
Abstract
This is our follow-up study carried out in an order to collect experimental evidence regarding the impact of heavy water (D2O) on the spontaneous oscillatory peptidization of the sulfur-containing proteinogenic α-amino acids and this time our target is l-methionine (l-Met). Our earlier study has been focused on l-cysteine (l-Cys) and it seemed interesting for us to compare the effect induced by D2O on these two structurally related compounds. In a broader sense though, our goal is to investigate if spontaneous oscillatory peptidization of various different proteinogenic α-amino acids might play any role in living organisms. As analytical techniques, we used high-performance liquid chromatography with the evaporative light-scattering detection (HPLC-ELSD), mass spectrometry (MS), scanning electron microscopy (SEM), and turbidimetry. The obtained results demonstrate that heavy water seriously hampers the oscillatory peptidization of l-Met, apparently due to the presence of the deuterium cation in the reaction medium and in the first instance, due to isotopic substitution of two functionalities participating in peptidization, i.e., –COOH and –NH2. This effect is fully analogous to that earlier reported for l-Cys. A cautious conclusion can be drawn that far more experimental evidence is still needed to be able to judge about an importance (or otherwise) of the oscillatory peptidization of proteinogenic α-amino acids for various different life processes.
Similar content being viewed by others


Introduction
The spontaneous oscillatory condensation of the low molecular weight carboxylic acids dissolved in organic-aqueous solvents was for the first time observed by our research group upon the examples of d- and l-phenylglycine (d- and l-Phg) dissolved in 70% aqueous ethanol and reported [1]. In another paper [2], the first theoretical model was proposed to explain the observed phenomenon. Since that time, spontaneous oscillatory peptidization of different proteinogenic α-amino acids was reported in a number of papers [3,4,5,6,7,8,9]. The main analytical tools in all these studies were the liquid chromatographic techniques (thin-layer chromatography, TLC, and high-performance liquid chromatography, HPLC), but also other instrumental techniques like polarimetry, spectroscopy of circular dichroism in the UV range (CD), mass spectrometry (MS), scanning electron microscopy (SEM), turbidimetry, and other. Periodicity of spontaneous oscillatory peptidization was checked with the use of the Fourier transform approach applied to the non-monotonous time series of the concentration changes with the selected monomeric α-amino acids. In certain cases, the circadian rhythm of the oscillations was observed (equal to ca. 20 h for l-proline (l-Pro) [4] and to ca. 24 h for l-cysteine (l-Cys) [9]), whereas for l-hydroxyproline (l-Hyp), the lack of the circadian rhythm was confirmed [4]. Moreover, the circadian rhythm of spontaneous oscillatory peptidization (equal to ca. 24 h) was revealed based on turbidimetric results for l-serine (l-Ser) and the lack of this characteristics was confirmed for d-serine (d-Ser) [10]. The discovery and then investigation of spontaneous oscillatory chiral inversion [11,12,13] and spontaneous oscillatory condensation [1,2,3,4,5,6,7,8,9,10] with the low molecular weight chiral carboxylic acids taking place in the abiotic liquid systems seemed to be a strong indication that these phenomena might have some relevance to biological systems as well, or even to evolutionary processes.
Although a number of theoretical models [2, 4, 13, 14] have been developed in an effort to rationalize the experimental results provided in the aforementioned studies, obtaining proper kinetic results which might ultimately confirm the validity of these models is hardly possible, basically due to the lack of adequate analytical tools able to register rapid concentration changes of α-amino acids and oligopeptides in a continuous mode, i.e., in very short (e.g., sub-minute) time intervals. Thus in an effort to check if spontaneous oscillatory peptidization might have a biological importance, we decided to dissolve certain proteinogenic α-amino acids in solvents containing heavy water (D2O) and to check their influence on oscillatory peptidization. On the one hand, it is well known that D2O exerts lethal effect on higher organisms such, as fishes, birds and mammals [15,16,17,18,19], and with certain mammals such effect has been established at the 30% D2O level per the organism’s weight [18]. On the other hand, some experiments have been performed on an impact of D2O on the most well-known oscillatory processes, i.e., the Belousov–Zhabotinsky (BZ) [20,21,22] and the Bray–Liebhafsky (BL) reaction [23, 24]. The most up-to-the-date information on the impact of D2O on the BZ reaction provided in paper [22] points out to an increased induction period in a cerium-catalyzed BZ reaction, in pace with progressive substitution of the deuterated reactants. Moreover, a delayed onset and reduced amplitude of oscillations were also observed. In separate works [23, 24], the authors revealed a considerable perturbation of the mechanism and kinetics of the BL reaction, caused by replacement of bulk H2O as a reaction medium with D2O.
