
Abstract
Far-UV circular dichroism (CD) spectroscopy is a classical method for the study of the secondary structure of polypeptides in solution. It has been the general view that the α-helix content can be estimated accurately from the CD spectra. However, the technique was less reliable to estimate the β-sheet contents as a consequence of the structural variety of the β-sheets, which is reflected in a large spectral diversity of the CD spectra of proteins containing this secondary structure component. By taking into account the parallel or antiparallel orientation and the twist of the β-sheets, the Beta Structure Selection (BeStSel) method provides an improved β-structure determination and its performance is more accurate for any of the secondary structure types compared to previous CD spectrum analysis algorithms. Moreover, BeStSel provides extra information on the orientation and twist of the β-sheets which is sufficient for the prediction of the protein fold.
The advantage of CD spectroscopy is that it is a fast and inexpensive technique with easy data processing which can be used in a wide protein concentration range and under various buffer conditions. It is especially useful when the atomic resolution structure is not available, such as the case of protein aggregates, membrane proteins or natively disordered chains, for studying conformational transitions, testing the effect of the environmental conditions on the protein structure, for verifying the correct fold of recombinant proteins in every scientific fields working on proteins from basic protein science to biotechnology and pharmaceutical industry. Here, we provide a brief step-by-step guide to record the CD spectra of proteins and their analysis with the BeStSel method.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
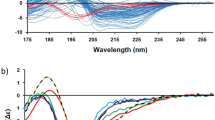
Circular dichroism (CD) corresponds to the differential absorption between left and right circularly polarized light (Fig. 1). In the far-UV region between 170 and 250 nm, mostly the electronic transitions of the peptide bonds contribute to the CD spectrum of proteins [1, 2]. Depending on the local geometry, environment, and H-bond pattern of the peptide bonds, the polypeptide chains with different conformations can exhibit distinct, characteristic spectral profiles, which is manifested in the CD spectra of proteins of different structural classes (Fig. 2). This observation initiated the development of algorithms for the secondary structure estimation from the CD spectra. In the last 30 years, a dozen CD spectrum analysis algorithms made attempts to accurately estimate the secondary structure composition of the proteins. These methods use reference CD spectra of proteins with known structure to make an estimation of different types of secondary structure elements (most often helix, β-sheet, turn, and disordered). The mathematical background and performances of these methods are reviewed and compared [3, 4]. Generally, they predict the helix content more or less accurately, while often fail to properly predict the β-sheet content due to the large spectral diversity of β-structured proteins (Fig. 3). In the background of this spectral diversity, there must be the variety of β-sheets in the orientation (parallel–antiparallel), the length and number of strands, and their twists, which made difficult to estimate this component from the CD spectrum and was believed to be an intrinsic limitation of the technique [5].

The phenomenon of circular dichroism. Light is an electromagnetic wave which can be characterized by the electric and magnetic fields that are perpendicular to each other and the direction of the travel of the light. Linearly polarized light is characterized by the electric field vector oscillating in one plane (a), while the electric field vector of circularly polarized light is rotating around the axis of propagation by maintaining a constant amplitude (b). Looking into the light propagating toward the observer, electric field vector rotating counterclockwise or clockwise depict the left and right circularly polarized lights, respectively. The summation of left and right circularly polarized light of equal amplitudes results in linearly polarized light while different amplitudes result elliptically polarized light (c). Optical active material (which should have chiral properties) interacts with light in a polarization dependent manner which can be manifested in optical rotation of the plane of polarization (a, and angle α in c) and in circular dichroism which is the differential absorption of the left and right circularly polarized light (b, c). For details of the theory of circular dichroism see [1]. At the practical level, the differential absorption of the left and right circularly polarized light can be expressed as the difference in the extinction coefficients, Δε = εL − εR, or as the ellipticity of the summation of the left and right circularly polarized lights of different amplitudes, tgΘ = a/b = (ER − EL)/(ER + EL), where ER and EL are the amplitudes of the electric field vectors. Θ will be negative if ER is smaller than EL. Measured ellipticity is usually given as Θ in the unit of mdeg. When Δε is in M−1·cm−1 units and Θ is also normalized to the molar number of residues (more precisely, to the number of peptide bonds) and pathlength in cm, denoted as [Θ] and given in the traditional unit of deg·cm2·dmol−1, the value of Δε is equal to [Θ]/3298 (we have to note, that for the correct equation, the factor of 3298 is not dimension-less)

