Post-transcriptional pseudouridylation in mRNA as well as in some major types of noncoding RNAs. Biochim Biophys Acta Gene Regul Mech 1862(3):230–239. https://doi.org/10.1016/j.bbagrm.2018.11.002
Hamma T, Ferre-D'Amare AR (2006) Pseudouridine synthases. Chem Biol 13(11):1125–1135. https://doi.org/10.1016/j.chembiol.2006.09.009
Yu YT, Meier UT (2014) RNA-guided isomerization of uridine to pseudouridine--pseudouridylation. RNA Biol 11(12):1483–1494. https://doi.org/10.4161/15476286.2014.972855
Carlile TM, Martinez NM, Schaening C, Su A, Bell TA, Zinshteyn B, Gilbert WV (2019) mRNA structure determines modification by pseudouridine synthase 1. Nat Chem Biol 15(10):966–974. https://doi.org/10.1038/s41589-019-0353-z
Karijolich J, Yu YT (2011) Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 474(7351):395–398. https://doi.org/10.1038/nature10165
Nakamoto MA, Lovejoy AF, Cygan AM, Boothroyd JC (2017) mRNA pseudouridylation affects RNA metabolism in the parasite toxoplasma gondii. RNA 23(12):1834–1849. https://doi.org/10.1261/rna.062794.117
Eyler DE, Franco MK, Batool Z, Wu MZ, Dubuke ML, Dobosz-Bartoszek M, Jones JD, Polikanov YS, Roy B, Koutmou KS (2019) Pseudouridinylation of mRNA coding sequences alters translation. Proc Natl Acad Sci U S A 116(46):23068–23074. https://doi.org/10.1073/pnas.1821754116
Mauger DM, Cabral BJ, Presnyak V, Su SV, Reid DW, Goodman B, Link K, Khatwani N, Reynders J, Moore MJ, McFadyen IJ (2019) mRNA structure regulates protein expression through changes in functional half-life. Proc Natl Acad Sci U S A 116(48):24075–24083. https://doi.org/10.1073/pnas.1908052116
Svitkin YV, Cheng YM, Chakraborty T, Presnyak V, John M, Sonenberg N (2017) N1-methyl-pseudouridine in mRNA enhances translation through eIF2alpha-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res 45(10):6023–6036. https://doi.org/10.1093/nar/gkx135
Pardi N, Hogan MJ, Weissman D (2020) Recent advances in mRNA vaccine technology. Curr Opin Immunol 65:14–20. https://doi.org/10.1016/j.coi.2020.01.008
Pardi N, Hogan MJ, Porter FW, Weissman D (2018) mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov 17(4):261–279. https://doi.org/10.1038/nrd.2017.243
Leppek K, Das R, Barna M (2018) Functional 5' UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 19(3):158–174. https://doi.org/10.1038/nrm.2017.103
Kudla G, Lipinski L, Caffin F, Helwak A, Zylicz M (2006) High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol 4(6):e180. https://doi.org/10.1371/journal.pbio.0040180
Lima SA, Chipman LB, Nicholson AL, Chen YH, Yee BA, Yeo GW, Coller J, Pasquinelli AE (2017) Short poly(A) tails are a conserved feature of highly expressed genes. Nat Struct Mol Biol 24(12):1057–1063. https://doi.org/10.1038/nsmb.3499
Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, Schlake T (2015) Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther 23(9):1456–1464. https://doi.org/10.1038/mt.2015.103
Kariko K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, Weissman D (2008) Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 16(11):1833–1840. https://doi.org/10.1038/mt.2008.200
Nowakowska M, Kowalska J, Martin F, d'Orchymont A, Zuberek J, Lukaszewicz M, Darzynkiewicz E, Jemielity J (2014) Cap analogs containing 6-thioguanosine--reagents for the synthesis of mRNAs selectively photo-cross-linkable with cap-binding biomolecules. Org Biomol Chem 12(27):4841–4847. https://doi.org/10.1039/c4ob00059e
Kore AR, Charles I (2010) Synthesis and evaluation of 2'-O-allyl substituted dinucleotide cap analog for mRNA translation. Bioorg Med Chem 18(22):8061–8065. https://doi.org/10.1016/j.bmc.2010.09.013
Kowalska J, Lewdorowicz M, Zuberek J, Grudzien-Nogalska E, Bojarska E, Stepinski J, Rhoads RE, Darzynkiewicz E, Davis RE, Jemielity J (2008) Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS. RNA 14(6):1119–1131. https://doi.org/10.1261/rna.990208
Grudzien-Nogalska E, Stepinski J, Jemielity J, Zuberek J, Stolarski R, Rhoads RE, Darzynkiewicz E (2007) Synthesis of anti-reverse cap analogs (ARCAs) and their applications in mRNA translation and stability. Methods Enzymol 431:203–227. https://doi.org/10.1016/S0076-6879(07)31011-2
Kopcial M, Wojtczak BA, Kasprzyk R, Kowalska J, Jemielity J (2019) N1-propargylguanosine modified mRNA cap analogs: synthesis, reactivity, and applications to the study of cap-binding proteins. Molecules 24(10). https://doi.org/10.3390/molecules24101899