Inspired by a well documented impact of D2O on physiology of living organisms on the one hand and the dynamics of the two widely recognized oscillatory reactions (BZ and BL) on the other, we performed a reconnaissance research on the influence of D2O on peptidization of the selected α-amino acids (l-Phe, l-His, l-Pro and l-Cys) using two analytical techniques, i.e., mass spectrometry (MS) and turbidimetry [25]. Based on these preliminary results, an immediate conclusion could be drawn as to an evident hampering effect of D2O on the dynamics and peptidization yields. Then we decided to investigate an impact of D2O on the oscillatory peptidization of the most important proteinogenic α-amino acids more systematically and with a wider range of analytical techniques. Earlier [9], we presented the results of our study on the influence of D2O on peptidization of l-Cys obtained with use of HPLC, MS, SEM, and turbidimetry. It was shown that dissolution of l-Cys in pure D2O fully stopped the oscillatory concentration changes of the monomeric l-Cys, as well as formation and accumulation of the l-Cys-derived oligopeptides. We also used a number of the aqueous organic solvents composed of 70% acetonitrile and 30% H2O, 30% D2O, and the 30% H2O–D2O mixtures in the different H2O/D2O volume proportions to show that even 30% D2O mixed with 70% acetonitrile was enough to stop oscillatory peptidization and formation of the oligopeptides almost completely.
In this study, we continue our investigations on the impact of D2O on spontaneous oscillatory peptidization and our present target is another sulfur-containing proteinogenic α-amino acid, i.e., l-methionine (l-Met). It plays an important role in living organisms as an inevitable substrate in formation of choline, creatine and adrenalin, and actively participates in the process of transmethylation [26]. Besides, it is abundantly present in casein and egg white, where it has been in fact first discovered [27]. The employed measuring techniques are HPLC with the evaporative light-scattering detection (HPLC-ELSD), MS, SEM and turbidimetry.
Experimental
Reagents and samples
In our experiments, we used l-Met of analytical purity (Reanal, Budapest, Hungary), acetonitrile (ACN) of HPLC purity (J.T. Baker, Deventer, The Netherlands) and heavy water (D2O) (Cambridge Isotopic Laboratories, Andover, MA, USA; 99% purity). Water (H2O) was de-ionized and double distilled in our laboratory by means of the Elix Advantage model Millipore System.
For the HPLC/ELSD experiment, we prepared the l-Met solution at a concentration of 1 mg mL−1 (i.e., 6.70 × 10−3 mol L−1) in ACN + H2O, 70:30 (v/v). The chromatographic analysis of the l-Met sample was carried out in the 10-min intervals for the period of 100 h at 21 ± 0.5 °C.
For mass spectrometry, scanning electron microscopy and turbidimetry, we prepared five l-Met samples at a concentration of 1 mg mL−1 in the binary liquid mixtures ACN + X, 70:30 (v/v), where X: the binary mixture of H2O + D2O in the changing volume proportions: 30:0, 25:5, 20:10, 10:20 and 0:30. The mass spectrometric, scanning electron microscopic and turbidimetric measurements were also carried out for the sixth sample, i.e., the l-Met solution (1 mg mL−1) prepared in pure heavy water (D2O).
