
Abstract
Virus-induced gene silencing (VIGS) has emerged as a powerful method for studying gene function. VIGS is induced by infecting a plant with a plant virus that has had its genome modified to include a sequence from the host gene to be silenced. DNAβ and DNA1 are satellite and single-stranded DNA molecules associated with begomoviruses (family Geminiviridae). We converted DNAβ and DNA1 into gene-silencing vectors. The VIGS vectors can induce silencing efficiently in many solanaceous plants. Here, we describe procedures for the use of these two gene-silencing vectors for VIGS in different hosts.
Similar content being viewed by others


Key words
1 Introduction
Virus-induced gene silencing (VIGS) is a technique used to harness the posttranscriptional gene silencing (PTGS) phenomenon for the generation of null mutants in functional genomics. It has emerged as a very attractive reverse-genetics tool and a rapid alternative method for suppression of gene expression without the need of genetic transformation of plants (1). In addition, VIGS can be applied in the analysis of genes whose functional knockout mutations are lethal, and can be targeted to silence multiple genes in the same family and thus overcome functional redundancy (2). Using this method, a recombinant virus genome, carrying a partial nucleotide sequence of a host gene, is used to infect a plant. When the virus spreads systemically, the endogenous gene transcripts, which are homologous to the insert in the viral vector, are degraded via a PTGS pathway (3).
In the last decade, many viruses and viral satellites have been modified into VIGS vectors, including RNA viruses such as Tobacco mosaic virus (4), Potato virus X (5), Tobacco rattle virus (TRV) (6, 7), Tomato bushy stunt virus (8), Turnip yellow mosaic virus (TYMV) (9), Apple latent spherical virus (10), and Poplar mosaic virus (11); DNA viruses such as Tomato golden mosaic virus (12, 13), Cabbage leaf curl virus (CaLCuV) (14), African cassava mosaic virus (15), Pepper huasteco yellow vein virus (16), and Cotton leaf crumple virus (17); and viral satellites such as Tobacco mosaic satellite virus (18), Tomato yellow leaf curl China virus (TYLCCNV) DNAβ (19), Tobacco curly shoot virus (TbCSV) DNAβ (20), Tomato leaf curl virus satellite DNA (21), and TbCSV DNA1 (22). VIGS system has been widely used in solanaceous plant species, such as tomato (7, 23), petunia (22–24), tobacco (10, 18), pepper (25–28), potato (29), and a few other Nicotiana species (19, 28). In recent years, some VIGS systems have been constructed to induce gene silencing in some important economic, ornamental, and/or crop plants, such as pea (30, 31), soybean (32, 33), cotton (17), cassava (15), orchids (34), barley (35), wheat (35, 36), rice and maize (37). In the dicotyledonous model plant Arabidopsis thaliana, VIGS has also been optimized with TRV (38, 39), CaLCuV (14), and TYMV (9) vectors. These vectors have been used to study gene functions in plants and functions of over hundreds of genes and cDNA clones have been identified in recent years. The identified genes are involved in plant abiotic and biotic stress, plant development, basic cell function, metabolic pathways, senescence and programmed cell death, and bacterium–plant, insect–plant, virus–plant, and fungal–plant interactions (40).
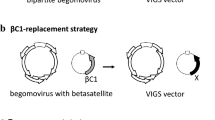
Satellite DNA, referred to as DNAβ and DNA1, are single-stranded DNA molecules associated with begomoviruses (family Geminiviridae). We have converted two begomovirus satellite molecules (DNAβ and DNA1) into gene-silencing vectors, and the DNAβ and DNA1 vectors can induce efficient silencing of a transgene and endogenous genes in tobacco, tomato, and petunia when co-inoculated with helper virus (19, 22). By comparing silencing efficiency induced by DNAβ or DNA1 vector and helper virus, we obtained high silencing efficiency in Nicotiana benthamiana, N. glutinosa, and Petunia hybrid by DNA1 vector and TYLCCNV as helper virus, N. tabacum by DNA1 vector and TbCSV as helper virus, and N. benthamiana, N. glutinosa, P. hybrid, and Solanum lycopersicum by DNAβ vector and TYLCCNV as helper virus. The vectors have been used to investigate gene functions (19, 22, 23, 41, 42). We also found that, compared with other vectors, DNAβ- and DNA1-induced gene silencing is insensitive to high temperature, and VIGS efficiency was not significantly different between 22 and 32°C (41). Now, we describe a detailed DNAβ- and DNA1-based VIGS procedure for identification of gene functions in plants.
