
Abstract
Natural deep eutectic solvents (NADES) are promising green solvents for the extraction of compounds from natural products. This is the first study to use a sample preparation method based on NADES for the extraction of flavonoids (rutin, hesperidin, neohesperidin, naringenin, naringin, quercetin, hesperetin, and chrysin) from fruits, vegetables, and spices. In total, 17 types of NADES based on choline chloride, acetylcholine chloride, choline tartrate, betaine, and carnitine with different compositions were tailored to test their extraction efficiency. Operational conditions such as water content in NADES and liquid/solid ratios were also studied. A response surface methodology was used for multivariate optimization of some extraction parameters. Efficient recovery of extracted flavonoids (higher than 70%) was achieved using a 30% water solution of acetylcholine chloride/lactic acid ratio (2:1) as an extraction solvent. Other conditions for SLE-NADES were as follows: liquid/solid ratio 3:1, extraction temperature 60 °C, extraction time 30 min, and stirring speed 1400 rpm. Each extract was analyzed by UHPLC with UV and MS/MS detection. The developed method was applied for analysis of flavonoids in fruits (cranberry, fruits of Lycium barbarum L., grape, plum, and orange peel), vegetables (onion and broccoli), and spices (mustard, rosemary, and black pepper).
Similar content being viewed by others

Introduction
Plants are potential sources of natural bioactive compounds such as secondary metabolites and antioxidants. Secondary metabolites are chemicals produced by plants whose functions in growth, photosynthesis, reproduction, and other primary processes are not yet fully understood. Flavonoids and phenolic acids are the most important groups of secondary metabolites and bioactive compounds in plants (Kim et al. 2003a; Ghasemzadeh and Ghasemzadeh 2011). Natural antioxidants such as phenolic acids and flavonoids are increasingly attracting attention because of their natural qualities as disease-preventing, health-promoting, and anti-aging substances (Saxena et al. 2012). Antioxidants may reduce oxidative damage induced by free radicals and reactive oxygen species under oxidative stress conditions in humans. Moreover, well-known and traditionally used natural antioxidants from tea, wine, fruits, vegetables, and herbs have already been exploited commercially either as antioxidant additives or as nutritional supplements (Pandey and Rizvi 2009; Lobo et al. 2010).
Sample preparation is the first step in analysis of plants, because the desired chemical components must be extracted from the herbal materials for further separation and characterization (Sasidharan et al. 2011; Locatelli et al. 2012). Plant metabolites may differ in their polarities, stabilities, boiling points, and quantities. The wide range of polarities and physical properties of natural compounds makes a one-step, single solvent extraction process for all metabolites from biomass virtually impossible (Kerton 2009). Solvents of different polarities are needed for extraction, separation, and purification of medicines. Solvents such as alcohols, chloroform, and ethyl acetate are commonly used for this purpose. However, organic solvents are often toxic, flammable, explosive, and poorly biodegraded. Thus, development of new sample-preparation techniques for extraction and analysis of plant compounds are necessary to ensure that high-quality herbal products are available for consumers worldwide (Huie 2002; Diuzheva et al. 2017). Advances that are needed include reduction in organic solvent consumption and sample degradation, elimination of additional sample clean-up and concentration steps before chromatographic analysis, ease of automation, and improvement in extraction efficiency, selectivity, and/or kinetics.