High-performance liquid chromatography with evaporative light scattering detection (HPLC-ELSD)
The analysis of the freshly prepared l-Met solution was performed in with use of the Varian model 920 liquid chromatograph equipped with the Varian 900-LC autosampler, the gradient pump, the Varian Pro Star 510 model column oven, the Varian 380-LC ELSD detector, the ThermoQuest Hypersil C18 column (150 mm × 4, 6 mm i.d.; 5 μm particle size) and the Galaxie software. The column was thermostatted at 35 °C. The chromatographic analyses were carried out for the 3 μL sample aliquots, using methanol–water (20:80, v/v) as mobile phase at the flow rate of 0.8 mL min−1 in the isocratic mode.
Scanning electron microscopy (SEM)
Visualization of nano- and microparticles was performed with the JEOL JSM-7600F model scanning electron microscope, SEM (Jeol Ltd., Tokyo, Japan). Series of micrographs were recorded for the six investigated l-Met samples after 1 month sample storage period at 22.0 ± 0.5 °C. Visualization was performed for the nano- and microparticles obtained from the respective solutions evaporated to dryness.
Mass spectrometry (MS)
The mass spectra were recorded for the six l-Met solutions, which contained the soluble peptide fraction (the insoluble microparticle suspensions self-separated by sedimentation). Mass spectrometric detection was carried out straight away after a 7-day sample storage period. All the obtained mass spectra were recorded in the positive ionization mode with use of the Varian MS-100 mass spectrometer (the extended ESI–MS scan, positive ionization, the spray chamber temperature 50 °C, the drying gas temperature 250 °C, the drying gas pressure 25 psi, the capillary voltage 50 V, and the needle voltage 5 kV).
Turbidimetry
Turbidity measurements were carried out using the turbidity sensor (TRB-BTA, Vernier Software & Technology, Beaverton, OR, USA) that allowed continuous monitoring of the changes in turbidity. Measurements were performed for the six investigated l-Met samples. Approximately 15-mL aliquots of these solutions were freshly prepared and placed in the instrument vials. The turbidity changes were registered for the period of 7 days (in the 1-min intervals) under the thermostatic conditions at 25.0 ± 0.5 °C. To confirm qualitative reproducibility of the results, the turbidity measurements were repeated twice.
Moreover, the stability of turbidity was controlled for D2O, H2O, acetonitrile and 70% aqueous acetonitrile as the reference solvents and it was established as equal to 91.8, 0.0, 80.1 and 40.1 NTU (nephelometric turbidity units), respectively, over the period of 20 h. In each case, the turbidity has proved as quite stable.
Results and discussion
Let us start our considerations from schematic presentation of the processes of spontaneous oscillatory chiral inversion, spontaneous oscillatory peptidization and these two processes running in the parallel (Supplementary Material, Fig. S1). They were discovered in our earlier studies on the low molecular weight chiral carboxylic acids and are mentioned in the Introduction part. Schemes given in Fig. S1 illustrate an up-to-the-date understanding of the molecular-level mechanisms of these two processes upon an example of l-Met. It is evident that they largely depend on proton transfer and at least partial replacement of H2O by D2O in the reaction environment can result in an isotopic H+/D+ exchange in the –COOH and –NH2 functionalities. Each part of our further discussion will focus on tracing an impact of D2O on the dynamics of peptidization of l-Met with use of different analytical techniques.