2 Materials
-
1.
Clones containing helper virus genome: pBINPLUS TYLCCNV-Y10 1.7A (TYLCCNV), pBINPLUS TbCSV-Y35 1.9A (TbCSV), pGEM T-Easy TYLCCNV-Y10 1.7A (TYLCCNV); DNAβ vector: pBINPLUS2mDNAβ (DNAβ); DNA1 vector: pBINPLUS2mDNA1 (DNA1) (see Note 1).
-
2.
Agrobacterium tumefaciens strain EHA105.
-
3.
Appropriate antibiotic.
-
4.
Injection buffer: 10 mM MgCl2, 10 mM 2-(n-Morpholino) ethanesulfonic acid (MES), and 200 μM 3′–5′ Dimethoxy 4′-hydroxy acetophenone (acetosyringone).
-
5.
The silencing vector contains an insert of Su gene fragment that can be used as a positive control for DNA1 and DNAβ vectors (see Note 2).
-
6.
1-mL syringe with beveled needle.
-
7.
DNA extraction buffer: 0.1 M Tris–HCl pH 8.0, 0.02 M ethylene diamine tetra-acetic acid (EDTA), 1.4 M NaCl, 2% (w/v) cetyl trimethyl ammonium bromide (CTAB), 2% (v/v) 2-β-mercaptoethanol. 2-β-mercaptoethanol must be added immediately before use.
-
8.
Nanodrop ND 1000 UV spectrophotometer.
-
9.
First Strand cDNA Synthesis Kit (MBI).
-
10.
Light Cycler Fast Start DNA Master SYBR Green I mix (Roche).
-
11.
Luria broth (LB) medium (1.0 L): 10 g bacto-tryptone, 5 g Bacto yeast extract, 10 g NaCl and adjust pH to 7.0, sterilize by autoclaving at 121°C for 20 min; YEP medium (1.0 L): 10 g Bacto tryptone, 10 g Bacto yeast extract, 5 g NaCl and adjust pH to 7.0. Sterilize by autoclaving at 121°C for 20 min.
-
12.
PCR primers to allow the specific detection of targeted genes and a control gene.
3 Methods
3.1 Insertion of Gene Sequences into the DNAβ or DNA1 Vector
To silence a gene in plant, a DNA fragment of the target gene should be amplified and cloned into a DNAβ or DNA1 vector by using appropriate restriction enzyme sites. Generally, target gene fragment with lengths of 100–1,000 bp can be amplified from plant cDNA by using specific PCR primers and then the amplified fragment can be inserted into a pGEM T-Easy vector (Promega). After DNA sequencing, the fragment containing appropriate restriction enzyme sites is digested by those enzymes and inserted into a DNAβ or DNA1 vector using the corresponding enzyme sites and the recombinant plasmid is transformed into A. tumefaciens strain EHA105 by electroshock.
3.1.1 Amplification of the Target Gene Fragment and Ligation into a pGEM T-Easy Vector
-
1.
Amplify a fragment (100–1,000 bp in length) from a host cDNA using specific primers (see Notes 3 and 4) under predetermined RT-PCR conditions. An approximately 50 μL PCR reaction mix is needed for each fragment.
-
2.
Purify the PCR product by using QIAquick PCR Purification Kit (QIAGEN) according to experimental protocol.
-
3.
Set up a ligation reaction by mixing 5 μL of PCR product (approximately 200 ng of DNA) with 0.5 μL of pGEM T-Easy Vector (50 ng), 1 μL of 10× ligation buffer, 2.5 μL of nuclease-free H2O, and 1 μL of T4 DNA ligase (Takara) in a microfuge tube. Mix the contents by flicking the tube several times. Spin the tube briefly to collect the ligation mixture at the bottom of the tube and incubate the tube for more than 3 h at 16°C.
-
4.