To address these problems, alternative solvents are required which are biodegradable, have low toxicity, and at the same time are able to dissolve a broad range of compounds of different polarities. In 2003, Abbott et al. presented a new class of solvents–deep-eutectic solvents (DES). A DES is a mixture of two or more compounds with a melting point lower than its individual components that includes both a quaternary ammonium salt as a hydrogen bond acceptor (HBA) and a hydrogen bond donor (HBD) that associate with each other by hydrogen bonding (Abbott et al. 2003; Francisco et al. 2013, Shishov et al. 2017). Recently, Dai et al. (2013) reported the preparation of several DES reagents of natural origin termed natural deep eutectic solvents (NADES). NADES solvents are obtained exclusively from natural components (sugars, organic acids, amino acids) which are commonly present in the cells of living organisms, unlike ordinary DES (Dai et al. 2013; Choi et al. 2011). These solvents have the following environmental and economic advantages: they are non-volatile, have a low toxicity, are biodegradable, sustainable, and inexpensive, and are made using simple preparation methods. They also show very good physicochemical properties: liquid state below 0 °C, adjustable viscosity, a broad range of polarities, and ability to dissolve a wide range of compounds (Paiva et al. 2014; Faggian et al. 2006; Dai et al. 2015). NADES therefore have good potential for the extraction of valuable secondary metabolites for use in the food and pharmaceutical industries.
Recently, NADES have been applied for extraction of flavonoids (Wei et al. 2015a; Radošević et al. 2016; Bakirtzi et al. 2016; Dai et al. 2016), alkaloids, phenolic acids, anthraquinones, saponins (Duan et al. 2016), phenolic compounds (Wei et al. 2015b), xanthonoids (Mulia et al. 2015), rutin (Huang et al. 2017), flavonoid glycosides, and aglycones (Zhuang et al. 2017) from plant materials. The mentioned publications describe the extraction of flavonoids other than those described in our research and the application of different NADES solvents for sample preparation. Moreover, NADES extraction has never been applied to cranberry, fruits of Lycium barbarum L., grape, plum, orange peel, onion, broccoli, mustard, rosemary, and black pepper. The number of reports on application of DES for extraction of natural products is limited, and the efficiency of NADES for extraction of other types of active natural compounds remains unknown.
As there is insufficient knowledge concerning NADES, research is needed to provide a better understanding of the interactions and complex nature of NADES compositions, as well as their applicability for extraction of biologically active compounds from plant material.
In spite of the advantages of NADES, to the best of our knowledge, no extraction procedure based on these solvents has been reported for the determination of selected flavonoids in food samples. Thus, the main purpose of this work is utilization of NADES in a solid–liquid extraction method (SLE-NADES) for rutin, hesperidin, neohesperidin, naringenin, naringin, quercetin, hesperetin, and chrysin from fruits (cranberry, fruits of Lycium barbarum L., grape, plum, and orange peel), vegetables (onion and broccoli), and spices (mustard, rosemary, and black pepper).
Materials and Methods
Chemicals and Reagents
Flavonoid standards, including rutin (RUT), hesperidin (HSD), neohesperidin (NHSD), naringenin (NAR), naringin (NARG), quercetin (QUE), hesperetin (HST), and chrysin (CHS) (IS, internal standard), were purchased from Sigma-Aldrich (St. Louis, MO). HPLC grade acetonitrile (ACN) and trifluoroacetic acid (TFA) were obtained from Merck (Darmstadt, Germany). Double-distilled water obtained from a laboratory purification system was used in all experiments. Analytical grade methanol, hydrochloric acid, and ammonia were purchased from Stanlab (Lublin, Poland). Choline chloride, citric acid, choline bitartrate, betaine, D,L-malic acid, L(+)-tartaric acid, L-lactic acid, chlorocholine chloride, acetylcholine chloride, and D,L-carnitine hydrochloride were purchased from Alfa Aesar (Lancashire, UK).
Individual standard stock solutions of flavonoids (1 mg/mL) were prepared in methanol and stored in dark flasks at 4 °C. Working solutions were prepared daily by dilution of stock solutions with HPLC grade methanol.
Plant Material
The selected fruits (cranberry, fruits of Lycium barbarum L., grape, plum, and orange peel), vegetables (onion and broccoli), and spices (mustard, rosemary, and black pepper) were purchased from a supermarket in Gliwice, Poland, in 2017. After the samples were cleaned with tap water, the plant material was powdered. The edible part of the plants was used for investigation. Samples were dried to constant weight using a WPS 30S moisture analyzer (RADWAG, Radom, Poland). Dried plants were milled, packed in paper bags, and stored at ambient temperature for 4–5 months before use.