Characteristic far-UV CD spectra of different protein architectures. Proteins of distinct secondary structures such as α-helix (red), parallel β-sheet (blue), antiparallel β-sheet (green), polyproline-helix (orange), and disordered chain (purple) exhibit characteristic spectral shapes indicating that CD spectroscopy can be useful for the determination of the secondary structure of proteins

The spectral diversity of β-structures. (a) α-helical proteins have uniform spectral shape as shown as demonstrated here by proteins having ~50% α-helix content. (b) Despite their similar (~50%) β-sheet content, β-structured proteins show a large spectral diversity making secondary structure estimation a difficult challenge
Recently, we have shown that the spectral contribution of β-sheets depends on the parallel-antiparallel orientation and the twist of the β-sheets [4]. Based on this observation, we have developed a new method named BeStSel (Beta Structure Selection) for the secondary structure estimation of proteins from the CD spectra that takes into account the orientation and twist of the β-sheets. The method defines eight structural components: regular and distorted α-helices, left-handed, relaxed (slightly right-hand twisted) and right-hand twisted antiparallel β-sheets, parallel β-sheet, turn. and “others” (Table 1, and for detailed definitions see Micsonai et al. [4]).
BeStSel provides an improved accuracy on a broad range of protein structures including β-sheet-rich proteins, membrane proteins, protein aggregates, and amyloid fibrils.
As a results of the detailed structural information gained from the CD spectrum, BeStSel is capable of predicting the protein fold down to the homology level using the CATH fold classification (Fig. 4) [9, 10].

The BeStSel method. Schematic representation of the secondary structure components of BeStSel (see also Table 1) and the pipeline of structure estimation. Obtaining the fractions of the eight components from the CD spectrum by BeStSel, the protein fold can be predicted
A web server was constructed at http://bestsel.elte.hu making the BeStSel method freely accessible for the scientific community.
In the Materials section completed with extended Notes we briefly describe the essential sample preparation steps for a reliable CD measurement that are necessary for an accurate secondary structure estimation. In the Methods section we give a step-by-step guide for the modules of the BeStSel webserver to analyze protein CD spectra.
2 Materials
A lot of buffer compounds and salts have high absorption in the far-UV region. Their use should be avoided or their concentration should be kept at the minimum that is acceptable for the protein. Phosphate buffer (not PBS ) is suitable for CD spectroscopy with as low salt added as possible. However, it might be incompatible with other buffer components to be used, for example with calcium, or with the protein. High absorption of the buffer limits the usable wavelength range and can be avoided by choosing a shorter pathlength cell which requires increased protein concentration (see Subheading 3.1 and Notes 1–3).
Depending on the instrument and the cell holder used, cylindrical or rectangular quartz cells can be used in the >180 nm wavelength region. Below 180 nm, or in the case of low sample volume, demountable calcium fluoride cells can be used.
3 Methods
3.1 Sample Preparation
-
1.
The CD spectrum shows the average spectrum of the components having CD signal in the sample. It is important to have a pure, homogenous protein sample free of contaminations of other proteins or other chiral biomolecules such as nucleic acids. Check the purity of the sample by SDS-PAGE , mass spectrometry, absorption spectroscopy (for nucleic acid contamination), and other complementary methods. Take into consideration that the CD spectrum is also affected by expression tags often used in the case of recombinant proteins (see Note 1).
-
2.
The inhomogeneity and light scattering also affect the CD signal causing shrinking of the amplitude and distorting the spectrum, which may have caused by protein aggregation and precipitation (see Note 2).
