
Abstract
Development of liposome-based formulations as vaccine adjuvants has been intimately associated with, and dependent on, and informed by, a fundamental understanding of biochemical and biophysical properties of liposomes themselves. The Walter Reed Army Institute of Research (WRAIR) has a fifty-year history of experience of basic research on liposomes; and development of liposomes as drug carriers; and development of liposomes as adjuvant formulations for vaccines. Uptake of liposomes by phagocytic cells in vitro has served as an excellent model for studying the intracellular trafficking patterns of liposomal antigen. Differential fluorescent labeling of proteins and liposomal lipids, together with the use of inhibitors, has enabled the visualization of physical locations of antigens, peptides, and lipids to elucidate mechanisms underlying the MHC class I and class II pathways in phagocytic APCs. Army Liposome Formulation (ALF) family of vaccine adjuvants, which have been developed and improved since 1986, and which range from nanosize to microsize, are currently being employed in phase 1 studies with different types of candidate vaccines.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


1 Introduction
The concept of using vesicles constructed from purified lipids, now known as liposomes, as models of cell membranes was introduced by (Bangham et al. 1965). Although many types of lipids can form liposomes, because of the similarity of liposomal phospholipid membranes to those of cells, the term “liposome” originally referred to vesicles which contained bulk lipids comprising phospholipids, with or without cholesterol. The discovery of liposomes led to three major directions of subsequent research: basic research on membranes containing lipid bilayers (Pick et al. 2018); development of liposome-encapsulated drugs for enhanced treatment of enzyme deficiencies, cancer, diabetes, or other diseases (Gregoriadis 1978; Li et al. 2019); and development of liposome-based vaccines (Schwendener 2014; De Serrano and Burkhart 2017; Wang et al. 2019; Alving et al. 2020). Thus, liposomes have been used both for prophylactic purposes as carriers of vaccine adjuvants and for therapeutic purposes as drug carriers (Nisini et al. 2018).
For use of liposomes as a vaccine formulation, the net charge of the particle should be considered, and the liposomes are generally either negative (anionic liposomes) or positive (cationic liposomes). Cationic liposomes are not addressed in this review but are reviewed in (Pedersen et al. 2018; De Serrano and Burkhart 2017). Early in the liposome field, two important discoveries dominated the direction of translational research: First, upon exposure to human serum, liposomes bound huge amounts of serum proteins, especially complement proteins; and second, upon intravenous injection into experimental animals, liposomes were taken up almost exclusively by macrophages in the liver and spleen.
The first application of liposomes as a vaccine adjuvant formulation in humans utilized a recombinant malaria protein which was encapsulated in multilamellar liposomes containing saturated neutral and anionic phospholipids, cholesterol, and monophosphoryl lipid A (MPLA) as an adjuvant (Fries et al. 1992). Although it was believed at that time that the liposomal protein was fully encapsulated, it is now believed that some of the proteins were also located both as transmembrane protein and as surface-adsorbed protein (Verma et al. 1991). Because of this, in this review we refer to proteins either in or on liposomes only as “liposome-associated” proteins. These liposomes represented the first iteration of an adjuvant composition that is now further refined and known as Army Liposome Formulation (ALF) (Beck et al. 2015b). The above liposomes containing malaria antigen were adsorbed to aluminum salt as a second adjuvant, and the formulation was injected intramuscularly (Fries et al. 1992). This approach, utilizing a mixture containing a range of large (microsize) and small (nanosize) liposomal particles, together with two different adjuvants for stimulation of innate immunity, seemed suitable for achieving vaccine potency in humans, and in three phase 1 clinical trials it did lead to high titers of antibodies to two different recombinant malaria protein antigens (Fries et al. 1992; Heppner et al. 1996) and to a recombinant HIV envelope protein, gp120 (Rao et al. 2018). These early studies utilizing liposomes as carriers of vaccines along with adjuvants laid the groundwork for future development of licensed liposomal vaccines.
A commercial GlaxoSmithKline (GSK) liposome formulation known as AS01B, which contains bulk lipids consisting of unsaturated neutral phospholipids and cholesterol, and which has both MPLA and a triterpenoid saponin known as QS21 as immunostimulants, has been included in an EMA-approved malaria vaccine in Africa (Mosquirix®, GSK) and in an FDA-licensed shingles vaccine (Shingrix®, GSK) in the US (Didierlaurent et al. 2017; Alving et al. 2020). The evolution of liposomes containing phospholipids, cholesterol, and MPLA and other immunostimulants as adjuvants thus presents a useful model for rational development of liposomes as carriers of antigens and adjuvants for vaccines. Various physical factors of liposomes, such as: surface charge (neutral, anionic, cationic); size (nano vs. micro); phospholipid fatty acyl composition (saturated vs. unsaturated); molar ratios of cholesterol and phospholipid in the lipid bilayer; number of lipid bilayer lamella (unilamellar, oligolamellar, or multilamellar vesicles); targeting molecules for attachment to immune cells; and the type, composition and number, if any, of attached adjuvant(s); have all been identified as factors related to the ease and cost of manufacture, and potential for unwanted toxicity. Putting all of these variables together has generated a complex number of ingenious physical immunostimulant compositions (Wang et al. 2019). Nonetheless, regardless of the structures of different types of liposomes, they are all foreign materials in the body, and each of the different liposome formulations faces the challenge of activation of innate immunity which has as its main goal the removal of the foreign particle.
2 Binding of Complement and Other Proteins to Liposomes Leading to Opsonization
In 1968, Kinsky and colleagues demonstrated that antigen-specific antibodies could bind to a liposomal glycolipid membrane antigen, resulting in complement-dependent damage and release of encapsulated liposomal glucose (Haxby et al. 1968, 1969; Kinsky et al. 1969). Further work demonstrated that the concentrations, by weight, of liposome-bound proteins were huge (as much as 800 µg of bound protein per µmole of liposomal phospholipid), and the amount of protein was easily measured by a Lowry protein assay on the surface of washed liposomes (Alving and Kinsky 1971). The above studies initiated the field of immunological aspects of liposomes; and they also introduced the concept that serum proteins (such as antibodies and complement) could stick to liposomes and accumulate in large amounts on the membrane surface.
In the early days of research on immunological aspects of liposomes, complement was thought to consist mainly of nine serum components (Nelson et al. 1966), and “complement” was, and often is today, defined informally as “fresh, unheated serum.” Incubation of liposomes containing an antigen, which were then coated with specific antibodies bound to the antigen, and then exposed to complement (fresh unheated serum), resulted in such a spectacular degree of phagocytic uptake in vitro by mouse peritoneal macrophages that the cells became rounded and bulging with slowly degrading intracellular vesicles (Wassef and Alving 1987).
In 1986, it was shown that addition of suspensions of liposomes lacking an antigen to human serum resulted in a more complex protein coating that included the nonspecific binding of immunoglobulins, complement proteins, albumin, fibrinogen, fibronectin, C-reactive protein, certain serum clotting factors, and scores of other proteins (Bonte and Juliano 1986; Chonn et al. 1991, 1992). In addition, it was also demonstrated that normal human serum contains naturally occurring antibodies to numerous phospholipids and cholesterol, and that the natural antibodies can bind to liposomes containing those components (Alving 1984; Alving and Wassef 1999; Matyas and Alving 2011). Upon intravenous injection of liposomes for the purpose of delivering drugs, the proteins that were adsorbed to the surface of liposomes in vivo resulted in the uptake of the injected particles by phagocytic cells, mainly Kupffer cells in the liver and splenic macrophages (Segal et al. 1974; Tyrrell et al. 1976). From those studies, it became apparent that incubation of liposomes in fresh human serum results in opsonization of the liposomes with numerous types of proteins, particularly the complement proteins, resulting in uptake of the liposomes by phagocytic cells.
In 1974, it was demonstrated that liposomes could serve as carriers of protein antigens, or even haptens, for immunization (Allison and Gregoriadis 1974; Uemura et al. 1974). The affinity of liposomes for uptake by phagocytic cells as antigen presenting cells has served as a major justification and rationale for using liposomes as vehicles for vaccines (Alving 1991). For liposome formulations as vaccine constituents, delivery of liposomal antigen to macrophages, dendritic cells, and T and B lymphocytes may be required for effectiveness.
It is now known from research on nanoparticles which are used for pharmaceutical and medical applications that exposure of liposomes and other types of particles to biological fluids in vivo or in vitro results in adsorption of proteins that alter the chemical and physical characteristics of the particles, a surface coating phenomenon that is referred to in the nanomedicines field as a “protein corona” (Cedervall et al. 2007; Ke et al. 2017). As noted by Ke et al. (2017): “… the protein corona owes its presence to thermodynamics in an aqueous environment, especially to the minimization of free enthalpy, and is mediated by Coulombic and van der Waals forces, hydrogen bonding, and hydrophobic interactions. Upon introduction into a biological fluid, a nanoparticle first assumes a transient or “soft” corona rendered by proteins of high abundance and is subsequently coated over time by a “hard” corona or proteins of high affinity according to the Vroman effect.”