High-performance liquid chromatography with evaporative light scattering detection (HPLC-ELSD)
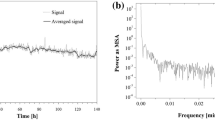
The chromatographic experiment was carried out for 100 h in the achiral HPLC-ELSD mode. Sampling of the l-Met solution was performed in the 10-min intervals, in order to gain a semi-kinetic insight in the dynamics of spontaneous peptidization of l-Met dissolved in ACN + H2O, 70:30 (v/v). Our goal was to separate the monomeric l-Met from the spontaneously formed peptides and to trace changing amounts of the monomeric l-Met (represented by changing peak heights of the monomer), due to the alternate processes of spontaneous peptidization and hydrolytic degradation of peptides. Fig. S2a presented in the Supplementary Material shows the time series of the changing l-Met peak heights in the function of the sample storage period. However, Fourier transform applied to this data set did not disclose periodicity within the time series, as it comes out from Fig. S2b, which shows the lack of predominant peak implying periodicity. In this sense, the spontaneous oscillatory peptidization of l-Met differs from the analogous process earlier observed for the other sulfur atom containing proteinogenic α-amino acid, i.e., l-Cys, for which the experiment was performed under fully analogous working conditions and revealed the circadian rhythm of the oscillations characterizing with the period of 23.8 h [9].
The analogous experiment of storing l-Met in pure D2O for the period of 48 h resulted in practically unchanged chromatographic peak heights of this compound, equivalent to an unchanged l-Met amount throughout the whole storage period. Thus, on the basis of the HPLC results we were able to assume that the environment of D2O—unlike that consisting of ACN + H2O (70:30, v/v)—practically hampers the process of spontaneous oscillatory peptidization of l-Met.
Scanning electron microscopic (SEM) tracing of spontaneous peptidization of l-Met
On the measuring pin of the microscope, we deposited drops of the investigated solutions upon shaking the vials, in order to deposit the suspensions including not only less soluble peptides, but the floating insoluble particles of higher peptides as well, which in the course of sample storage precipitated and accumulated at the bottom of the storage vials. Then these drops were evaporated to dryness, so that by means of SEM we targeted both, the lower soluble peptides (registered also by mass spectrometry) and the higher insoluble ones (which could not be registered by mass spectrometry). The selected results are shown in Fig. 1a–d.

Scanning electron micrographs recorded for the l-Met-derived peptides retrieved from the samples dissolved in ACN + X, 70:30 (v/v), where X: the binary mixture of H2O + D2O in the changing volume proportions; a 0% D2O, × 550; b 5% D2O, × 3700; c 20% D2O, × 7500; d 100% D2O, × 10,000 (by × the magnification is indicated)
Fig. 1a–c show selected micrographs obtained for the l-Met samples containing 0, 5 and 20% D2O, respectively. Fig. 1d shows the micrographs of l-Met dissolved in pure D2O. It can be seen that with the growing proportions of heavy water in solution, peptide aggregates become perceptibly smaller both in size and yields. Peptides formed in an absence of D2O (Fig. 1a give extensive, flat and band-wise structures, with the length of some of them exceeding even 100 μm. The 5% proportion of D2O in the l-Met solution results in a drastic change of the peptide formation pattern. Now we observe flat and tile-shaped peptides (Fig. 1b), and a considerable shortening of their band-wise segments (Fig. 1b). An increase of the D2O content in solution to 20% results in single, flat and tile-shaped peptides mostly a few micrometers in length and in yet smaller structures, not exceeding 1 μm in size (Fig. 1c). No flat, tile-shaped peptides can be seen on the micrographs obtained for l-Met dissolved in pure D2O, but the small and dispersed structures only, below 1 μm in size (Fig. 1d). In order to get a better insight in an impact of D2O on peptide formation, in Fig. S3 (Supplementary Material) we provided two magnification ranges for each proportion of D2O to better show the details in the zoomed in picture.
Based on a comparison of the micrographs shown in Fig. 1a–d and also in Fig. S3a–d presented in the supplementary material, it seems reasonable to assume that the considerably large and band-wise structures originate from insoluble particles of higher peptides, whereas the small and largely dispersed structures originate from the lower and soluble peptides. It is also evident that the increasing proportions of D2O in solution tend to obstruct formation of higher insoluble peptides, shifting the peptidization process toward the lower and soluble structures.