-
3.
Transfer the sample into a buffer suitable for CD measurements. The best method for this is dialysis where the dialysis buffer can be used for baseline measurement. A lyophilized protein powder often contains contaminations, so it is advised not only to dissolve it in the proper buffer but dialyze it. An alternative method can be a transfer of the protein to the buffer of the measurement by using a filtration spinning tube or desalting column.
-
4.
Determination of the accurate protein concentration is crucial for the correct normalization and quantitative analysis of the CD spectra. Select the pathlength of the cuvette depending on the concentration in a way that the product of the pathlength in mm and the concentration in mg/ml should be ~0.1 (it means that for a solution of 0.1 mg/ml concentration, a 1 mm cell is optimal for use). Selecting the appropriate buffer and pathlength, CD spectroscopy is capable of studying the protein structure in a wide concentration range of 0.05–20 mg/ml, which is a significant advantage over the other techniques used for protein structure determination, such as NMR, infrared spectroscopy, vibrational CD or RAMAN spectroscopy (see Note 3 for concentration determination).
-
5.
The instrumentation of CD spectroscopy is well-developed, the users routinely can measure the spectra in the 190–260 nm range with some considerations on the buffer and salt compositions of the sample. Choose shorter pathlengths (10–50 μm) and high protein concentrations (2–10 mg/ml) to record the spectra down to 180 nm on conventional instruments. Synchrotron radiation CD (SRCD) stations can collect spectra at even shorter wavelengths [11]. To collect high quality CD spectra suitable for quantitative structural analysis, the instrumental parameters should be carefully chosen following the instrument manual. Note 4 discusses the preferable measurement parameters. For quantitative measurements, calibrate the instrument occasionally for amplitude and wavelength accuracy (see Note 5).
3.2 Wavelength Range, Baseline Subtraction, and Data Normalization
-
1.
CD spectroscopy is a type of absorption spectroscopy and the CD signal is measured above the overall absorption of the sample which should be kept at low for the good signal-to-noise ratio and linearity of the detector. The voltage (high tension, HT) of the detector is adjusted to this overall absorption and should not exceed a limit (e.g., ~600 V limit in the case of a detector having 900–1000 V maximum HT). Discard the data measured at HT values over this limit.
-
2.
Correct the sample spectrum by subtracting the baseline measurement of the same buffer that is used for the protein. A moderate smoothing can be applied on the spectrum by taking care not to change significantly any sharp component or steep part of the spectrum.
-
3.
Normalize the CD spectrum for the concentration, pathlength and number of peptide bonds. The mean residue molar ellipticity (deg·cm2·dmol−1) is defined as follows:
$$ {\left[\varTheta \right]}_{MRE}=\varTheta /\left(10\cdot {c}_r\cdot l\right) $$where Θ, the measured ellipticity, is in mdeg, cr is the molar concentration per residue, and l is the pathlength in cm. The also commonly used extinction coefficient difference, Δε = [Θ]/3298.2, its unit is M−1·cm−1. Although BeStSel can handle the baseline subtracted raw data, it is important to understand the normalization procedure because the output of BeStSel and the proper form of CD spectra for publication is the normalized data.
3.3 Single Spectrum Analysis
At the starting page of the BeStSel webserver, by default, data can be uploaded for single spectrum analysis in the form of a text file or can be copied into the window in two data columns, separator can be space, tab, comma or semicolon. Upload the data either as normalized in Δε or [Θ]MRE, or as measured, baseline subtracted data. In the latter case, you have to provide the concentration (μM), pathlength (cm), and the number of residues. The page is protected by a captcha against malicious use. In all cases, the program normalizes or converts the uploaded data to Δε, which can be verified in the next, Data Examination page. Note, that the numeric format uses dot as decimal point. If the spectrum in the Data examination page contains steps, probably the decimal sign is incorrect. Starting the calculation, the results will appear in a graphical image with all the useful information provided: wavelength range, the estimated secondary structure content, the curve and error of the fitted spectrum, and user provided information. At first, data is analyzed in the possible widest wavelength range of the uploaded data. However, we strongly suggest to choose an appropriate wavelength range where the PMT voltage was below a limit (e.g., 600 volts) determined by the manufacturer upon the measurement (see Subheading 3.2). See Notes 2 and 4 for buffer selection and experimental setup. Below the results, change the output format for your convenience. Results can be saved as a graphical image. For further data processing by the users, result can be shown in text format with the predicted secondary structure contents at the top and the experimental, fitted, and the residual data in columns below. Transfer the data by copying it to any data processing software to make your own plots, etc.