From these new insights, it is useful to consider the influence of surface-bound protein on liposomal carriers of antigens and adjuvants for vaccines. Although numerous proteins can attach to liposomes to form a protein corona, many with variable affinities, it seems reasonable from the earliest liposome research, and from recent research on nanoparticles (Vu et al. 2019), to suspect that IgG and IgM immunoglobulins, whether natural antibodies to the lipids or to other attached proteins in the corona, would activate the complement cascade. As described by Bohlson et al.: “The complement system is composed of more than 50 different molecules and cleavage products, including, but not limited to, pattern recognition molecules, proenzymes, proteases, anaphylatoxins, opsonins, receptors, regulators, and multi-molecular complexes that are critical to host defense and maintenance of normal tissue homeostasis.” (Bohlson et al. 2019). Three pathways to complement activation have been identified, each initiated by different recognition molecules and mechanisms: (A) classical pathway, mainly initiated by antibody binding to an antigen, or binding other molecules, leading to recognition by C1q; (B) lectin pathway, initiated by recognition of ligands by mannose-binding lectin (MBL) and several other molecules; and (C) the alternative pathway, which can be activated spontaneously, although properdin may play a role (Lubbers et al. 2017). In an evolutionary sense, complement is thought to have evolved as a mechanism of innate immunity that can be activated by appearance of foreign invaders to produce protective effector mechanisms. It is likely that the natural antibodies to autologous lipids, proteins, or nucleic acids exist as markers to recognize cellular debris, or for recognizing the appearance of epitopes that are normally hidden due to steric hindrance; and the antibodies serve as agonists that attract scavenger cells (Alving and Wassef 1999).
The protein corona can pose a difficult problem for targeted intravascular delivery of liposomal therapeutic drugs, for example, to tumor cells, because the liposomes are rapidly coated with opsonins, including complement, resulting in uptake by phagocytic cells. However, from a vaccine standpoint, delivery of liposomes that are coated with opsonins (such as complement) to cells in the immune system, including phagocytic cells, represents a natural advantage and also highlights a rational mechanism for using liposomes for adjuvant development.
The composition of the protein corona that will associate with the liposomes after injection in vivo depends on several factors. First, the initial protein corona, which is the only protein or peptide initially associated with the liposomes, is the antigen itself. The affinity of the immunogenic liposome-associated peptide or protein for the liposomal surface can be influenced by the liposomal surface charge, by the hydrophobicity of the antigen and by its access to the hydrophobic region of the lipid bilayer, or by the composition of the liposomal phospholipid headgroup.
3 Intramuscular Fate of Injected Liposomal Adjuvant Formulations
Among the many mechanisms by which liposomal vaccine adjuvants work, targeting of the liposomes to phagocytic cells, and activation of the cells can be an important and sometimes a critical first step for initiation of the adaptive immune response. The original paradigm in the complement field held that complement was a system of proteins that were present in serum. However, certain locations in the body exist where serum proteins do not have ready access, but where complement or other types of opsonins are still needed, for example, in skeletal muscle or subcutaneous tissue, or in other areas that include interstitial spaces. By the same token, skeletal muscle contains very few immune cells such as macrophages, monocytes, dendritic cells, and neutrophils (Liang et al. 2015). Recent research has demonstrated that various complement components, regulators, and receptors are synthesized by immune cells themselves, including polymorphonuclear leukocytes, mast cells, monocytes, macrophages, dendritic cells, NK cells, and B and T lymphocytes (Lubbers et al. 2017). In addition, a “hidden arsenal” of complement and complement-related proteins exists inside T lymphocytes, and perhaps in many, and perhaps even most cells (Liszewski et al. 2017). However, the major problem for vaccine development is that immune cells are only few in number in muscle tissue. For liposomes that bind directly to cells, such as certain types of targeted liposomal (virosome) vaccines (Zurbriggen 2003; Herzog et al. 2009), or for cationic liposomes that attach to negatively charged cell surfaces to form a depot (Henriksen-Lacey et al. 2010), this might not be a great problem.
It is interesting to speculate that perhaps complement (or natural antibodies), which are synthesized by even a few cells in skeletal muscle, might accumulate in the local extracellular environment; and these materials might bind to injected liposomes, resulting in complement activation and generation of chemotactic factors and other attractants that could result in increased numbers of immune cells through chemotaxis. However, because of the paucity of immune cells in skeletal muscle, and the paucity of opsonins in muscle that attach to liposomes, in many instances anionic liposomes by themselves lacking adjuvants do not increase the immunogenicities of certain antigens, and the liposomal formulation requires addition of an adjuvant, such as monophosphoryl lipid A (Alving et al. 1986; White et al. 1995). Thus, it is more likely that an immunostimulator that serves as an adjuvant must be added to many types of vaccines, including neutral or anionic liposomal vaccines, to cause a local inflammation in order to achieve a significant adaptive immune response to the antigen after injection in skeletal muscle (Liang and Lore 2016). Even second-generation virosomal virus-like particle types of liposomal vaccines are greatly improved by the addition of an adjuvant (Kamphuis et al. 2012; Moser et al. 2013; Smith et al. 2013). Regardless of the mechanism by which liposomal formulations containing adjuvants attract immune cells to the intramuscular injection site, the intracellular fate of liposomes in phagocytic cells is often a critical step in the adaptive immune response.
4 Intracellular Fate of Liposomes in Phagocytic Cells
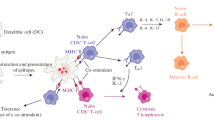
As mentioned above, there is an influx of phagocytic cells to the intramuscular injection site and, after uptake of the liposome-associated antigen by a phagocyte the antigen is ultimately transported to the draining lymph nodes for interaction with T-cells. One of the main roles of phagocytic antigen presenting cells (APC) is to sample the environment and present antigens, and then to start appropriate immune signaling once the danger is identified. The first step in this process is the internalization of the liposomal antigen by phagocytic cell for processing followed by presentation of the peptides on the surface of the antigen presenting cell in the context of the major histocompatibility complex (MHC) class I or class II molecules to interact with the T-cell receptor. Antigen processing is highly complex, involving a number of different components, some of which overlap between MHC class I and class II pathways. In conjunction with the secondary stimulus consisting of costimulatory molecules, an antigen-specific cellular (Th1) or a humoral (Th2) response is induced along with the corresponding cytokines.
Liposomes, both anionic and cationic, induce mixed Th1/Th2 responses (Rao et al. 1999a, 2002; Jafari et al. 2018). Antigen processing through both the MHC class I and class II pathways can account for the mixed Th1/Th2 responses induced by liposomes containing MPLA-associated antigen. Typically, endogenous antigens are processed through the MHC class I pathway, while exogenous antigens are processed through the MHC class II pathway (Bevan 1987; Germain and Margulies 1993). Liposomes containing antigen and MPLA are unique in that, although liposomes are exogenous particles, liposomal antigen can escape to the cytoplasm, as shown by the presence of gold-labeled antibody–antigen complex in the cytoplasm (Fig. 1). The antigen can be processed through both the MHC class I (Fig. 2) and class II pathways (Fig. 3). The antigen presentation pathway leading to the loading of exogenous antigens on MHC class I molecules is called cross-presentation. Cross-presentation can occur by two main pathways; the cytosolic pathway, in which antigen processing occurs in the cytosol, and the vacuolar pathway, in which antigen processing occurs within endocytic compartments (Joffre et al. 2012). Because of the close contact between the phagosomes and the endoplasmic reticulum (ER), it has been speculated that specific lipids might act as second messengers promoting a cross-presentation pathway, although no such lipids have been identified so far (Nunes-Hasler and Demaurex 2017). In contrast to macrophages, the endocytic compartments of DCs prevent destruction of internalized antigens due to limited proteolysis; and because of this, DCs, but not macrophages, favor cross-presentation.