Transformation of competent E. coli cells with ligated product. Place competent cells (DH5α or JM109, 200 μL cell in a 1.5-mL microfuge tube) from −80°C freezer on ice for 5–10 min, then add 5 μL ligation product into the tube, mix gently by flicking the tube several times, incubate the tube on ice for 20 min, and put the tube to a 42°C metal bath with heat-shock treatment for 60 s followed by a 5-min incubation on ice. Add 800 μL of LB medium into the tube and incubate the tube in a 37°C shaker for 1 h, centrifuge and plate transformed cell culture onto an LB medium plate containing 100 mg/L ampicillin, and incubate the plate overnight in a 37°C incubator.
-
5.
Screen positive clones by PCR using primers specific for target gene. Add the following reagents to microtubes individually (0.2 or 0.5 mL): 2 μL of 10× reaction buffer (contains 25 mM MgCl2), 0.5 μL of 10 mM dNTPs, 0.25 μL of each 10 μM primer solution, 0.5 μL of Taq polymerase, and 16.5 μL of double-distilled H2O (final volume: 20 μL). Pick colonies from the plate with toothpicks and dip them in reagent solution. Program the thermocycler for the following conditions of cycle: initial step at 94°C for 3 min, 30 cycles with the following parameters: denaturation at 94°C for 1 min, annealing at 50°C for 2 min, DNA extension at 72°C for 1 min, and an additional extension step of 10 min at 72°C after the last cycle.
-
6.
Pick positive colonies from the plate with toothpicks and inoculate them into individual test tubes containing 5 mL of LB liquid medium with 100 mg/L ampicillin. Then, incubate the test tubes in a 37°C shaker set at 250 rpm overnight.
-
7.
Obtain pGEM-T Easy plasmid containing target gene fragment through QIAquick Plasmid Extraction Kit (QIAGEN) according to experimental protocol and sequence it.
3.1.2 Insertion of Target Gene Fragment into a DNAβ or DNA1 Vector and Transformation of the Recombined Plasmid into A. tumefaciens EHA105
-
1.
Use appropriate restriction enzyme(s) to cut the fragment from pGEM T-Easy to generate either blunt ends or overhangs that could be cloned into the multi-clone sites (MCSs) of DNAβ or DNA1 vector.
-
2.
Reaction products were electrophoresed on 1% agarose gels and extracted target DNA fragments from gels using QIAquick Gel Extraction Kit (QIAGEN).
-
3.
Set up a ligation reaction by mixing 7 μL of enzyme digestion product (approx 200 ng of DNA) with 1 μL of DNAβ or DNA1 vector (50 ng), 1 μL of 10× ligation buffer, and 1 μL of T4 DNA ligase (Takara) in a microfuge tube and incubate it overnight at 16°C.
-
4.
Transform the ligated product into E. coli-competent cells by heat shock method as described in step 4 of Subheading 3.1.1, spread onto LB plates supplemented with antibiotic kanamycin (50 mg/L), and incubate overnight at 37°C.
-
5.
Screen positive clones as described in step 5 of Subheading 3.1.1, inoculate a positive E. coli clone into 5 mL of LB supplemented with kanamycin (50 mg/L), and grow at 37°C for 12–16 h with 250–300-rpm vigorous shaking.
-
6.
Isolate the recombinant vector by using QIAquick Plasmid Extraction Kit. If using DNA1 vector in tobacco and petunia, directly go to step 11. If using DNAβ vector in N. glutinosa, P. hybrid, and tomato, the help virus genome (TYLCCNV) should be inserted into DNAβ vector following steps 7–10 (see Note 5).
-
7.
Digest the DNAβ recombinant vector containing target gene and pGEM T-Easy TYLCCNV-Y10 1.7A containing TYLCCNV genome with SalΙ; digestion products were electrophoresed on 1% agarose gels and TYLCCNV genome (approximately 4.6 kb) and recombinant DNAβ fragment were extracted from gels using QIAquick Gel Extraction Kit (QIAGEN).
-
8.
Set up a ligation reaction by mixing 7 μL of TYLCCNV genome (approximately 200 ng of DNA) with 1 μL of DNAβ recombinant fragment (50 ng), 1 μL of 10× ligation buffer, and 1 μL of T4 DNA ligase (Takara) in a microfuge tube and incubate it overnight at 16°C.