On the left side of the Results page, the wavelength range can be chosen and the analysis can be recalculated. Different wavelength ranges will provide slightly different results; however, in the case of using correct concentrations and normalization, the difference is within the estimation error. A scale factor can be chosen for recalculation, as well. The CD amplitude is multiplied with this factor. The “Best factor” function carries out a series of analysis by changing the current scaling factor automatically in the range of 0.5–2. The dependence of the individual secondary structure components on the CD amplitude is plotted. This can be informative in the case of uncertainties in the protein concentration or pathlength. In case of CD data in a wide wavelength range (down to at least 180 nm), the alteration of the factor with the lowest fitting NRMSD from 1 is a good indicator of incorrect concentration or pathlength values.
3.4 Fold Recognition
The eight secondary structure components of BeStSel bear sufficient information that is characteristic to the protein fold and makes possible its prediction. At first, twenty closest structures based on Euclidean distance are searched on the entire PDB . In case of single domain proteins, a fold prediction using the CATH protein fold classification [10, 12] can be done. The single domain PDB subset is a nonredundant collection of chains containing single CATH domains or homodomains filtered for <=95% sequence homology and resolution better than 3.0 Angströms. This dataset contains 55,350 single domains covering 4 classes, 41 architectures, and 1310 topologies and 5398 homologies [9]. The fold can be predicted by searching for the closest structures based on the Euclidean distance in the eight components. While this method does not take into account the possible error of the secondary structure estimation from CD , it can be used even if the secondary structural space is rarely populated by structures around the estimated result. Another method is surveying all the structures within the expected error of the CD results and sort them by their fold and the frequency of that fold [4]. At the level of architecture and topology , the ten most populated groups are presented. The most sophisticated way of fold prediction is a weighted K-nearest neighbors search using the chain length as extra parameter. Fold prediction can be initiated from within the Single Spectrum Analysis after getting the secondary structure contents or from a separate block at the starting page by manually providing the Secondary structure contents and chain length [9].
Use the Fold recognition module to find structures in the PDB and fold domains in CATH that are similar to the experimentally investigated protein. This function can be especially useful to verify the correct fold of recombinant proteins or search for the fold of proteins having low sequence homology to the proteins in the PDB .
3.5 Multiple Spectra Analysis
In this module, upload a series of spectra in a text file or copy into the window from a worksheet to analyze the CD spectra as a function of temperature, ligand concentration, etc. In the uploaded data, the first row should contain the values of the variable as the function of which the spectra were recorded. Below, there are columns. The first column contains the wavelength values and the others columns contain the corresponding spectral data. Therefore, the total number of columns should be equal to the number of values in the first row plus one. Data separator can be either tab, comma, semicolon, or space. The units of the input data can be chosen similarly to Single Spectrum Analysis . After the checkup of the uploaded data as a series of spectra in Δε, starting the calculation, the estimated secondary structure contents will be shown on the Result page as the function of the given parameter (temperature, ligand concentration, etc.). The wavelength range can be changed or the results can be recalculated with using a scaling factor applied for all the spectra. The results can be saved as image or copied out as data text. We have to note that Multiple Spectra Analysis is developed for analysis of a series of related CD spectra with the same number of data points and wavelength ranges. Unrelated spectra should be evaluated separately in Single Spectrum Analysis .