Immunogold electron microscopy of macrophages after phagocytosis of liposomes containing lipid A (isolated from Salmonella minnesota R595 LPS) and recombinant R32NS1 malaria antigen. Murine bone marrow-derived macrophages were fixed 6 h after incubation with liposomes containing malaria antigen. The malaria antigen was detected by a specific antibody to the malaria antigen followed by treatment with gold-labeled secondary antibody. Please note that the gold-labeled antibody–antigen complex has escaped into the cytoplasm. Bar = 0.5 µm. From Verma et al. (1991)

MHC class I liposomal antigen trafficking pathway. (1) Protein encapsulated in liposomes is phagocytosed into antigen presenting cells; and (2) enters an endocytic compartment. (3) Protein, lipid-protein or lipopeptide are released into the cytosol of the cell, and (4) are processed by either a proteasome or immunoproteasome complex. (5) Peptide or lipopeptide is then transported by the heterodimeric TAP complex into the endoplasmic reticulum and loaded onto MHC class I-β2 microglobulin molecules. (6) The MHC class I-loaded molecule traffics to the cis-Golgi. (7) From the cis-Golgi, the complex traffics through trans-Golgi complex. (8) After exiting the trans-Golgi, the MHC class I-peptide complex is transported to the cell surface where it binds to CD8+ T-cells via the T-cell receptor. (9) The TCR-MHC-I/peptide complex activates the CD8+ cell, which in turn releases cytokines such as IFN-γ

MHC class II liposomal antigen trafficking pathway. (1) Liposome-associated antigen is phagocytosed into antigen presenting cells, and (2) enters an endocytic compartment. (3) The heterotrimeric α and β chains of the MHC class II molecule along with the invariant chain (Ii) are synthesized in the endoplasmic reticulum, and traffic to the (4) cis- and (5) trans-Golgi complex. (6) Following maturation in the Golgi complex, the MHC class II heterotrimeric complex with the Ii chain bound to the MHC class II groove then enters the MHC class II compartment (MIIC) where the invariant chain is degraded into the Class II Invariant chain Peptide (CLIP) which is still bound to the MHC-II groove. Peptides generated in the endosomes from exogenous liposomal antigens also enter the MIIC compartment. In this compartment, CLIP (present in the MHC class II groove) is replaced by the peptides (generated in the endosomes) that also traffic to the MIIC compartment. (6) The peptide-MHC-II complexes then move to the cell surface, (7) for interaction with the T-cell receptor of CD4+ T-cells. (8) The TCR-MHC-II/peptide complex activates the CD4+ T-cells and orchestrates a range of immune responses, which includes stimulation of B cells for antibody production
At one time, it was proposed that “pH-sensitive” liposomes were a requirement for the delivery of exogenous antigen into the cytoplasmic compartment of the APCs and for the induction of cytotoxic T-cells (CTLs) in vivo (Reddy et al. 1991; Collins et al. 1992). However, it is now well established that liposomes which are pH-insensitive can also deliver the antigen to the cytosol (Fig. 1) and can induce good CTL responses against a variety of antigens, ranging from viruses to encapsulated or surface-bound recombinant proteins and peptides (Engler et al. 2004; Rao et al. 2002, 2004; White et al. 1993, 1995; Laborde et al. 2017). To demonstrate the presence of liposomal antigens in the various cellular compartments, we utilized bone marrow-derived macrophages as pure populations of APCs and studied the intracellular trafficking of liposome-encapsulated antigen in vitro, using fluorescently labeled lipids and proteins (Rao and Alving 2000).
4.1 MHC Class I Processing Pathway
Multiple steps within the MHC class I and II pathways have been characterized for liposome-associated antigen uptake and processing. The first step in each pathway is the internalization of the liposome-associated antigen either through pinocytosis, endocytosis, or phagocytosis, depending upon the size of the liposomes. For further details on a variety of internalization mechanisms of nanostructured systems, see (Voltan et al. 2017). These different routes of uptake could result in the antigen entering different processing compartments such as the endosomes, lysosomes, or the cytosol, as shown pictorially in Figs. 2 and 3 (steps 1 and 2). The presence of liposomal antigen in the cytoplasmic compartment has been shown for ovalbumin, ferritin or a malaria antigen by electron microscopy or by the use of fluorescent liposomes (Zhou et al. 1994; Perry and Martin 1995; Verma et al. 1991; Rao and Alving 2000). At the point of contact between either a soluble or liposomal antigen with the cell membrane, a rearrangement of the cytoskeleton occurs to facilitate the uptake of the antigens. Further trafficking of the liposomal antigen requires a functional microtubule-dependent translocation system (Peachman et al. 2004). The protein, lipoprotein or lipopeptide that escapes into the cytoplasm (Fig. 2 step 3) is ubiquitinated and then channeled through the proteasome complex (Fig. 2, step 4) for further hydrolyses into short peptides (Rothwell et al. 2000; Steers et al. 2008).
Peptides that are generated by the proteasome cleavage step are transported across the membrane of the ER and the cis- and trans-Golgi in an ATP-dependent manner by specific trimeric transporters associated with antigen processing (TAP) (Fig. 2 steps 5–7 and Fig. 4A). TAP proteins consist of TAP1 and TAP2, tapasin [an ER resident chaperone, also known as TAP-binding protein (TAPBP)], ERp57 (an oxidoreductase), and calreticulin (a sugar-binding chaperone) (Margulies et al. 2020; Androlewicz et al. 1992; Neefjes et al. 1993; Kleijmeer et al. 1992; Li et al. 1999). These molecules interact with the short peptides mainly due to specific determinants found at the carboxyl termini of the peptides (Androlewicz et al. 1992; Neefjes et al. 1993; Kleijmeer et al. 1992; Li et al. 1999). In the absence of TAP proteins, the liposomal peptides are not transported to the ER or to the Golgi complex and remain in the cytoplasmic compartment (Fig. 4A, panels 1–3) (Rao et al. 1997). The peptide–lipopeptides are loaded onto empty MHC class I-β2 microglobulin molecules in the ER and then transported to the cell surface via the Golgi complex (Fig. 2, steps 6–8). A schematic three-dimensional representative drawing of the structure of the Golgi can be found in Pagano (1990) and Krstic (1979) in which the Golgi fields of a composite Golgi apparatus were drawn from thick sections examined in a high-voltage electron microscope. Branching tubules connecting the Golgi cisternae, which create a complex network with numerous pores on the inner surface of the Golgi cisternae, can be seen in the drawing. One important post-translational modification that mainly occurs in the Golgi is glycosylation, which is driven by glycosyltransferases including sialyltransferase (Bao et al. 2015). The peptides bound to MHC class I molecules are then transported to the cell surface to interact with CD8+ T-cells resulting in the activation of the T-cell and secretion of IFNγ (Fig. 2, step 9). Since the peptide has to traverse through the Golgi complex before being transported to the cell surface, and because this is an important step in the intracellular trafficking pathway, localization of the peptide/lipopeptide in the trans-Golgi can be visualized by the congruence of the overlaid fluorescence of the labeled protein and a vital stain for the Golgi apparatus, C6NBD-ceramide, that is specific to the trans-Golgi (Lipsky and Pagano 1985; Pagano et al. 1989). The trans-Golgi is labeled with C6NBD-ceramide in all of the figures presented in this article.

Liposomal antigen trafficking in murine bone marrow-derived cells. A Importance of TAP complex. (1) Lack of concentration in the trans-Golgi of Texas red-labeled liposomal conalbumin in live bone marrow-derived macrophages from TAP1 knock-out mice. (2) Trans-Golgi was labeled with C6NBD-ceramide (green color). Concentration of the labeled liposomal ovalbumin in macrophages from (4) wild-type mice in the (5) trans-Golgi. (3) and (6) The corresponding bright field images of the respective macrophages are shown. From Rao et al. (1999b). B Human macrophages do not concentrate soluble antigen. (1) Diffuse staining of Texas red-labeled Ebola envelope peptide in the absence of liposomes and lack of concentration in the (2) trans-Golgi labeled with C6NBD-ceramide (green color) as seen with the (3) overlay image. (4) The bright field image is shown. C Concentration of liposomal Ebola peptide. (5) Concentration and localization of Texas red-labeled Ebola envelope peptide encapsulated in liposomes in the (6) trans-Golgi as evidenced by the (7) overlay image. The regions of complete identity appear yellowish orange. (8) The bright field image is shown. The white arrows indicate the (5) area of antigen concentration and (6) in the trans-Golgi area in panel D, respectively. Scale bar, 10 mm. From Peachman et al. (2005)
Although both free antigens and liposome-associated antigens (in this case, an Ebola antigen) can be internalized by macrophages, as shown in Fig. 4B and c, soluble Ebola peptide remained diffuse and punctate (Fig. 4B, panel 1) while the liposome-associated Texas red-labeled Ebola glycoprotein peptide [L(TR-Ebola peptide)] was concentrated (Fig. 4C, panel 5). The Golgi staining (Fig. 4B, panel 2 and Fig. 4C, panel 6) and the antigen distributions are shown as overlays in Fig. 4B, panel 3 and Fig. 4C, panel 7, respectively (Peachman et al. 2005). Thus, unlike the liposomal antigen, soluble free antigens did not localize to the area of the trans-Golgi. Furthermore, antigen concentration within the trans-Golgi (Fig. 5B, E, H) required both the lipid and the protein components (Fig. 5D, G), since the absence of either component, the protein (Fig. 5A) or the lipid (Fig. 4B, panel 1), resulted in the absence of localization to the trans-Golgi area or to any particular region of the cell. Therefore, when the liposomes were associated with an antigen, the fluorescent lipids mainly localized in the Golgi (Fig. 5G, H); and conversely, when the liposomes were associated with an antigen that was fluorescently labeled, the antigen also colocalized in the region of the trans-Golgi (Fig. 5D, E). This pattern of intracellular trafficking of liposomal lipids was affected neither by the MHC haplotype nor by the chain length of the fatty acid in the phospholipid of the liposomes (Rao and Alving 2000). In the absence of protein in the liposomes, only 5.3% of the cells showed localization of the lipids in the Golgi. However, with liposome-associated protein, the lipids localized predominately in the Golgi in about 70% of the cells. Thus, the presence of a protein in the liposomes had a remarkable “targeting” effect on the liposomal lipid, resulting in the deposition of most of the lipid within the Golgi. Likewise, the presence of the lipid associated with the protein also resulted in the trafficking of the fluorescent peptides to the trans-Golgi (Rao and Alving 2000; Rao et al. 1999b). These results have profound implications for both protein and lipid processing pathways and presentation to T-cells. At the present time, the intracellular pathway for lipid antigens has not been fully studied and this could be a fruitful subject for future analysis.