Mass spectrometric (MS) tracing of spontaneous peptidization of l-Met
With use of the SEM technique, we gained a cumulative picture of the impact of D2O on spontaneous peptidization of l-Met and we were able to trace changing proportions between the yields of the higher insoluble peptides on the one hand and the lower soluble ones on the other, depending on the amount of D2O in solution. Mass spectrometric results are going to show an impact of D2O on formation of the lower soluble peptides only. For the needs of our current discussion, we first have to pre-define the notions of the lower and the higher peptides (and their respective m/z values). Let us make it clear that the peptides recorded in the range up to m/z 3500 (Fig. 2a–d) were soluble and therefore we have labeled all of them as lower peptides. Presence of higher insoluble peptides in the aged l-Met solutions (i.e., of those with molecular weights above 3500 Da) was documented by the scanning electron microscopy (SEM) alone.

Mass spectra recorded for l-Met dissolved in ACN + X, 70:30 (v/v), where X: the binary mixture of H2O + D2O in the changing volume proportions; a 5% D2O; b 20% D2O; c 30% D2O and dl-Met dissolved in pure D2O
Mass spectra for the selected l-Met solutions are presented in Fig. 2. With the l-Met sample containing 5% D2O (Fig. 2a), mass spectrum shows relatively low numbers of the lower-frequency signals (contained mostly in the m/z range below 2000 and with the highest signal intensities below 10 kCounts). Similar characteristics of the mass spectrum is valid for the l-Met sample dissolved in ACN + H2O, 70: 30 (v/v), i.e., in complete absence of heavy water (the result not shown). With the growing amounts of D2O in the investigated solutions, the m/z range of signal appearance is extended to 3500, with the signal yields and intensities growing. Illustration of this trend is provided for the l-Met solutions containing 20% D2O (Fig. 2b) and 30% D2O (Fig. 2c). In these two cases, the intensities of the highest signals are below 15 and 20 kCounts, respectively. Mass spectrum recorded for l-Met dissolved in pure D2O characterizes with very high signal intensities (up to 1300 kCounts), and specifically in the m/z range 1500–3500 (Fig. 2d).
Summing up, mass spectrometric results remain in good agreement with those originating from the scanning electron microscopy and they complement the SEM data by showing that the higher is the amount of D2O in the l-Met solution, the higher are the yields of the lower soluble peptides also, apparently at an expense of the higher insoluble ones. Although mass spectrometry is predominantly an identification technique, in this study, we showed its usefulness as a fingerprinting tool able to provide a convincing enough overall insight in the role of D2O in the process of peptidization. In order to emphasize the peptide nature of the mass spectrometric signals, we tentatively identified the most intense signal encountered in this study (m/z 2754.7; Fig. 2d) and valid for the l-Met solution in pure D2O as [Met21–H2O]+, i.e., as a peptide assembled of 21 monomeric l-Met units.
Turbidimetric tracing of spontaneous peptidization of l-Met
Turbidity changes of all l-Met samples were recorded in one-minute intervals (i.e., in a quasi-continuous mode) for the period of 7 days. Fig. 3 shows the plots of turbidity changes for the selected l-Met solutions, which illustrate the observed trends the best. A sudden initial drop of signal intensity in Fig. 3a and sudden initial growth of signal intensity in Fig. 3b are due to adjusting sample temperature to that of the turbidity measuring device set at 25.0 ± 0.5 °C.

Turbidity changes (in nephelometric turbidity units, NTU) for the l-Met solution in ACN + X, 70:30 (v/v), where X: the binary mixture of H2O + D2O with the changing volume proportions: a 0% D2O and b 10% D2O
For l-Met dissolved in ACN + H2O with 0% D2O, the turbidity values started step-wise dropping after the first day of relative stability (indicated with an arrow in Fig. 3a). In the fourth day of sample storage, the step-wise growth of the turbidity values began, soon followed by further non-monotonous changes. Summing up, the turbidity changes of l-Met stored in an absence of D2O were the most robust, with clearly pronounced ups and downs of the plot. The amount of 5% D2O did not extend the preliminary stability period (figure not shown). The 10% content of D2O only resulted in an extension of the initiation period to 2 days (as indicated with an arrow in Fig. 3b), after which a slow and gradual increase of sample turbidity started. A further increase of the D2O content in the l-Met solutions did not extend the initiation period (plots not shown) and yet the plot valid for an absence of D2O and given in Fig. 3a characterized with the most pronounced curvature.