-
9.
Transform the ligated product into E. coli-competent cells by heat shock method as described in step 4 of Subheading 3.1.1, spread onto LB plates supplemented with antibiotic kanamycin (50 mg/L), and incubate overnight at 37°C.
-
10.
Screen positive clones by using the primers F (5′-ATGGATTCACGCACCGGGGAAC-3′)/R (5′-TTAATAAATATTAAATTTTATATCATG-3′) as described in step 5 of Subheading 3.1.1.
-
11.
Incubate a positive E. coli clone and extract plasmid following steps 5 and 6 of 3.1.2 to get recombinant vector containing TYLCCNV genome and DNAβ carrying target gene.
-
12.
Transform the recombinant vector into A. tumefaciens strain EHA105-competent cells using electroporation. A 0.2 mL volume of A. tumefaciens strain EHA105-competent cells was mixed with 0.5 μg plasmid DNA and was placed in 0.2 cm cuvettes in Gene Pulser (Biorad). The instrument was set at the voltage of 2,500 V and 25 μF with the pulse controller set at 200 Ω.
-
13.
Immediately after electroporation, 1 mL YEP was added and the mixture was placed on ice for 3 min followed by incubation at 28°C for 2 h.
-
14.
Collect the A. tumefaciens cells by centrifugation at 6,000 × g in a microcentrifuge for 30 s, discard the supernatant, resuspend cells in 200 μL YEP, spread on LB plates supplemented with 50 mg/L kanamycin and 50 mg/L rifampicin, and incubate at 28°C for more than 48 h.
-
15.
Screen positive clones as described in step 5 of subheading in 3.1.1 to select A. tumefaciens containing the VIGS vector carrying the target gene.
3.2 Plant Inoculation
As DNAβ and DNA1 are entirely dependent on their helper viruses for replication and (or) movement, DNAβ and DNA1 vector should be co-inoculated with helper virus for induction of gene silencing in plants.
3.2.1 Preparation of the Inoculum
-
1.
Inoculate individually colony of each A. tumefaciens culture containing DNAβ vector, DNA1 vector, or helper virus into individual 10-mL test tubes containing 5 mL of YEP medium supplemented with kanamycin (50 mg/L) and rifampicin (50 mg/L) and shake the tubes at 250 rpm (28°C) for more than 24 h.
-
2.
Use the 2 mL overnight cultures to inoculate 100-mL flasks containing 25 mL of YEP medium with the same antibiotics and shake flasks at 250 rpm (28°C) overnight.
-
3.
Harvest the bacterial cells in sterile disposable 50-mL conical tubes by centrifugation at 2,800 × g for 10 min and resuspend in injection buffer.
-
4.
Adjust the concentration of each culture to OD600 of 0.8–1.0 and incubate at room temperature for 3 h (see Note 6).
3.2.2 Plant Inoculation
-
1.
Plant preparation: The plants can be inoculated when the first five leaves have emerged, but are not fully expanded.
-
2.
Use a syringe with beveled needle to inject approximately 300 μL of bacterial suspension into the phloem of stem. Three to five inoculations should be done, one into the phloem of the stem just above the base of the plant, the next one approximately 3 mm above this, and a third injection which is done vertically down the stem toward the base of the plant. The bacterial suspension can also be inoculated into a petiole joining the stem.
-
3.
Move the seedlings onto a clean tray and make sure to place each batch of infiltrated seedlings on a separate tray. Maintain seedlings in an insect-free growth chamber at appropriate temperature.
3.3 Analysis of Virus Infection
To ensure that the virus has infected the injected plants, the assay of virus infection should be employed at 12–15 days post inoculation (dpi).
-
1.
Grind freshly harvested leaf tissue (as little as 50 mg) with 500 μL of DNA extraction buffer in a 1.5-mL microtube using a tissue lyser (Qiagen).
-
2.
Vortex for 2 min and incubate at 65°C for 1 h.
-
3.
Add 500 μL of chloroform, vortex for 30 s, incubate at room temperature for 10 min, and spin for 10 min at 6,000 × g in a microcentrifuge.
-
4.