3.6 Secondary Structure Composition from PDB Structures
In this module of BeStSel, provide the four letters codes of atomic resolution structures deposited in the PDB to list out their secondary structure contents. Besides the eight secondary structure components of BeStSel, the six components of SELCON/CONTIN/CDSSTR methods [8] and the eight components of DSSP [6] are also shown for the entire molecule or selected subunits. Upon selecting the chain, the protein fold classification is also provided using the CATH classification [10]. This module of the BeStSel server is useful to compare the secondary structure results to the available reference protein structures.
3.7 Limitations of the BeStSel Method
The eight secondary structure components of BeStSel do not account for some special secondary structure types. Polyproline-II helix, different type of turns, 310-helices are not distinguished by BeStSel and thus analysis for such structures is not adequate. BeStSel does not handle the aromatic contributions (other algorithms neither do) which gives some uncertainty when the number of aromatic residues is high in the protein. The spectra of highly disordered proteins somewhat remind the highly right-twisted antiparallel β-sheets (Anti3 component), and partly might be counted as Anti3 instead of “Others” [9].
4 Notes
-
1.
Sample purity and preparation
The CD spectrum shows the average spectrum of the components having CD signal in the sample. Thus, it is important to have a pure, homogenous protein sample free of contaminations of other proteins or other chiral biomolecules such as nucleic acids. The purity of the sample should be checked by SDS-PAGE , mass spectrometry, absorption spectroscopy (for nucleic acid contamination) and other complementary methods. Recombinant proteins are often expressed using fused protein tags that provide higher expression or used for efficient purification (N-terminal extension of Met or more residues, His-, GST-, or other tags on either terminal) or stabilize the protein structure. These extensions or tags can affect the structure and stability of the proteins and contribute to the CD spectrum, as well. It is advised to have them removed from the protein. When removal of these extensions is not possible, it is important to take them into account in the analysis of the CD spectrum (number of residues, molecular weight, and presumed contribution to the estimated secondary structure contents).
CD spectroscopy is sensitive for light scattering effects which may have caused by protein aggregation and precipitation. To remove any precipitates, the sample should be spun down at least in a table top centrifuge at >10,000 × g force. To remove small oligomers of a protein, ultracentrifuge around ~100,000 × g could be used. In all cases the protein concentration should be determined after centrifugation.
In the case of measuring protein aggregates and amyloid fibrils, no centrifugation is applied or only a short centrifugation at low force can be used to remove the large aggregates which cause inhomogeneity and light scattering of the sample. Amyloid samples should be well homogenized by thorough pipetting or even using a slight ultrasonication.
-
2.
Buffer selection
A lots of buffer compounds and salts have high absorption in the far-UV region. Their use should be avoided or their concentration should be kept at the minimum that is acceptable for the protein. Using shorter pathlengths (that needs higher protein concentrations) can decrease the buffer absorption. Table 2 shows the usable wavelength range for CD of the different buffer compounds and salts. Denaturants such as GdnHCl and urea which are usually used at high concentrations have especially high absorptions which often make impossible the quantitative analysis of the CD spectrum in the lack of sufficient usable wavelength range. Instead of them dodine could be used [14], which denatures the protein at orders of magnitude lower concentrations. Sodium and reducing agents such as dithiothreitol or mercaptoethanol also have high absorption. These compounds should be dialyzed out from the sample prior to the measurement. Tris(2-carboxyethyl)phosphine (TCEP) is better as reducing agent for CD because of its lower effective concentration range and somewhat lower extinction coefficient. Short peptides or other organic chemicals are often dissolved in dimethyl sulfoxide (DMSO) which is noncompatible with CD spectroscopy even after ten thousand-fold dilution.
-
3.