Goldstrohm AC, Wickens M (2008) Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol 9(4):337–344. https://doi.org/10.1038/nrm2370
Gampe C, White ACS, Siva S, Zecri F, Diener J (2018) 3'-modification stabilizes mRNA and increases translation in cells. Bioorg Med Chem Lett 28(14):2451–2453. https://doi.org/10.1016/j.bmcl.2018.06.008
Pardi N, Hogan MJ, Naradikian MS, Parkhouse K, Cain DW, Jones L, Moody MA, Verkerke HP, Myles A, Willis E, LaBranche CC, Montefiori DC, Lobby JL, Saunders KO, Liao HX, Korber BT, Sutherland LL, Scearce RM, Hraber PT, Tombacz I, Muramatsu H, Ni H, Balikov DA, Li C, Mui BL, Tam YK, Krammer F, Kariko K, Polacino P, Eisenlohr LC, Madden TD, Hope MJ, Lewis MG, Lee KK, Hu SL, Hensley SE, Cancro MP, Haynes BF, Weissman D (2018) Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J Exp Med 215(6):1571–1588. https://doi.org/10.1084/jem.20171450
Andries O, Mc Cafferty S, De Smedt SC, Weiss R, Sanders NN, Kitada T (2015) N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Control Release 217:337–344. https://doi.org/10.1016/j.jconrel.2015.08.051
Boros G, Miko E, Muramatsu H, Weissman D, Emri E, Rozsa D, Nagy G, Juhasz A, Juhasz I, van der Horst G, Horkay I, Remenyik E, Kariko K, Emri G (2013) Transfection of pseudouridine-modified mRNA encoding CPD-photolyase leads to repair of DNA damage in human keratinocytes: a new approach with future therapeutic potential. J Photochem Photobiol B 129:93–99. https://doi.org/10.1016/j.jphotobiol.2013.09.010
Kariko K, Muramatsu H, Keller JM, Weissman D (2012) Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol Ther 20(5):948–953. https://doi.org/10.1038/mt.2012.7
Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, Kariko K (2011) Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res 39(21):9329–9338. https://doi.org/10.1093/nar/gkr586
Anderson BR, Muramatsu H, Nallagatla SR, Bevilacqua PC, Sansing LH, Weissman D, Kariko K (2010) Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res 38(17):5884–5892. https://doi.org/10.1093/nar/gkq347
Miedziak B, Dobiezynska A, Darzynkiewicz ZM, Bartkowska J, Miszkiewicz J, Kowalska J, Warminski M, Tyras M, Trylska J, Jemielity J, Darzynkiewicz E, Grzela R (2020) Kinetic analysis of IFIT1 and IFIT5 interactions with different native and engineered RNAs and its consequences for designing mRNA-based therapeutics. RNA 26(1):58–68. https://doi.org/10.1261/rna.073304.119
Strenkowska M, Grzela R, Majewski M, Wnek K, Kowalska J, Lukaszewicz M, Zuberek J, Darzynkiewicz E, Kuhn AN, Sahin U, Jemielity J (2016) Cap analogs modified with 1,2-dithiodiphosphate moiety protect mRNA from decapping and enhance its translational potential. Nucleic Acids Res 44(20):9578–9590. https://doi.org/10.1093/nar/gkw896
Wu MZ, Asahara H, Tzertzinis G, Roy B (2020) Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA 26(3):345–360. https://doi.org/10.1261/rna.073858.119
Kariko K, Muramatsu H, Ludwig J, Weissman D (2011) Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res 39(21):e142. https://doi.org/10.1093/nar/gkr695
Hoernes TP, Heimdorfer D, Kostner D, Faserl K, Nussbaumer F, Plangger R, Kreutz C, Lindner H, Erlacher MD (2019) Eukaryotic translation elongation is modulated by single natural nucleotide derivatives in the coding sequences of mRNAs. Genes (Basel) 10(2). https://doi.org/10.3390/genes10020084
Liao S, Sun H, Xu C (2018) YTH domain: a family of N(6)-methyladenosine (m(6)A) readers. Genomics Proteomics Bioinformatics 16(2):99–107. https://doi.org/10.1016/j.gpb.2018.04.002
Acknowledgments
I would like to thank Monica Z. Wu for help with the illustrations and New England Biolabs Inc. for supporting the basic research on mRNA modifications. Funding for open access was provided by New England Biolabs Inc.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this protocol

Cite this protocol
Roy, B. (2021). Effects of mRNA Modifications on Translation: An Overview. In: McMahon, M. (eds) RNA Modifications. Methods in Molecular Biology, vol 2298. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1374-0_20
Download citation
DOI: https://doi.org/10.1007/978-1-0716-1374-0_20
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-1373-3
Online ISBN: 978-1-0716-1374-0
eBook Packages: Springer Protocols