Transfer 450 μL of the supernate to a clean microfuge tube avoiding the tissue debris. Repeat the centrifugation if any debris is left in the supernate.
-
5.
Add 0.5 volume of isopropanol, vortex, and centrifuge for 10 min at 12,000 rpm in a microcentrifuge.
-
6.
Carefully remove and discard the supernate.
-
7.
To the pellet, add 1 mL of 70% (v/v) ethanol, vortex, centrifuge at 12,000 rpm for 5 min, and carefully remove as much of the supernatant as possible with a pipette.
-
8.
Vacuum-dry the pellet for 5 min.
-
9.
Dissolve the DNA pellet in 35 μL of sterile double-distilled water.
-
10.
Use 1–5 μL of the DNA as a template for a PCR reaction with specific primers for detecting the presence of DNAβ or DNA1 (see Note 7).
3.4 Analysis of Gene Silencing in Plants
The level of plant-target-transcript silencing can be monitored through quantitative RT-PCR using primers specific for the target gene, complementary or identical to sequences outside the region of the gene fragment inserted in DNAβ or DNA1 vector, and the gene encoding Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) can be used as an internal control (see Notes 8 and 9). Relative transcript levels for the target gene between various treatments are estimated after multiple PCR cycle numbers and normalizing the intensities according to estimates of the substrate RNA levels based on results from the internal control.
3.4.1 RNA Extraction and Reverse Transcription
-
1.
Harvest tissue from plants infected with the silencing vector containing, or not containing, the plant gene insert at 2–3 weeks after inoculation. The harvested tissue can be used for RNA isolation immediately or stored at −70°C for future use.
-
2.
Take 0.1 g of tissue from each sample, add liquid nitrogen, and grind to a powder using a mortar and pestle. Add 1 mL of TRIzol reagent (Invitrogen) to each mortar and extract the RNA according to the manufacturer’s protocol.
-
3.
Dissolve the RNA pellet in 30 μL of nuclease-free double-distilled water.
-
4.
Determine the RNA concentration for each sample by loading 1 μL of isolated RNA onto a Nanodrop ND 1000 UV spectrophotometer. Acceptable A 260/A 280 ratios for RNA are between 1.6 and 1.8.
-
5.
Add 1 μL of RNase-free DNase I (Takara) to each RNA sample and incubate the samples at 37°C for 15 min to remove any contaminating DNA.
-
6.
Set up a reverse transcription reaction as follows: Mix the total RNA 1 μg and oligo (dT) 1 μL of 18-mer primer (0.5 μg/μL) and add DEPC-treated water to 12 μL on ice. Incubate the mixture at 70°C for 5 min, chill on ice, and collect any condensed water by brief centrifugation. Place the tube on ice and add 4 μL of 5× reaction buffer, 1 μL of Ribonuclease Inhibitor (20 U/μL), 2 μL of 10 mM dNTP mixture, and 1 μL of M-MuLV Reverse Transcriptase (200 U/μL). Incubate the mixture at 42°C for 1 h.
3.4.2 Quantitative PCR
PCR amplification and analysis were achieved using a LightCycler 480 instrument (Roche) and software version 1.5.0 (Roche), respectively.
-
1.
The optimized reaction was carried out in a 20 μL final reaction volume. Add forward and reverse primers to 0.4 μmol/L concentration, 2 μL of DNA solution, 6.4 μL of distilled water, and 10 μL of kit-supplied SYBR® PCR master mix (Roche, including HotStart Ex Taq HS DNA polymerase, reaction buffer, dNTP mix, and SYBR Green I).
-
2.
All templates were amplified using the following LightCycler protocol. The thermal profile for the real-time PCR was 95°C for 5 min, followed by 40 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 20 s. Fluorescent data were acquired during each extension phase.
-
3.
After 40 cycles, a melting curve is generated by heating the sample to 95°C programmed for 10 s followed by cooling down to 60°C for 15 s and slowly heating the samples at 0.11°C/s to 95°C while the fluorescence was measured continuously.
-
4.
Obtain the result by using the LightCycler software automatically (see Note 10).
4 Notes
-
1.