We should not forget that in view of a lack of standardization in turbidity units, measurement devices and calibration techniques, usage of turbidimetry to analytical determinations can only be empirical and rather qualitative (as directly cited from [28]). However, we can assume that the presented plots of turbidity changes illustrate the overlapping yet opposed effects of spontaneous peptidization and hydrolytic degradation of the peptides in the examined solutions and have a tentative importance only. In an approximative manner though, they confirm our earlier observations regarding the hampering effect of D2O on these processes. An increase of turbidity is interpreted as an increase in the amount of nano- and microparticles suspended in the solution and resulting from the peptidization process, whereas a decrease in turbidity may result from dissociation of higher (insoluble) peptides to lower (soluble) ones and/or from sedimentation of higher insoluble peptides on the bottom of the measuring vial.
Conclusion
In this study, we examined an influence of heavy water (D2O) on spontaneous oscillatory peptidization of l-Met. With use of HPLC-ELSD, we demonstrated oscillatory nature of this process in an absence of D2O and its practical stillstand in pure D2O. With use of SEM, we demonstrated the influence of the increasing proportions of D2O in solution on gradual shift of peptidization equilibria toward the lower and soluble peptides and with use of MS, we confirmed these findings. Turbidimetry provided yet another and purely qualitative insight in the hampering effect of D2O on the dynamics of peptidization. Obviously, these results cannot provide an answer to the question of the importance (or otherwise) of spontaneous oscillatory peptidization of the proteinogenic α-amino acids for various different life processes. However, the similarity between the hampering effect of D2O on peptidization and its lethal effect on many living organisms involuntarily raises such so far unanswered question.
References
Sajewicz M, Gontarska M, Kronenbach D, Kowalska T (2009) On the spontaneous abiotic peptization of phenylglycine in an aqueous medium. Acta Chromatogr 21:151–160
Sajewicz M, Gontarska M, Kronenbach D, Leda M, Kowalska T, Epstein IR (2010) Condensation oscillations in the peptidization of phenylglycine. J Syst Chem. https://doi.org/10.1186/1759-2208-1-7
Sajewicz M, Gontarska M, Kowalska T (2014) HPLC/DAD evidence of the oscillatory chiral conversion of phenylglycine. J Chromatogr Sci 52:329–333
Sajewicz M, Dolnik M, Kowalska T, Epstein IR (2014) Condensation dynamics of l-proline and l-hydroxyproline in solution. RSC Adv 4:7330–7339
Sajewicz M, Godziek A, Maciejowska A, Kowalska T (2015) Condensation dynamics of the l-Pro-l-Phe and l-Hyp-l-Phe binary mixtures in solution. J Chromatogr Sci 53:31–37
Godziek A, Maciejowska A, Talik E, Sajewicz M, Kowalska T (2016) Scanning electron microscopic evidence of spontaneous heteropeptide formation in abiotic solutions of selected a-amino acid pairs. Isr J Chem 56:1057–1066
Maciejowska A, Godziek A, Talik E, Sajewicz M, Kowalska T, Epstein IR (2016) Spontaneous pulsation of peptide microstructures in an abiotic liquid system. J Chromatogr Sci 54:1301–1309
Godziek A, Maciejowska A, Talik E, Wrzalik R, Sajewicz M, Kowalska T (2016) On spontaneously pulsating proline-phenylalanine peptide microfibers. Curr Prot Pept Sci 17:106–116
Fulczyk A, Łata E, Dolnik M, Talik E, Kowalska T, Sajewicz M (2018) Impact of D2O on peptidization of l-cysteine. Reac Kinet Mech Cat 125:555–565