Concentration determination
An advantage of CD spectroscopy is the usable wide protein concentration range which starts at least an order of magnitude lower concentration than the minima for NMR, infrared, RAMAN and other spectroscopies used for the study of protein secondary structure. It can be as low as 0.05 mg/ml in a 2 mm cell and as high as 20 mg/ml in a 5 μm cell. Thus, it is a complementary method for the other spectroscopy techniques to check whether at high concentration the protein still exhibits the same conformation as it does at low, more physiological concentrations. A lot of proteins aggregate at higher protein concentrations undermining the results of other, often expensive and time consuming methods. Using CD spectroscopy, the conformational state of the protein as a function of the concentration, pH and other parameters can be easily verified. At short pathlengths, CaF2 cells are often used instead of quartz cells. Using very short pathlengths of few micrometers may result orientation of long molecules such as amyloid fibrils in the cell which should be taken into consideration.
The method considered to be the most accurate for concentration determination is quantitative amino acid analysis. In case the protein contains tryptophan and tyrosine residues, the concentration can be determined by measuring the absorbance at 280 nm. The extinction coefficient at 280 nm can be calculated from the primary sequence using the ProtParam tool (https://web.expasy.org/protparam/ ) [15]. In the absence of these amino acids, the concentration can be determined by the absorbance at 205 nm [16] or 214 nm [17]. An advantage of measuring at these two wavelengths is that, because of the high extinction coefficients, the CD samples can be directly measured. If the spectropolarimeter is capable of accurately converting the HT values to absorbances, then the concentrations can be determined right from the CD measurements after subtracting the baseline absorptions. Extinction coefficients at 205 and 214 nm can be calculated from the amino acid sequence at the BeStSel homepage (http://bestsel.elte.hu).
-
4.
Instrument settings
Although the CD spectra of the protein do not contain sharp peaks, the bandwidth should not be set to more than 2 nm, preferably, it is 1 nm. In case of continuous scanning mode when the wavelength is continuously changed at a scanning rate, the response/data integration time and the scanning rate should be harmonized in a way that during averaging of one data point, the wavelength should not be shifted more than the value of the bandwidth. It means that at a rate of 100 nm/min 0.5 or at most 1 s integration time should be used and these values are 1–2 s for 50 nm/min, 2–4 s for 20 nm/min and 4–8 s for 10 nm/min scanning rates. Depending on the amplitude and noise, several scans should be accumulated (averaged) at the convenience of the user. Usually a spectrum recording for 15 min overall time (~10 scans averaged at 50 nm/min scanning rate) is sufficient for an acceptable quality. To double the signal-to-noise ratio, four times more scans are needed. The baseline spectrum of the buffer should be collected with using the same parameters.
To collect as much information as possible, the CD spectra should be recorded in the widest usable wavelength range limited by the sample absorption at the low end, down to at least 200 nm but favorably to 190 or 180 nm. SRCD instruments can provide the CD spectra down to 175 nm. The recommended starting wavelength is 260 nm. In the 260–250 nm region (after baseline subtraction), a flat signal, close to zero, is an indication of a good baseline subtraction and the lack of light scattering effects and nucleic acid or other contaminations. Normally, the baseline CD spectrum of the buffer solution is recorded first and the usable wavelength range is estimated from the HT values which should not exceed the 50–60% of the maximum value. It is better to collect a fast protein sample spectrum first to determine the usable wavelength range and then carry out the high quality measurement only in the appropriate wavelength range to save time.
-
5.
Instrument calibration
Conventional benchtop instruments are usually calibrated by the manufacturer and the calibration can be repeated occasionally following the instruction manual. In the case of SRCD beamlines, the spectra can be corrected by a reference measurement of 1S-(+)-10-camphorsulfonic acid (CSA) which provides a negative and a positive peak at 192.5 and 290.5 nm having Δε values −4.72 and 2.36 M−1·cm−1, respectively [18]. The concentration of the CSA can be determined at 280 nm using an extinction coefficient of 34.58 ± 0.18 M−1·cm−1 [19].