We found that when the TYLCCNV genome and DNAβ are on the same binary vector, silencing efficiency in N. glutinosa, P. hybrid, and tomato can be increased remarkably. The TYLCCNV genome can be obtained from the pGEM T-Easy TYLCCNV-Y10 1.7A plasmid after digestion with SalΙ and then insertion into the DNAβ vector. The detailed process is described in steps 7–10 of Subheading 3.1.2.
-
2.
In different plants, we use different positive controls. We insert a 351-bp Su fragment from N. benthamiana, 170-bp Su fragment from P. hybrid, and 351-bp Su fragment from S. lycopersicum into the silencing vector as positive controls for tobacco, petunia, and tomato plants, respectively.
-
3.
Choose appropriate fragment of gene insert into vector; if you want to silence a gene of gene family specially, use 3′ UTR region of this gene, and if you want to silence all or most members of a given family, use a targeting sequence derived from the most highly conserved region of this gene family.
-
4.
BamHI, SamI, and XbaI sites are available in these vectors, so appropriate enzyme sites can be added at the 5′ end of the forward and reverse primers.
-
5.
For inducing efficient gene silencing in N. glutinosa, P. hybrid, and S. lycopersicum by DNAβ, the TYLCCNV genome and DNAβ should be on one binary vector; so the TYLCCNV genome needs be inserted into DNAβ to generate TYLCCNV + DNAβ. If silencing a gene by DNA1, or in N. benthamiana by DNAβ, steps 7–10 are not necessary.
-
6.
Except for induction of gene silencing in N. glutinosa, P. hybrid, and S. lycopersicum by DNAβ and TYLCCNV, inocula should consist of mix equal volumes of A. tumefaciens cultures containing helper virus and the DNAβ or DNA1 vector.
-
7.
Generally, we use primers β01(5′-GTAGGTACCACTACGCTACGCAGCAGCC-3′) and β02 (5′-AGTGGTACCTACCCTCCCAGGGGTACAC-3′) to detect DNAβ, and UN101 (5′-AAGCTTGCGACTATTGTATGAAAGAGG-3′) and UN102 (5′-AAGCTTCGTCTGTCTTACGAGCTCGCTG-3′) to detect DNA1 in plants; PCR products for DNAβ and DNA1 (not including inserted fragment) are approximately 1.0 and 1.3 kb, respectively.
-
8.
Primers are designed to detect control gene and target gene expression by quantitative RT-PCR. Primers that anneal outside the region targeted for silencing are necessary. Primers can be designed using Primer Premier (version 5.0, Premier Biosoftware) software. The calculated annealing temperature for primers is about 60°C.
-
9.
Plant mRNAs may serve as internal gene expression controls. The best internal control genes (such as actin or ubiquitin) are those whose levels do not fluctuate during virus infection.
-
10.
For monitoring gene silencing efficiency, absolute quantification is not required. Instead, the relative quantity of target can be obtained by comparing differences in fractional cycle numbers between samples using an internal control gene as a reference.
References
Robertson D (2004) VIGS vectors for gene silencing: many targets, many tools. Annu Rev Plant Biol 55:495–519
Burch-Smith TM, Anderson JC, Martin GB, Dinesh-Kumar SP (2004) Applications and advantages of virus-induced gene silencing for gene function studies in plants. Plant J 39: 734–746
Baulcombe DC (1999) Fast forward genetics based on virus-induced gene silencing. Curr Opin Plant Biol 2:109–113
Kumagai MH, Donson J, Dellacioppa G, Harvey D, Hanley K, Grill LK (1995) Cytoplasmic inhibition of carotenoid biosynthesis with virus-derived RNA. Proc Natl Acad Sci U S A 92:1679–1683
Ruiz MT, Voinnet O, Baulcombe DC (1998) Initiation and maintenance of virus-induced gene silencing. Plant Cell 10:937–946
Ratcliff F, Martin-Hernandez AM, Baulcombe DC (2001) Tobacco rattle virus as a vector for analysis of gene function by silencing. Plant J 25:237–245