Maciejowska A, Godziek A, Sajewicz M, Kowalska T (2017) Circadian rhythm of spontaneous non-linear peptidization with proteinogenic amino acids in abiotic solutions versus homochirality. Acta Chromatogr 29:135–142
Sajewicz M, Piętka R, Pieniak A, Kowalska T (2005) Application of thin-layer chromatography (TLC) to investigating oscillatory instability of the selected profen enantiomers. Acta Chromatogr 15:131–149
Sajewicz M, Piętka R, Pieniak A, Kowalska T (2005) Application of thin-layer chromatography (TLC) to investigate oscillatory instability of the selected profen enantiomers in dichloromethane. J Chromatogr Sci 43:542–548
Sajewicz M, Gontarska M, Wojtal Ł, Kronenbach D, Leda M, Epstein IR, Kowalska T (2008) Experimental and model investigation of the oscillatory transenantiomerization of l-a-phenylalanine. J Liq Chromatogr Relat Technol 31:1986–2005
Sajewicz M, Matlengiewicz M, Leda M, Gontarska M, Kronenbach D, Kowalska T, Epstein IR (2010) Spontaneous oscillatory in vitro chiral conversion of simple carboxylic acids and its possible mechanism. J Phys Org Chem 23:1066–1073
Katz JJ, Crespi HL, Czajka DM, Finkel AJ (1962) Course of deuteriation and some physiological effects of deuterium in mice. Am J Physiol 203:907–913
Bachner P, McKay DG, Rittenberg D (1964) The pathological anatomy of deuterium intoxication. Proc Natl Acad Sci USA 51:464–471
Richter CP (1976) A study of taste and smell of heavy water (98%) in rats. Proc Soc Exp Biol Med 152:677–684
Richter CP (1977) Heavy water as a tool for study of the forces that control length of period of the 24-hour clock of the hamster. Proc Natl Acad Sci USA 74:1295–1299
Kanto U, Clawson AJ (1980) Use of deuterium oxide for the in vivo prediction of body composition in female rats in various physiological states. J Nutr 110:1840–1848
Kreuels T, Martin W, Brinkmann K (1980) Influence of D2O on the Belousov–Zhabotinsky reaction. Ber Bunsenges Phys Chem 84:411–412
Karavaev AD, Kazakov VP, Tolstikov GA (1986) Deuteration effect in auto-oscillation chemiluminescencje of the Belousov–Zhabotinskii reaction. Reac Kinet Catal Lett 32:21–26
Rossi F, Rustici M, Rossi C, Tiezzi E (2007) Isotopic effect on the kinetics of the Belousov–Zhabotinsky reaction. Int J Mol Sci 8:943–949
Stanisavljev D, Begović N, Vukojević V (1998) Influence of heavy water in the Bray–Liebhafsky oscillating reaction. J Phys Chem A 102:6887–6891
Stanisavljev DR, Vukojević VB (2002) Investigation of the influence of heavy water on kinetic pathways in the Bray–Leiebhafsky reaction. J Phys Chem A 106:5618–5625
Godziek A, Łągiewka A, Kowalska T, Sajewicz M (2018) The influence of heavy water as a solvent on the spontaneous oscillatory reactions of α-amino acids. Reac Kinet Mech Cat 123:141–153
Willke T (2014) Methionine production: a critical review. Appl Microbiol Biotechnol 98:9893–9914
Mueller JH (1923) A new sulphur-containing amino-acid isolated from the hydrolytic products of protein. J Biol Chem 56:157–169
Lawler DM (2005) Turbidity and nephelometry. In: Worsfold P, Townshend A, Poole C (eds) Encyclopedia of analytical science, 2nd edn. Elsevier Academic, San Diego, pp 343–352
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fulczyk, A., Łata, E., Talik, E. et al. Impact of D2O on the peptidization of l-methionine. Reac Kinet Mech Cat 126, 939–949 (2019). https://doi.org/10.1007/s11144-019-01538-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-019-01538-4