Synthesis of Natural Deep Eutectic Solvents
Natural deep eutectic solvents can be prepared according to several known procedures (Dai et al. 2013; Bajkacz and Adamek 2017). However, ultrasound-assisted synthesis of NADES, described below, is easier and more effective than previously reported procedures.
A two- or three-component mixture with a calculated amount of deionized water was added to a glass vial sealed with a screw-cap and exposed to ultrasound (37 kHz, 30 W) at 50 °C until a homogeneous liquid was formed (10–15 min). Seventeen different NADES systems with two or three components were obtained and examined. Table 1 shows the composition, molar ratios, and symbols of NADES used in this study. Five HBAs were tested: choline chloride, chlorocholine chloride, acetylcholine chloride, betaine, and carnitine. HBAs were mixed with various types of HBD (citric acid, malic acid, tartaric acid, and lactic acid) at different molar ratios.
Natural Deep Eutectic Solvent Microextraction Procedure
A selection process was performed to select the best experimental conditions for microextraction of flavonoids. Factors such as the type of NADES components, water content in NADES, and the ratio of NADES volume to sample amount were studied. Vortex time, speed, and temperature were optimized with the help of a response surface methodology (RSM) design.
A powdered mustard sample (used as a blank sample due to its low flavonoid content) spiked with a known amount of compounds was used to select the extraction procedure. Mustard samples were dried to a constant weight using a WPS 30S moisture analyzer (RADWAG, Radom, Poland). The samples used for selection of the NADES extraction procedure were spiked with flavonoids. Fortified samples were left to stand at room temperature for 48 h before analysis. The concentration of flavonoids in all procedures was 10 μg/g, and each experiment was performed three times.
For method optimization, a real sample (onion) was treated as the fortified mustard sample described above to mimic the real extraction process. In this way, mass transfer is taken into account in addition to the solubility of the fortified compounds.
Finally, dried plant material (200 mg) was mixed with 600 μL of natural deep eutectic solvent (NADES) and stirred at 60 °C for 45 min at 1400 rpm in an Eppendorf tube using a thermomixer comfort (Eppendorf AG, Hamburg, Germany). The mixture was then centrifuged at 2000×g for 5 min in an IKA mini G centrifuge (IKA, Staufen, Germany), and 300 μL of the liquid supernatant was moved to another tube. The solutions were then diluted 1:1 with methanol and centrifuged for 5 min at 2000×g. All extractions were performed in triplicate.
Additionally, the sample preparation procedure was compared with conventional extraction approaches based on shaking the sample with methanol and water at 60 °C for 45 min at 1400 rpm.
The extraction efficiency of the proposed NADES procedure was also compared with that of the UAE-NADES method. Plant material (200 mg) was extracted with 600 μL NADES solvent in an ultrasonic bath at 60 °C for 45 min using 700 W (Sonorex Digital 10P, Bandelin Electronic). The extract was centrifuged and diluted as described above.
Development of NADES Extraction Procedure
Central composite design (CCD) was used to find the optimal values for three independent variables (extraction time, temperature, and stirring speed) and identify interactions between variables. The matrix design included 18 experiments to evaluate factors, including four replicates at the center point to evaluate experimental error. Experiments were performed in a random order to avoid the effects of extraneous variables. The center values were 39 min for extraction time, 45 °C for extraction temperature, and 768 rpm for stirring speed. Preliminary experiments allowed us to identify the model variables at five coded levels: −α (= − 1.68), − 1, 0, + 1, and +α (= + 1.68). Model adequacy and significant terms were examined using analysis of variance (ANOVA) combined with Fisher’s statistical test (p < 0.05) (Statistica 12 software, StatSoft, Poland).
Instrumentation and Chromatographic Conditions
UHPLC-UV Analysis
Chromatography was performed on an UHPLC system (Merck Hitachi, Germany) with a model L-2160U binary pump, a model L-2350U column oven, a model L-2200, and a model L-2400U UV detector. EZ Chrom Elite System Manager software was used for control and data handling.