Lipid and protein requirement for Golgi-specific localization. (A) Diffuse staining pattern of Texas red-labeled liposomes (no protein) in live murine bone marrow-derived macrophages with a distinct absence of localization of the labeled lipid in the trans-Golgi area (B). The trans-Golgi (white arrow) was labeled with C6NBD-ceramide (Lipsky and Pagano 1985; Pagano et al. 1989). (D) When the liposomes contained an antigen, Texas red-labeled conalbumin, the liposomal antigen localized to the trans-Golgi (E) as depicted by the white arrows. (G) Localization of the liposomal lipids (labeled lipids) was observed (white arrow) (H) When the labeled liposomes contained unlabeled antigen. Thus, both protein and lipids are essential for liposomal antigens to localize to the trans-Golgi area. The corresponding bright field images are shown respectively in (C, F, and I). Scale bar, 10 nm. From Rao et al. (1999b)
The peptide-MHC-I complex probably utilizes the microtubules or shuttle vesicles to reach the cell surface. The presence of this complex was visualized either by flow cytometry (Fig. 6A) or by fluorescence microscopy (Fig. 6B) with an antibody specific for the MHC class I-peptide complex, in this case the SIINFEKL complex (Porgador et al. 1997; Rothwell et al. 2000). Once on the surface, it was accessible for interaction with the TCR of CD8+ T-cells (Fig. 2 step 8) (Rao et al. 1999a, b). This interaction caused cell activation and the release of cytokines such as IFN-γ (Fig. 2 step 9). Liposome-associated protein and exogenous particulate antigens are noted for their propensities to induce IFN-γ secretion and to induce cytotoxic T lymphocytes (CTLs) (Schmidt et al. 2019; Ramakrishnan et al. 2019a, b, c; Alving et al. 1995; Lopes and Chain 1992; Reddy et al. 1992; White et al. 1993; Rao et al. 2004). Entry into the MHC class I pathway was demonstrated by the induction of conalbumin-specific MHC class I-restricted CD8 CTL responses in mice (Fig. 6C) after intraperitoneal injection of liposomal conalbumin (Rao and Alving 2000). The data generated in vitro thus predicted the possible outcomes in vivo. Although, there are differences in the proteins required for MHC class I and MHC class II processing and presentation, antigens processed through one pathway can also be presented by the other pathway (Giodini and Albert 2010).

Detection of peptide-MHC class I complexes on the cell surface and induction of cytotoxic T-cells. Peptide-MHC class I complexes were detected following incubation of murine bone marrow-derived hybridoma C2.3 cells with either SIINFEKL peptide (an OVA peptide) or liposomal ovalbumin L(OVA) or buffer (control) followed by an antibody specific for the peptide–MHC complex and examined either by A flow cytometry or B fluorescence microscopy. C Cytotoxic T lymphocyte recall responses in murine splenic lymphocytes immunized with liposomal conalbumin, cultured with conalbumin peptides, CTL epitope of malaria CSP antigen (negative control), or media. Standard 4-hr CTL assays were performed using 51Cr-labeled peptide pulsed target cells, and percentage specific lysis was determined. From Rothwell et al. (2000), Rao and Alving (2000)
4.2 MHC Class II Processing Pathway
MHC class II molecules are expressed on professional APCs, including DCs, monocytes, macrophages, and B cells. Exogenous antigens such as liposomal antigens can enter the endosomal compartment through endocytosis, phagocytosis (Fig. 3 steps 1 and 2), or by autophagy (Munz 2010). The endosomal compartments contain numerous cathepsins and other proteases, which are cell-type specific, and can be differentially expressed based on the activation status of the cell (Stern et al. 2006). Dendritic cells and nonactivated cells contain low concentrations of cathepsins, which allows for the preservation of the antigens. In contrast, macrophages and activated cells contain high concentrations of cathepsins that rapidly degrade antigens. The enzymes cleave endocytosed antigens and generate a peptide array containing both MHC class I and MHC class II epitopes. The peptides can follow two routes. They are either retro-translocated into the cytosol where they can then enter the classical MHC class I pathway and be processed by the proteasome complex, or they enter the MHC class II compartment (MIIC) where they encounter the MHC class II molecules.
The α and β glycoprotein chains of the MHC class II molecule are synthesized in the ER and associate with the invariant chain (Ii) to form a heterotrimer complex. The Ii chaperone molecule stabilizes the MHC heterodimer and also prevents endogenous peptides from binding to the groove of the MHC molecule. The heterotrimer complex then transits from the ER (Fig. 3 step 3) to the cis and trans-Golgi (Fig. 3 steps 4, 5) and enters the MIIC compartment, where the Ii chain is degraded by cathepsins and other proteases. A part of the Ii chain, the class II invariant chain peptide (CLIP), is retained in the MHC-II groove (Couture et al. 2019). The peptides containing the MHC class II epitopes enter the MIIC compartment where they are loaded onto class II molecules after removal of CLIP (Fig. 3 step 6) (Roche and Cresswell 1991; Boes et al. 2005) and then transported to the cell surface (Fig. 3 step 7) as a 12–15 amino acid epitope bound to an MHC class II molecule (Stern et al. 2006; Chapman 2006). The peptide-MHC-II complex then interacts with the T-cell receptor on CD4+ T-cells and in conjunction with the costimulatory signals, activates the T-cell (Fig. 3 step 8) and orchestrates a broad range of immune responses, which include providing help to the B cells for antibody production (Rao et al. 2018).
HIV-1 Env-A244 gp120 protein, one of the protein boost subunits of the RV144 phase 3 vaccine trial, was shown to be resistant to proteasomes but susceptible to cathepsin cleavage, and the resulting peptides were identified by mass spectrometry. The peptide fragments induced a polyfunctional cytokine response including the generation of IFN-γ from CD4+ T-cell lines derived from RV144 vaccinated volunteers (Steers et al. 2012). In vivo studies with both liposome-encapsulated as well as liposome-associated antigens have demonstrated robust induction of antibody and cellular immune responses (Rao et al. 2018; Cawlfield et al. 2019; Ramakrishnan et al. 2019a, b, c; Beck et al. 2015b, 2018; Genito et al. 2017; Seth et al. 2017; Alving et al. 2016; Wieczorek et al. 2015) thus proving that liposomal antigens can enter both MHC class I and class II pathways in vitro and in vivo.
As shown above, anionic liposomes containing MPLA undergo a complex choreographed intracellular trafficking pattern in phagocytes leading to generation of Th1-type and Th2-type immunity. It is important to note that ALF-like liposomes lacking MPLA often have little or no immunostimulant adjuvant activity (Alving et al. 1986; White et al. 1995). Furthermore, the liposomal lipid serves as a physical chaperone to deliver the processed antigen through the Golgi to present the peptide together with MHC class I and class II molecules on the cell surface, a trafficking pattern that is not available to the antigen in the absence of the liposome.
5 Rational Selection of Liposomal Human Vaccine Adjuvant Formulations
Immunization of human volunteers with experimental vaccine formulations has long been viewed as a benchmark for evaluation and rational selection of constituents likely to achieve scientific and commercial success (Alving 2002). Certainly, for analysis of the potential for toxicity, feedback from human recipients is a major requirement; and challenge of human volunteers with the target pathogen can lead to even more rigorous analysis. Obviously, caution is required in any type of human immunization, and animal models are often critically important both for preclinical evaluation of potential vaccine efficacy and toxicity. However, in the absence of a rigorous animal model of the human disease, such as is the case for a therapeutic vaccine to prostate cancer which is unique to humans, human experimentation is required (Alving 2002).