Liu YL, Schiff M, Dinesh-Kumar SP (2002) Virus-induced gene silencing in tomato. Plant J 31:777–786
Pignatta D, Kumar P, Turina M, Dandekar A, Falk BW (2007) Quantitative analysis of efficient endogenous gene silencing in Nicotiana benthamiana plants using tomato bushy stunt virus vectors that retain the capsid protein gene. Mol Plant Microbe Interact 20:609–618
Pflieger S, Blanchet S, Camborde L, Drugeon G, Rousseau A, Noizet M, Planchais S, Jupin I (2008) Efficient virus-induced gene silencing in Arabidopsis using a ‘one-step’ TYMV-derived vector. Plant J 56:678–690
Igarashi A, Yamagata K, Sugai T, Takahashi Y, Sugawara E, Tamura A, Yaegashi H, Yamagishi N, Takahashi T, Isogai M, Takahashi H, Yoshikawa N (2009) Apple latent spherical virus vectors for reliable and effective virus-induced gene silencing among a broad range of plants including tobacco, tomato, Arabidopsis thaliana, cucurbits, and legumes. Virology 386:407–416
Naylor M, Reeves J, Cooper JI, Edwards ML, Wang H (2005) Construction and properties of a gene-silencing vector based on poplar mosaic virus (genus Carlavirus). J Virol Methods 124:27–36
Kjemtrup S, Sampson KS, Peele CG, Nguyen LV, Conkling MA, Thompson WF, Robertson D (1998) Gene silencing from plant DNA carried by a Geminivirus. Plant J 14:91–100
Peele C, Jordan CV, Muangsan N, Turnage M, Egelkrout E, Eagle P, Hanley-Bowdoin L, Robertson D (2001) Silencing of a meristematic gene using geminivirus-derived vectors. Plant J 27:357–366
Turnage MA, Muangsan N, Peele CG, Robertson D (2002) Geminivirus-based vectors for gene silencing in Arabidopsis. Plant J 30:107–114
Fofana IBF, Sangare A, Collier R, Taylor C, Fauquet CM (2004) A geminivirus-induced gene silencing system for gene function validation in cassava. Plant Mol Biol 56:613–624
Abraham-Juarez MD, Rocha-Granados MD, Lopez MG, Rivera-Bustamante RF, Ochoa-Alejo N (2008) Virus-induced silencing of Comt, pAmt and Kas genes results in a reduction of capsaicinoid accumulation in chili pepper fruits. Planta 227:681–695
Tuttle JR, Idris AM, Brown JK, Haigler CH, Robertson D (2008) Geminivirus-mediated gene silencing from cotton leaf crumple virus is enhanced by low temperature in cotton. Plant Physiol 148:41–50
Gossele V, Fache I, Meulewaeter F, Cornelissen M, Metzlaff M (2002) SVISS - a novel transient gene silencing system for gene function discovery and validation in tobacco plants. Plant J 32:859–866
Tao XR, Zhou XP (2004) A modified viral satellite DNA that suppresses gene expression in plants. Plant J 38:850–860
Qian YJ, Mugiira RB, Zhou XP (2006) A modified viral satellite DNA-based gene silencing vector is effective in association with heterologous begomoviruses. Virus Res 118:136–142
Li D, Behjatnia SA, Dry IB, Walker AR, Randles JW, Rezaian MA (2008) Tomato leaf curl virus satellite DNA as a gene silencing vector activated by helper virus infection. Virus Res 136: 30–34
Huang CJ, Xie Y, Zhou XP (2009) Efficient virus-induced gene silencing in plants using a modified geminivirus DNA1 component. Plant Biotechnol J 7:254–265
Tao XR, Qian YJ, Zhou XP (2006) Modification of a viral satellite DNA-based gene silencing vector and its application to leaf or flower color change in Petunia hybrida. Chin Sci Bull 51: 2208–2213
Chen JC, Jiang CZ, Gookin TE, Hunter DA, Clark DG, Reid MS (2004) Chalcone synthase as a reporter in virus-induced gene silencing studies of flower senescence. Plant Mol Biol 55:521–530
Chung E, Seong E, Kim YC, Chung EJ, Oh SK, Lee S, Park JM, Joung YH, Choi D (2004) A method of high frequency virus-induced gene silencing in chili pepper (Capsicum annuum L. cv. Bukang). Mol Cells 17:377–380
Kim YC, Kim SY, Paek KH, Choi D, Park JM (2006) Suppression of CaCYP1, a novel cytochrome P450 gene, compromises the basal pathogen defense response of pepper plants. Biochem Biophys Res Commun 345:638–645