Separation was carried out on a Poroshell 120 EC-C18 analytical column (100 mm × 3.0 mm; 2.7 μm, Agilent Technologies, USA). The column temperature was maintained at 25 °C. Analysis was performed with gradient elution using (A) ACN and (B) 0.05% TFA in water as the mobile phase. The gradient program was as follows: linear gradient from 20% A to 60% A in 6 min, increased to 75% A in 1 min, changed to 20% A immediately, and held at 20% A for 3 min to equilibrate the column. The flow rate was 1 mL/min. The injection volume was 2 μL. For quantitative analyses, selective detection was performed at λ = 254 nm for RUT, QUE, and CHS and at λ = 285 nm for NARG, NAR, HSD, NHSD, and HST.
Flavonoids were analyzed using UHPLC-UV. Identified polyphenols were quantified with their respective standard calibration curves.
UHPLC-MS/MS Analysis
For confirming molecule identity, UHPLC-MS/MS analysis was performed on a Dionex UHPLC system (Dionex Corporation, Sunnyvale, CA, USA) coupled with an AB Sciex Q-Trap® 4000 mass spectrometer (Foster City, CA, USA). Chromatographic separation was performed using the column and gradient elution program described above, except without TFA in the mobile phase because it suppresses ionization in the negative ion mode. Instead, 0.1% formic acid (FA) in water was used for MS/MS acquisition.
MS/MS conditions were applied as described previously (Magiera et al. 2012). A mass spectrometer with a TurboIonSpray source was run in negative ion mode (ion spray voltage IS = − 4000 V; source temperature T = 550 °C; nebulizer gas GS1 = 50 psi; turbo heater gas GS2 = 50 psi; curtain gas CUR = 20 psi). Analyses were performed using multiple reaction monitoring (MRM) mode (Table 2). UHPLC-MS/MS was used to confirm the qualitative identification of flavonoids in fruit, vegetable, and spice samples.
Results and Discussion
Chromatographic Separation
The chromatographic conditions were selected to obtain better separation of analytes in shorter times. First, different types of columns were studied: Zorbax RRHD SB-C18 (50 mm × 2.1 mm, 1.8 μm), Poroshell 120 EC-C18 (100 mm × 3.0 mm, 2.7 μm), and Hypersil GOLD™ (100 mm × 2.1 mm, 1.9 μm). Effective separation of analytes was achieved on the Poroshell 120 EC-C18 column; therefore, this column was selected as the analytical column. Next, different mobile phase compositions were tested to achieve satisfactory analyte resolution. Given that the retention time of the aglycone of flavonoids (QUE, NAR, HST, and CHS) was longer than that of the glycosides (RUT, NARG, HSD, and NHSD) because of its higher polarity, a gradient elution method was applied. Different mobile phase compositions, including ACN–H2O and MeOH–H2O, with different gradient elution programs were compared. Additionally, different mobile phase additives were tested, such as TFA, acetic acid, and FA. The flavonoids could be resolved with a mobile phase of 0.05% trifluoroacetic acid in water and acetonitrile under the gradient program described in Section UHPLC-UV Analysis. The flow rate was maintained at 1 mL/min during analysis. The resulting gradient elution method had baseline resolution of all compounds within 10 min. Column temperature was maintained at 25 °C, and injection volume was 2 μL. Based on the ultraviolet (UV) spectra of the analyzed compounds, detection wavelengths of 254 and 280 nm were chosen. As shown in Fig. 1, good chromatographic elution of flavonoids was obtained under these conditions.

UHPLC-UV chromatogram of a standard solution containing the analyzed flavonoids and IS
Selection of NADES Extraction Parameters
In this section, eight flavonoids were chosen as the variables for selecting NADES-based extraction parameters. These representative components included four flavonoid glycosides (rutin, naringin, hesperidin, and neohesperidin) and four flavonoid aglycones (quercetin, naringenin, hesperetin, and chrysin). The selected compounds represented the entire range of major flavonoid polarities and structures.