Adjuvant research, and particularly malaria vaccine adjuvant research, has been a major beneficiary of the availability of human challenge models, such as the controlled human malaria infection (CHMI) for which curative drugs are available (Chulay et al. 1986). In 1984, the gene encoding the circumsporozoite (CS) protein on the outer surface of P. falciparum was cloned and sequenced (Dame et al. 1984; Enea et al. 1984). Of particular importance for vaccine development was a region in the middle of the protein composed of 37 Asn-Ala-Asn-Pro tetrapeptide repeats interspersed with four Asn-Val-Asp-Pro tetrapeptides. Although murine antibodies to recombinant proteins comprising multiple copies of the tetrapeptides recognized the complete CS protein, higher titers of the antibodies in experimental animals required the presence of either aluminum hydroxide or Freund’s adjuvant (Young et al. 1985). In the first phase 1 human trial of a recombinant protein (R32tet32) adsorbed to aluminum hydroxide (falciparum sporozoite vaccine-1; known as FSV-1), six immunized volunteers were challenged by CHMI with the bite of infected mosquitoes. The good news was that protection was observed against infection; but the disappointing news was that fifty weeks after the first immunization, and after three subsequent boosts of vaccine, only 1 of 6 volunteers was protected, and protection was observed only in the volunteer with the highest antibody titer (Ballou et al. 1987; Young et al. 1987).
At the same time that the FSV-1 malaria vaccine was being developed, a new adjuvant, known as Walter Reed liposomes, and later known as Army Liposome Formulation (ALF), was being developed at WRAIR for additional human vaccine trials (Alving et al. 1986, 2020). Based on the previous disappointing results observed with aluminum-adsorbed FSV-1, the first use of a liposome-based vaccine in humans was undertaken, and a recombinant protein containing the CS protein tetrapeptides was used as the antigen (Fries et al. 1992). In this study, two adjuvants were present in the vaccine formulation: MPLA in the liposomal bilayer, and the liposomes were also adsorbed to aluminum hydroxide as a second adjuvant. In addition to strong immune potency, the liposomes completely blocked the inherent pyrogenicity of MPLA, the primary signal of toxicity, even at potentially lethal amounts (>2 mg of MPLA). When compared to the FSV-1 vaccine, which contained only aluminum salt as an adjuvant, anti-NANP titers induced by the aluminum-adsorbed liposomal MPLA formulation were tenfold higher. When later compared with six other adjuvants in a multicenter phase 1 trial with a single antigen (recombinant gp120 envelope protein from HIV-1) (AVEG 015), the aluminum-adsorbed ALF formulation containing encapsulated antigen had greater apparent potency and the lowest apparent toxicity (equivalent to aluminum hydroxide alone) (Mcelrath 1995). Recent analysis of sera obtained from individuals in the AVEG 015 trial immunized with aluminum-adsorbed antigen or aluminum-adsorbed ALF containing encapsulated antigen revealed dramatically increased anti-gp120 titers and increased antibody duration, and high titers of antibodies specific to V1/V2 loop of the gp120 envelope protein (Rao et al. 2018).
In our experience, the variables to be considered in construction of liposomal formulations are many, but first and foremost the ability to sterilize the preparation is critical. This can be achieved by sterile filtration of each of the components at some point during the manufacture through a 0.2-μm filter and maintaining sterility afterward (Matyas et al. 2003). Thus, liposomes that are nanoparticles can be easily sterile-filtered after manufacture. However, for anionic liposomes the requirement for nanosized particles potentially limits the use of phospholipids to those that have unsaturated fatty acids which automatically form nanoliposomes. Liposomes with saturated phospholipids (such as ALF) can be manufactured by the ethanol injection technique (Wagner and Vorauer-Uhl 2011), or by the use of microfluidization to create nanosized particles. The chemical composition of the liposomes, the physical chemistry of the liposomal membrane scaffold, and the type and mechanism of action of the liposome-associated adjuvant, are all important factors. For example, in studies in which the protective antigen (PA) of anthrax was adjuvanted with aluminum salt (as in the licensed anthrax vaccine) and then improved by using ALF instead of aluminum salt, it was discovered that liposome encapsulation of PA was not necessary, and the PA could simply be mixed with ALF in formulating the vaccine (Rao et al. 2011).
In the above-described first human liposomal vaccine trial with malaria antigen, and in the AVEG 015 trial with HIV-1 gp120, the liposomal adjuvant formulation (ALF) was manufactured at WRAIR using synthetic phospholipids and natural MPLA (MPL®) extracted from Salmonella bacterial lipopolysaccharide supplied by Ribi ImmunoChem Research, Inc. The liposomes contained a heterogeneous population of large (micro) and small (nano) liposomes, thus preventing sterile filtration with a 0.22 μm filter; and in this case, the liposomes required sterile manufacture. Because of the potency of the Ribi MPLA as a human vaccine adjuvant, the Ribi company became valuable as an adjuvant manufacturing company, and it now belongs to GSK. The ALF-type adjuvant was re-worked by GSK and expanded to include a formulation of nanoliposomes containing both MPLA and the saponin QS21 as adjuvants, which is now known as Adjuvant System 01 (AS01) (Vandepapeliere 2018; Alving et al. 2020). After numerous comparative clinical trials, a successful sporozoite malaria vaccine (RTS,S/AS01, also known as Mosquirix®) with a recombinant protein containing the repeat tetrapeptides of the CS protein (RTS,S) and the AS01 adjuvant emerged. The formulation received a positive scientific opinion by the European Medicines Agency exclusively for markets outside of the European Union and is currently being further tested with a four-injection regimen under World Health Organization auspices among hundreds of thousands of children 5–9 months of age in settings of moderate-to-high parasite transmission for malaria in sub-Saharan Africa (World Health Organization 2018). The AS01 adjuvant has also enabled a highly successful vaccine to Herpes zoster (Shingrix®) that is currently licensed for human use by the U.S. Food and Drug Administration for prevention of shingles (Lal et al. 2015).
In view of the above sequence of events, it seemed reasonable to presume that the AS01 adjuvant could serve as a rational model for further development of similar types of liposomal vaccine adjuvant formulations (Alving et al. 2020). To that end, ALFQ, namely ALF liposomes containing QS21 saponin, was created as a new entry in the ALF family of adjuvants (Beck et al. 2015a, b) (Fig. 7). ALFQ was created by adding QS21 to microfluidized nanoliposomes (50–100 nm diameter), thus allowing sterile filtration prior to this step. However, in contrast to the AS01 formulation, addition of sterile-filtered QS21 to the nanoliposomes caused a huge increase in the size of the liposomes, to produce large unilamellar or oligolamellar vesicles, as large as 30,000 nm in diameter (Beck et al. 2015b). This size change occurred through cannibalization of the nanoparticles to form microparticles. Although some nanoparticles remained, they were greatly reduced in number, and the vast majority of the total volume and total surface area of the ALFQ particles were likely contributed by microparticles instead of nanoparticles. To illustrate this by solid geometry, a single 30,000 nm spherical particle contains a volume that is >216 million times greater than that of a 50 nm particle, and the surface area of the single outer leaflet of the lipid bilayer of a 30,000 nm particle is approximately 360,000 times greater than that of a 50 nm particle. Further investigation revealed that the difference in the liposome size between AS01 and ALFQ was due to the use of unsaturated dioleoyl phosphatidylcholine (DOPC) as the bulk phospholipid in AS01, and of saturated dimyristoyl phosphatidylcholine and dimyristoyl phosphatidylglycerol as the bulk phospholipids in ALFQ (Singh et al. 2019). Preliminary experiments have suggested that the adjuvant potencies in mice of a lab-prepared AS01-like formulation and the micron-sized ALFQ liposome preparations are not significantly different (manuscript in preparation).

Schematic illustration of the Army Liposome Formulation containing QS21 (ALFQ). Liposomes containing 55% cholesterol and monophosphoryl lipid A are shown with QS-21 which is approaching the cholesterol where it binds tightly via hydrophobic regions. This enables the eight sugars of QS21 to be displayed at the liposomal surface. Monophosphoryl lipid A adds two more sugars at the water interface, thus forming a complex and diverse sugar display (a “sugar lawn”) by ALFQ on the liposome surface. From Alving et al. (2020)
As mentioned above, ALF liposomes completely lack the pyrogenic toxicity of MPLA (Fries et al. 1992). However, free QS21 by itself is toxic in that it causes considerable acute local pain immediately on injection which recedes over the course of thirty minutes (Waite et al. 2001; Edelman et al. 2002), and it causes necrosis at the injection site in muscles in rodents (Garcon et al. 2007). Although the exact mechanism of the acute pain experienced after injection of QS21 has never been fully studied, it is likely due to cytotoxicity, such as hemolysis, caused by irreversible binding of QS21 to cholesterol present in lipid bilayer membranes of local cells (Beck et al. 2015a). To mitigate this toxic effect of QS21, AS01 and ALFQ each contain cholesterol in their liposomal membranes, and the liposomal cholesterol serves as a sink that irreversibly and strongly binds to QS21, and liposomal cholesterol thus blocks the binding of QS21 to cells in the vicinity of the injection site. Still, despite the absence of immediate pain, many who have received the Shingrix® vaccine that contains AS01B as an adjuvant have experienced local reactions at the injection site that occur hours after injection and which last for several days. If these delayed local effects are due to the AS01 adjuvant, it might seem unlikely that they are caused by the QS21 because of the strong and irreversible binding of QS21 to the liposomal cholesterol; and if this is so, then either the DOPC or the MPLA in the AS01 might somehow be the culprit. It is conceivable that MPLA that is present in AS01 might be expressing local toxicity due to incomplete blocking by the highly unsaturated phospholipid environment of AS01. Two ongoing clinical trials utilizing ALFQ adjuvant with two different malaria vaccines may help to determine whether reactogenicity is experienced by humans after injections of vaccines containing ALFQ (Alving et al. 2020).