References
Fasman GD (ed) (1996) Circular dichroism and the conformational analysis of biomolecules. Plenum Press, New York
Berova N, Nakanishi K, Woody RW (eds) (2000) Circular Dichroism: principles and applications, 2nd edn. Wiley, New York
Greenfield NJ (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1:2876–2890
Micsonai A, Wien F, Kernya L, Lee YH, Goto Y, Refregiers M, Kardos J (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci U S A 112:E3095–E3103
Khrapunov S (2009) Circular dichroism spectroscopy has intrinsic limitations for protein secondary structure analysis. Anal Biochem 389:174–176
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637
Sreerama N, Venyaminov SY, Woody RW (1999) Estimation of the number of alpha-helical and beta-strand segments in proteins using circular dichroism spectroscopy. Protein Sci 8:370–380
Sreerama N, Woody RW (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem 287:252–260
Micsonai A, Wien F, Bulyaki E, Kun J, Moussong E, Lee YH, Goto Y, Refregiers M, Kardos J (2018) BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res 46:W315–W322
Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM (1997) CATH—a hierarchic classification of protein domain structures. Structure 5:1093–1108
Miles AJ, Wallace BA (2006) Synchrotron radiation circular dichroism spectroscopy of proteins and applications in structural and functional genomics. Chem Soc Rev 35:39–51
Sillitoe I, Lewis TE, Cuff A, Das S, Ashford P, Dawson NL, Furnham N, Laskowski RA, Lee D, Lees JG, Lehtinen S, Studer RA, Thornton J, Orengo CA (2015) CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res 43:D376–D381
Creighton TE (ed) (1989) Spectral methods of characterizing protein conformation and conformational changes in protein structure: a practical approach. Oxford Universiy Press, Oxford
Guin D, Sye K, Dave K, Gruebele M (2016) Dodine as a transparent protein denaturant for circular dichroism and infrared studies. Protein Sci 25:1061–1068
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112:531–552
Anthis NJ, Clore GM (2013) Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Sci 22:851–858
Kuipers BJ, Gruppen H (2007) Prediction of molar extinction coefficients of proteins and peptides using UV absorption of the constituent amino acids at 214 nm to enable quantitative reverse phase high-performance liquid chromatography-mass spectrometry analysis. J Agric Food Chem 55:5445–5451
Chen GC, Yang JT (1977) 2-point calibration of circular dichrometer with D-10-Camphorsulfonic acid. Anal Lett 10:1195–1207
Miles AJ, Wien F, Wallace BA (2004) Redetermination of the extinction coefficient of camphor-10-sulfonic acid, a calibration standard for circular dichroism spectroscopy. Anal Biochem 335:338–339
Acknowledgments
This work was supported by the National Research, Development and Innovation Fund of Hungary [K120391, KH125597, 2017-1.2.1-NKP-2017-00002, FIEK16-1-2016-0005, TÉT16-1-2016-0134, TÉT16-1-2016-0197, 2019-2.1.11-TÉT-2019-00079]; SOLEIL Synchrotron, France [proposals 20191810, 20181890, 20181896, 20180805, 20171582]; and CampusFrance [Balaton-Programme Hubert Curien, 38642YK]. A.M. is supported by the Bolyai János Scholarship of the Hungarian Academy of Sciences, and the New National Excellence Program (ÚNKP-18-4-ELTE-833, ÚNKP-19-4-ELTE-790).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, a link is provided to the Creative Commons license and any changes made are indicated.
The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2021 The Author(s)
About this protocol

Cite this protocol
Micsonai, A., Bulyáki, É., Kardos, J. (2021). BeStSel: From Secondary Structure Analysis to Protein Fold Prediction by Circular Dichroism Spectroscopy. In: Chen, Y.W., Yiu, CP.B. (eds) Structural Genomics. Methods in Molecular Biology, vol 2199. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-0892-0_11
Download citation
DOI: https://doi.org/10.1007/978-1-0716-0892-0_11
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-0891-3
Online ISBN: 978-1-0716-0892-0
eBook Packages: Springer Protocols