Ryu CM, Anand A, Kang L, Mysore KS (2004) Agrodrench: a novel and effective agroinoculation method for virus-induced gene silencing in roots and diverse Solanaceous species. Plant J 40:322–331
Senthil-Kumar M, Hema R, Anand A, Kang L, Udayakumar M, Mysore KS (2007) A systematic study to determine the extent of gene silencing in Nicotiana benthamiana and other Solanaceae species when heterologous gene sequences are used for virus-induced gene silencing. New Phytol 176:782–791
Brigneti G, Martin-Hernandez AM, Jin HL, Chen J, Baulcombe DC, Baker B, Jones JDG (2004) Virus-induced gene silencing in Solanum species. Plant J 39:264–272
Constantin GD, Gronlund M, Johansen IE, Stougaard J, Lund OS (2008) Virus-induced gene silencing (VIGS) as a reverse genetic tool to study development of symbiotic root nodules. Mol Plant Microbe Interact 21:720–727
Constantin GD, Krath BN, MacFarlane SA, Nicolaisen M, Johansen IE, Lund OS (2004) Virus-induced gene silencing as a tool for functional genomics in a legume species. Plant J 40:622–631
Zhang C, Yang CL, Whitham SA, Hill JH (2009) Development and use of an efficient DNA-based viral gene silencing vector for soybean. Mol Plant Microbe Interact 22:123–131
Zhang CQ, Ghabrial SA (2006) Development of bean pod mottle virus-based vectors for stable protein expression and sequence-specific virus-induced gene silencing in soybean. Virology 344:401–411
Lu HC, Chen HH, Tsai WC, Chen WH, Su HJ, Chang DCN, Yeh HH (2007) Strategies for functional validation of genes involved in reproductive stages of orchids. Plant Physiol 143: 558–569
Holzberg S, Brosio P, Gross C, Pogue GP (2002) Barley stripe mosaic virus-induced gene silencing in a monocot plant. Plant J 30: 315–327
Scofield SR, Huang L, Brandt AS, Gill BS (2005) Development of a virus-induced gene-silencing system for hexaploid wheat and its use in functional analysis of the Lr21-mediated leaf rust resistance pathway. Plant Physiol 138: 2165–2173
Ding XS, Schneider WL, Chaluvadi SR, Mian MA, Nelson RS (2006) Characterization of a Brome mosaic virus strain and its use as a vector for gene silencing in monocotyledonous hosts. Mol Plant Microbe Interact 19:1229–1239
Burch-Smith TM, Schiff M, Liu YL, Dinesh-Kumar SP (2006) Efficient virus-induced gene silencing in Arabidopsis. Plant Physiol 142:21–27
Wang CC, Cai XZ, Wang XM, Zheng Z (2006) Optimisation of tobacco rattle virus-induced gene silencing in Arabidopsis. Funct Plant Biol 33:347–355
Godge MR, Purkayastha A, Dasgupta I, Kumar PP (2008) Virus-induced gene silencing for functional analysis of selected genes. Plant Cell Rep 27:209–219
Cai X, Wang C, Xu Y, Xu Q, Zheng Z, Zhou X (2007) Efficient gene silencing induction in tomato by a viral satellite DNA vector. Virus Res 125:169–175
He XX, Jin CW, Li GX, You GY, Zhou XP, Zheng SJ (2008) Use of the modified viral satellite DNA vector to silence mineral nutrition-related genes in plants: silencing of the tomato ferric chelate reductase gene, FRO1, as an example. Sci Chin Ser C 51:402–409
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grants No. 31000839) and the National Key Basic Research and Development Program (2012CB114004).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Zhou, X., Huang, C. (2012). Virus-Induced Gene Silencing Using Begomovirus Satellite Molecules. In: Watson, J., Wang, MB. (eds) Antiviral Resistance in Plants. Methods in Molecular Biology, vol 894. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-61779-882-5_4
Download citation
DOI: https://doi.org/10.1007/978-1-61779-882-5_4
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-61779-881-8
Online ISBN: 978-1-61779-882-5
eBook Packages: Springer Protocols