Regardless of the issues of reactogenicity and how to address them, the mechanisms underlying the extraordinary potency of AS01 as an adjuvant for Mosquirix® and Shingrix® are still subjects of ongoing research, but it seems clear that the combined constituents of AS01, namely DOPC, cholesterol, MPLA, and QS21, mobilizes multiple mechanisms of innate immunity leading to the adaptive immune response (Alving et al. 2012; Didierlaurent et al. 2014; Welsby et al. 2016). Nanoliposomes are clearly important as a practical means for achieving sterility of liposomal vaccine development; however, it should be noted that although ALFQ starts with nanoliposomes, QS21 interacting with the high concentrations of cholesterol transforms the vesicles from nanosize to microsize (Singh et al. 2019; Alving and Beck 2019). ALFQ is easily manufactured by using sterile-filtered QS21 that is added to the sterile-filtered ALF nanoliposomes. In summary, adjuvant research is still a dynamic and developing field in which successful adjuvant products, including liposomal adjuvants, that are present in licensed human vaccines provide important directions for rational adjuvant selection.
6 Conclusion
Early discoveries of liposome–protein complexes (such as complement protein complexes) emphasized the impact of exposure of liposomes to biological proteins such as complement on the delivery and uptake of liposome-associated antigens by APCs. This has been further informed by research from the nanomedicines field on the importance of a so-called protein corona of proteins attached to nanoparticles introduced to the biosphere. While the protein corona of a particle sometimes may have a detrimental effect on delivery of nanomedicines, in the vaccine adjuvant field a protein corona attached to liposomes can be an important factor for delivery of liposomal antigens to APCs. The intracellular trafficking patterns of liposomes and liposome-associated protein in APCs provided important insights into the mechanisms by which liposomes can influence the immune response. For example, these studies indicated that liposome-associated peptide, but not free peptide, can transit through the trans-Golgi, and a complex structure of lipid and protein thus appears to be an important element for the MHC class I processing pathway in phagocytic cells.
Because of the continuous fifty-year background of experience with liposome-associated proteins and different types of liposomes at WRAIR, there have been smooth transitions to translational applications (Fig. 8). The development of the Army Liposome Formulation (ALF) family of vaccine adjuvants is currently being employed in a variety of phase 1 studies with different types of candidate vaccines. As with the first liposome vaccine injected into humans in 1982, which comprised a candidate malaria vaccine, the first injections of candidate vaccines containing ALFQ as antigens are currently ongoing with two different types of candidate malaria vaccines (Alving et al. 2020). The overall safety and potency of the vaccines containing ALFQ will be evaluated over the next year.

Selected milestones of liposome research at WRAIR. Liposome research has been ongoing at WRAIR for fifty years, since 1970. It has been undertaken with three foci: a basic membrane biochemistry, cellular interactions, and immunological research; b drug delivery research; and c research on liposomes as adjuvants for many types of vaccines
References
Allison AG, Gregoriadis G (1974) Liposomes as immunological adjuvants. Nature 252:252
Alving CR (1984) Natural antibodies against phospholipids and liposomes in humans. Biochem Soc Trans 12:342–344
Alving CR (1991) Liposomes as carriers of antigens and adjuvants. J Immunol Methods 140:1–13
Alving CR (2002) Design and selection of vaccine adjuvants: animal models and human trials. Vaccine 20(Suppl 3):S56–S64
Alving C, Beck Z (2019) Non-toxic adjuvant formulation comprising a monophosphoryl lipid A (MPLA)-containing liposome composition and a saponin. US patent application, 8 Oct 2019
Alving CR, Kinsky SC (1971) The preparation and properties of liposomes in the LA and LAC states. Immunochemistry 8:325–343
Alving CR, Wassef NM (1999) Naturally occurring antibodies to cholesterol: a new theory of LDL cholesterol metabolism. Immunol Today 20:362–366
Alving CR, Richards RL, Moss J et al (1986) Effectiveness of liposomes as potential carriers of vaccines: applications to cholera toxin and human malaria sporozoite antigen. Vaccine 4:166–172
Alving CR, Koulchin V, Glenn GM et al (1995) Liposomes as carriers of peptide antigens: induction of antibodies and cytotoxic T lymphocytes to conjugated and unconjugated peptides. Immunol Rev 145:5–31
Alving CR, Peachman KK, Rao M et al (2012) Adjuvants for human vaccines. Curr Opin Immunol 24:310–315
Alving CR, Beck Z, Matyas GR et al (2016) Liposomal adjuvants for human vaccines. Expert Opin Drug Deliv 13:807–816
Alving CR, Peachman KK, Matyas GR et al (2020) Army Liposome Formulation (ALF) family of vaccine adjuvants. Expert Rev Vaccines 1–14
Androlewicz MJ, Browning JL, Ware CF (1992) Lymphotoxin is expressed as a heteromeric complex with a distinct 33-kDa glycoprotein on the surface of an activated human T cell hybridoma. J Biol Chem 267:2542–2547
Ballou WR, Hoffman SL, Sherwood JA et al (1987) Safety and efficacy of a recombinant DNA Plasmodium falciparum sporozoite vaccine. Lancet 1:1277–1281
Bangham AD, Standish MM, Watkins JC (1965) Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol 13:238–252
Bao L, Ding L, Yang M et al (2015) Noninvasive imaging of sialyltransferase activity in living cells by chemoselective recognition. Sci Rep 5:10947
Beck Z, Matyas GR, Alving CR (2015a) Detection of liposomal cholesterol and monophosphoryl lipid A by QS-21 saponin and Limulus polyphemus amebocyte lysate. Biochim Biophys Acta 1848:775–780
Beck Z, Matyas GR, Jalah R et al (2015b) Differential immune responses to HIV-1 envelope protein induced by liposomal adjuvant formulations containing monophosphoryl lipid A with or without QS21. Vaccine 33:5578–5587
Beck Z, Torres OB, Matyas GR et al (2018) Immune response to antigen adsorbed to aluminum hydroxide particles: effects of co-adsorption of ALF or ALFQ adjuvant to the aluminum-antigen complex. J Control Release 275:12–19
Bevan MJ (1987) Antigen recognition. Class discrimination in the world of immunology. Nature 325:192–194
Boes M, Van Der Wel N, Peperzak V et al (2005) In vivo control of endosomal architecture by class II-associated invariant chain and cathepsin S. Eur J Immunol 35:2552–2562
Bohlson SS, Garred P, Kemper C et al (2019) Complement nomenclature-deconvoluted. Front Immunol 10:1308
Bonte F, Juliano RL (1986) Interactions of liposomes with serum proteins. Chem Phys Lipids 40:359–372
Cawlfield A, Genito CJ, Beck Z et al (2019) Safety, toxicity and immunogenicity of a malaria vaccine based on the circumsporozoite protein (FMP013) with the adjuvant army liposome formulation containing QS21 (ALFQ). Vaccine 37:3793–3803
Cedervall T, Lynch I, Lindman S et al (2007) Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci U S A 104:2050–2055
Chapman HA (2006) Endosomal proteases in antigen presentation. Curr Opin Immunol 18:78–84
Chonn A, Cullis PR, Devine DV (1991) The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J Immunol 146:4234–4241
Chonn A, Semple SC, Cullis PR (1992) Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J Biol Chem 267:18759–18765
Chulay JD, Schneider I, Cosgriff TM et al (1986) Malaria transmitted to humans by mosquitoes infected from cultured Plasmodium falciparum. Am J Trop Med Hyg 35:66–68
Collins DS, Findlay K, Harding CV (1992) Processing of exogenous liposome-encapsulated antigens in vivo generates class I MHC-restricted T cell responses. J Immunol 148:3336–3341
Couture A, Garnier A, Docagne F et al (2019) HLA-Class II artificial antigen presenting cells in CD4(+) T cell-based immunotherapy. Front Immunol 10:1081
Dame JB, Williams JL, Mccutchan TF et al (1984) Structure of the gene encoding the immunodominant surface antigen on the sporozoite of the human malaria parasite Plasmodium falciparum. Science 225:593–599
De Serrano LO, Burkhart DJ (2017) Liposomal vaccine formulations as prophylactic agents: design considerations for modern vaccines. J Nanobiotechnol 15:83
Didierlaurent AM, Collignon C, Bourguignon P et al (2014) Enhancement of adaptive immunity by the human vaccine adjuvant AS01 depends on activated dendritic cells. J Immunol 193:1920–1930
Didierlaurent AM, Laupeze B, Di Pasquale A et al (2017) Adjuvant system AS01: helping to overcome the challenges of modern vaccines. Expert Rev Vaccines 16:55–63
Edelman R, Wasserman SS, Kublin JG et al (2002) Immediate-type hypersensitivity and other clinical reactions in volunteers immunized with a synthetic multi-antigen peptide vaccine (PfCS-MAP1NYU) against Plasmodium falciparum sporozoites. Vaccine 21:269–280
Enea V, Ellis J, Zavala F et al (1984) DNA cloning of Plasmodium falciparum circumsporozoite gene: amino acid sequence of repetitive epitope. Science 225:628–630
Engler OB, Schwendener RA, Dai WJ et al (2004) A liposomal peptide vaccine inducing CD8+ T cells in HLA-A2.1 transgenic mice, which recognise human cells encoding hepatitis C virus (HCV) proteins. Vaccine 23:58–68
Fries LF, Gordon DM, Richards RL et al (1992) Liposomal malaria vaccine in humans: a safe and potent adjuvant strategy. Proc Natl Acad Sci U S A 89:358–362
Garcon N, Chomez P, Van Mechelen M (2007) GlaxoSmithKline adjuvant systems in vaccines: concepts, achievements and perspectives. Expert Rev Vaccines 6:723–739
Genito CJ, Beck Z, Phares TW et al (2017) Liposomes containing monophosphoryl lipid A and QS-21 serve as an effective adjuvant for soluble circumsporozoite protein malaria vaccine FMP013. Vaccine 35:3865–3874
Germain RN, Margulies DH (1993) The biochemistry and cell biology of antigen processing and presentation. Annu Rev Immunol 11:403–450
Giodini A, Albert ML (2010) A whodunit: an appointment with death. Curr Opin Immunol 22:94–108
Gregoriadis G (1978) Liposomes in therapeutic and preventive medicine: the development of the drug-carrier concept. Ann N Y Acad Sci 308:343–370
Haxby JA, Kinsky CB, Kinsky SC (1968) Immune response of a liposomal model membrane. Proc Natl Acad Sci U S A 61:300–307
Haxby JA, Gotze O, Muller-Eberhard HJ et al (1969) Release of trapped marker from liposomes by the action of purified complement components. Proc Natl Acad Sci U S A 64:290–295
Henriksen-Lacey M, Bramwell VW, Christensen D et al (2010) Liposomes based on dimethyldioctadecylammonium promote a depot effect and enhance immunogenicity of soluble antigen. J Control Release 142:180–186
Heppner DG, Gordon DM, Gross M et al (1996) Safety, immunogenicity, and efficacy of Plasmodium falciparum repeatless circumsporozoite protein vaccine encapsulated in liposomes. J Infect Dis 174:361–366
Herzog C, Hartmann K, Kunzi V et al (2009) Eleven years of inflexal V-a virosomal adjuvanted influenza vaccine. Vaccine 27:4381–4387
Jafari I, Heravi Shargh V, Shahryari M et al (2018) Cationic liposomes formulated with a novel whole Leishmania lysate (WLL) as a vaccine for leishmaniasis in murine model. Immunobiology 223:493–500
Joffre OP, Segura E, Savina A et al (2012) Cross-presentation by dendritic cells. Nat Rev Immunol 12:557–569
Kamphuis T, Meijerhof T, Stegmann T et al (2012) Immunogenicity and protective capacity of a virosomal respiratory syncytial virus vaccine adjuvanted with monophosphoryl lipid A in mice. PLoS ONE 7:e36812
Ke PC, Lin S, Parak WJ et al (2017) A decade of the protein corona. ACS Nano 11:11773–11776
Kinsky SC, Haxby JA, Zopf DA et al (1969) Complement-dependent damage to liposomes prepared from pure lipids and Forssman hapten. Biochemistry 8:4149–4158
Kleijmeer MJ, Kelly A, Geuze HJ et al (1992) Location of MHC-encoded transporters in the endoplasmic reticulum and cis-Golgi. Nature 357:342–344
Krstic RE (1979) Ultrastructure of the Mammalian Cell. An Atlas. Springer-Verlag, New York
Laborde RJ, Sanchez-Ferras O, Luzardo MC et al (2017) Novel adjuvant based on the pore-forming protein sticholysin II encapsulated into liposomes effectively enhances the antigen-specific CTL-mediated immune response. J Immunol 198:2772–2784
Lal H, Cunningham AL, Godeaux O et al (2015) Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 372:2087–2096
Li S, Paulsson KM, Sjogren HO et al (1999) Peptide-bound major histocompatibility complex class I molecules associate with tapasin before dissociation from transporter associated with antigen processing. J Biol Chem 274:8649–8654
Li M, Du C, Guo N et al (2019) Composition design and medical application of liposomes. Eur J Med Chem 164:640–653
Liang F, Lore K (2016) Local innate immune responses in the vaccine adjuvant-injected muscle. Clin Transl Immunology 5:e74
Liang F, Ploquin A, Hernandez JD et al (2015) Dissociation of skeletal muscle for flow cytometric characterization of immune cells in macaques. J Immunol Methods 425:69–78
Lipsky NG, Pagano RE (1985) A vital stain for the Golgi apparatus. Science 228:745–747
Liszewski MK, Elvington M, Kulkarni HS et al (2017) Complement’s hidden arsenal: new insights and novel functions inside the cell. Mol Immunol 84:2–9
Lopes LM, Chain BM (1992) Liposome-mediated delivery stimulates a class I-restricted cytotoxic T cell response to soluble antigen. Eur J Immunol 22:287–290
Lubbers R, Van Essen MF, Van Kooten C et al (2017) Production of complement components by cells of the immune system. Clin Exp Immunol 188:183–194
Margulies DH, Jiang J, Natarajan K (2020) Structural and dynamic studies of TAPBPR and Tapasin reveal the mechanism of peptide loading of MHC-I molecules. Curr Opin Immunol 64:71–79
Matyas GR, Alving CR (2011) Antigen-specific enhancement of natural human IgG antibodies to phosphatidylcholine, phosphatidylglycerol, phosphatidylinositol-4-phosphate, cholesterol, and lipid A by a liposomal vaccine containing lipid A. Vaccine 29:5137–5144
Matyas GR, Muderhwa JM, Alving CR (2003) Oil-in-water liposomal emulsions for vaccine delivery. Methods Enzymol 373:34–50
Mcelrath MJ (1995) Selection of potent immunological adjuvants for vaccine construction. Semin Cancer Biol 6:375–385
Moser C, Muller M, Kaeser MD et al (2013) Influenza virosomes as vaccine adjuvant and carrier system. Expert Rev Vaccines 12:779–791
Munz C (2010) Antigen processing via autophagy–not only for MHC class II presentation anymore? Curr Opin Immunol 22:89–93
Neefjes JJ, Momburg F, Hammerling GJ (1993) Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science 261:769–771
Nelson RA Jr, Jensen J, Gigli I et al (1966) Methods for the separation, purification and measurement of nine components of hemolytic complement in guinea-pig serum. Immunochemistry 3:111–135
Nisini R, Poerio N, Mariotti S et al (2018) The multirole of liposomes in therapy and prevention of infectious diseases. Front Immunol 9:155
Nunes-Hasler P, Demaurex N (2017) The ER phagosome connection in the era of membrane contact sites. Biochim Biophys Acta Mol Cell Res 1864:1513–1524
Pagano RE (1990) The Golgi apparatus: insights from lipid biochemistry. Biochem Soc Trans 18:361–366
Pagano RE, Sepanski MA, Martin OC (1989) Molecular trapping of a fluorescent ceramide analogue at the Golgi apparatus of fixed cells: interaction with endogenous lipids provides a trans-Golgi marker for both light and electron microscopy. J Cell Biol 109:2067–2079
Peachman KK, Rao M, Palmer DR et al (2004) Functional microtubules are required for antigen processing by macrophages and dendritic cells. Immunol Lett 95:13–24
Peachman KK, Rao M, Alving CR et al (2005) Human dendritic cells and macrophages exhibit different intracellular processing pathways for soluble and liposome-encapsulated antigens. Immunobiology 210:321–333
Pedersen GK, Andersen P, Christensen D (2018) Immunocorrelates of CAF family adjuvants. Semin Immunol 39:4–13
Perry DG, Martin WJ 2nd (1995) Fluorescent liposomes as quantitative markers of phagocytosis by alveolar macrophages. J Immunol Methods 181:269–285
Pick H, Alves AC, Vogel H (2018) Single-vesicle assays using liposomes and cell-derived vesicles: from modeling complex membrane processes to synthetic biology and biomedical applications. Chem Rev 118:8598–8654
Porgador A, Yewdell JW, Deng Y et al (1997) Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 6:715–726
Ramakrishnan A, Schumack NM, Gariepy CL et al (2019a) Correction for Ramakrishnan et al., “Enhanced immunogenicity and protective efficacy of a Campylobacter jejuni conjugate vaccine coadministered with liposomes containing monophosphoryl lipid A and QS-21”. mSphere 4
Ramakrishnan A, Schumack NM, Gariepy CL et al (2019b) Enhanced immunogenicity and protective efficacy of a Campylobacter jejuni conjugate vaccine coadministered with liposomes containing monophosphoryl lipid A and QS-21. mSphere 4
Ramakrishnan A, Schumack NM, Gariepy CL et al (2019c) Erratum for Ramakrishnan et al., “Enhanced immunogenicity and protective efficacy of a Campylobacter jejuni conjugate vaccine coadministered with liposomes containing monophosphoryl lipid A and QS-21”. mSphere 4
Rao M, Alving CR (2000) Delivery of lipids and liposomal proteins to the cytoplasm and Golgi of antigen-presenting cells. mangala.rao@na.amedd.army.mil. Adv Drug Deliv Rev 41:171–188
Rao M, Rothwell SW, Wassef NM et al (1997) Visualization of peptides derived from liposome-encapsulated proteins in the trans-Golgi area of macrophages. Immunol Lett 59:99–105
Rao M, Matyas GR, Grieder F et al (1999a) Cytotoxic T lymphocytes to Ebola Zaire virus are induced in mice by immunization with liposomes containing lipid A. Vaccine 17:2991–2998
Rao M, Rothwell SW, Wassef NM et al (1999b) Trafficking of liposomal antigen to the trans-Golgi of murine macrophages requires both liposomal lipid and liposomal protein. Exp Cell Res 246:203–211
Rao M, Bray M, Alving CR et al (2002) Induction of immune responses in mice and monkeys to Ebola virus after immunization with liposome-encapsulated irradiated Ebola virus: protection in mice requires CD4(+) T cells. J Virol 76:9176–9185
Rao M, Matyas GR, Vancott TC et al (2004) Immunostimulatory CpG motifs induce CTL responses to HIV type I oligomeric gp140 envelope protein. Immunol Cell Biol 82:523–530
Rao M, Peachman KK, Li Q et al (2011) Highly effective generic adjuvant systems for orphan or poverty-related vaccines. Vaccine 29:873–877
Rao M, Onkar S, Peachman KK et al (2018) Liposome-encapsulated human immunodeficiency virus-1 gp120 induces potent V1V2-specific antibodies in humans. J Infect Dis 218:1541–1550
Reddy R, Zhou F, Huang L et al (1991) pH sensitive liposomes provide an efficient means of sensitizing target cells to class I restricted CTL recognition of a soluble protein. J Immunol Methods 141:157–163
Reddy R, Zhou F, Nair S et al (1992) In vivo cytotoxic T lymphocyte induction with soluble proteins administered in liposomes. J Immunol 148:1585–1589
Roche PA, Cresswell P (1991) Proteolysis of the class II-associated invariant chain generates a peptide binding site in intracellular HLA-DR molecules. Proc Natl Acad Sci U S A 88:3150–3154
Rothwell SW, Wassef NM, Alving CR et al (2000) Proteasome inhibitors block the entry of liposome-encapsulated antigens into the classical MHC class I pathway. Immunol Lett 74:141–152
Schmidt ST, Olsen CL, Franzyk H et al (2019) Comparison of two different PEGylation strategies for the liposomal adjuvant CAF09: towards induction of CTL responses upon subcutaneous vaccine administration. Eur J Pharm Biopharm 140:29–39
Schwendener RA (2014) Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines 2:159–182
Segal AW, Wills EJ, Richmond JE et al (1974) Morphological observations on the cellular and subcellular destination of intravenously administered liposomes. Br J Exp Pathol 55:320–327
Seth L, Bingham Ferlez KM, Kaba SA et al (2017) Development of a self-assembling protein nanoparticle vaccine targeting Plasmodium falciparum circumsporozoite protein delivered in three army liposome formulation adjuvants. Vaccine 35:5448–5454
Singh P, Beck Z, Matyas GR et al (2019) Saturated phospholipids are required for nano- to micron-size transformation of cholesterol-containing liposomes upon QS21 addition. J Liposome Res 29:247–250
Smith GE, Flyer DC, Raghunandan R et al (2013) Development of influenza H7N9 virus like particle (VLP) vaccine: homologous A/Anhui/1/2013 (H7N9) protection and heterologous A/chicken/Jalisco/CPA1/2012 (H7N3) cross-protection in vaccinated mice challenged with H7N9 virus. Vaccine 31:4305–4313
Steers NJ, Alving CR, Rao M (2008) Modulation of immunoproteasome subunits by liposomal lipid A. Vaccine 26:2849–2859
Steers NJ, Ratto-Kim S, De Souza MS et al (2012) HIV-1 envelope resistance to proteasomal cleavage: implications for vaccine induced immune responses. PLoS ONE 7:e42579
Stern LJ, Potolicchio I, Santambrogio L (2006) MHC class II compartment subtypes: structure and function. Curr Opin Immunol 18:64–69
Tyrrell DA, Heath TD, Colley CM et al (1976) New aspects of liposomes. Biochim Biophys Acta 457:259–302
Uemura K, Nicolotti RA, Six HR et al (1974) Antibody formation in response to liposomal model membranes sensitized with N-substituted phosphatidylethanolamine derivatives. Biochemistry 13:1572–1578
Vandepapeliere P (2018) Vaccine compositions comprising a saponin adjuvant, 4 Dec 2018
Verma JN, Wassef NM, Wirtz RA et al (1991) Phagocytosis of liposomes by macrophages: intracellular fate of liposomal malaria antigen. Biochim Biophys Acta 1066:229–238
Voltan AR, Alarcon KM, Fusco-Almeida AM et al (2017) Highlights in endocytosis of nanostructured systems. Curr Med Chem 24:1909–1929
Vu VP, Gifford GB, Chen F et al (2019) Immunoglobulin deposition on biomolecule corona determines complement opsonization efficiency of preclinical and clinical nanoparticles. Nat Nanotechnol 14:260–268
Wagner A, Vorauer-Uhl K (2011) Liposome technology for industrial purposes. J Drug Deliv 2011:591325
Waite DC, Jacobson EW, Ennis FA et al (2001) Three double-blind, randomized trials evaluating the safety and tolerance of different formulations of the saponin adjuvant QS-21. Vaccine 19:3957–3967
Wang N, Chen M, Wang T (2019) Liposomes used as a vaccine adjuvant-delivery system: from basics to clinical immunization. J Control Release 303:130–150
Wassef NM, Alving CR (1987) Complement-dependent phagocytosis of liposomes by macrophages. Methods Enzymol 149:124–134
Welsby I, Detienne S, N’kuli F et al (2016) Lysosome-dependent activation of human dendritic cells by the vaccine adjuvant QS-21. Front Immunol 7:663
White K, Krzych U, Gordon DM et al (1993) Induction of cytolytic and antibody responses using Plasmodium falciparum repeatless circumsporozoite protein encapsulated in liposomes. Vaccine 11:1341–1346
White WI, Cassatt DR, Madsen J et al (1995) Antibody and cytotoxic T-lymphocyte responses to a single liposome-associated peptide antigen. Vaccine 13:1111–1122
Wieczorek L, Krebs SJ, Kalyanaraman V et al (2015) Comparable antigenicity and immunogenicity of oligomeric forms of a novel, acute HIV-1 Subtype C gp145 envelope for use in preclinical and clinical vaccine research. J Virol 89:7478–7493
World Health Organization (2018) Malaria vaccine: WHO position paper, January 2016—recommendations. Vaccine 36:3576–3577
Young JF, Hockmeyer WT, Gross M et al (1985) Expression of Plasmodium falciparum circumsporozoite proteins in Escherichia coli for potential use in a human malaria vaccine. Science 228:958–962
Young JF, Ballou WR, Hockmeyer WT (1987) Developing a human malaria sporozoite vaccine. Microb Pathog 2:237–240
Zhou F, Watkins SC, Huang L (1994) Characterization and kinetics of MHC class I-restricted presentation of a soluble antigen delivered by liposomes. Immunobiology 190:35–52
Zurbriggen R (2003) Immunostimulating reconstituted influenza virosomes. Vaccine 21:921–924
Disclaimer
The views expressed are those of the authors and should not be construed to represent the positions of the US Army or the Department of Defense.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made.
Copyright information
© 2021 This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply
About this chapter

Cite this chapter
Rao, M., Peachman, K.K., Alving, C.R. (2021). Liposome Formulations as Adjuvants for Vaccines. In: Gill, H.S., Compans, R.W. (eds) Nanoparticles for Rational Vaccine Design. Current Topics in Microbiology and Immunology, vol 433. Springer, Cham. https://doi.org/10.1007/82_2020_227
Download citation
DOI: https://doi.org/10.1007/82_2020_227
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-85066-1
Online ISBN: 978-3-030-85067-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)