As mentioned in the experimental section, the study of select NADES extraction parameters was performed using a model sample (mustard sample fortified with flavonoids) and a real sample (onion which contained flavonoids). The resulting dependencies in both cases were the same. The studied parameters affected recovery of analytes from the model samples in the same way as from real samples. Due to ease of presentation, in the following sections, fortified mustard sample results are discussed.
Effect of NADES Composition
Several NADES were tested under unchanged extraction conditions, such as solid to solvent ratio 3:1, extraction time 45 min, extraction temperature 60 °C, and stirring speed 1400 rpm (Fig. 2). Significant differences in the extractability of flavonoids with tested NADES were reflected by UHPLC profiles and the calculated recovery of flavonoids.

a–c Effect of NADES composition (Table 1) on the extraction efficiency of flavonoids (molar ratio for all NADES 1:1) (highlighted NADES 4, NADES 8, and NADES 15 with the highest recoveries)
High recovery of selected flavonoids was obtained using NADES based on lactic acid and acetylcholine chloride (NADES 8) or lactic acid and choline chloride (NADES 4). The lowest recovery was obtained using mixtures based on D,L-carnitine hydrochloride and citric acid (1:1) (NADES 14).
NADES properties, such as polarity and extraction affinity, can be modulated (to some extent) by changing the molar ratios of their components. Therefore, NADES based on acetylcholine chloride/lactic acid and choline chloride/lactic acid ratios at different molar ratios were evaluated. In the case of NADES with choline chloride/lactic acid ratio, changes in molar ratios did not affect extraction efficiency significantly. More pronounced results were obtained for NADES including acetylcholine chloride and lactic acid (Fig. S1). An increase in the acetylcholine chloride content led to a higher extraction yield (mean recoveries 75.9% for molar ratio 1:1, 77.9% for molar ratio 1:2, 90.1% for molar ratio 2:1), especially for less polar flavonoids (e.g., chrysin). Hence, acetylcholine chloride/lactic acid ratio (2:1) NADES was used as the extraction solvent for further studies.
The developed procedure for solid–liquid extraction with NADES (SLE-NADES) was compared with a conventional extraction technique based on shaking the sample with water and methanol at 60 °C for 45 min at 1400 rpm. The proposed NADES approach improved the extraction efficiency of the target analytes compared to SLE for water and methanol extractions (Fig. S2).
Ultrasound-assisted extraction with NADES (UAE-NADES), based on sonicating the sample with a mixture of acetylcholine chloride/lactic acid ratio (2:1), was also evaluated. The extraction efficiency of UAE-NADES was comparable to that provided by the conventional solid–liquid extraction procedure (Fig. S2).
Effect of Water Content on NADES
The addition of water could lead to an effective decrease in the viscosity of NADES. So, NADES 8 including acetylcholine chloride and lactic acid (2:1) with different concentrations of water (10, 30, and 75%, w/w) was evaluated for extraction of flavonoids. An increase in water content to more than 10% increased flavonoid recovery. The best recoveries were observed for a 30% concentration of water in NADES. When the water content increases further (30–75%), extraction efficiencies of flavonoids markedly decreased (Fig. S3). Although addition of water effectively reduces viscosity, an excessively high water content negatively impacts interactions between NADES and target compounds and increases extraction media polarity. NADES with high water content provided better extraction efficiencies for polar compounds than for less polar compounds. Therefore, a concentration of 30% (v/v) water in NADES was selected as the optimal extraction system.
Effect of Liquid/Solid Ratio
To increase extraction efficiency and decrease solvent consumption, the liquid/solid ratio (from 1:1 to 5:1) was investigated (Fig. S4). The effect of liquid/solid ratio on extraction efficiency showed a similar trend for all target compounds. Extraction efficiencies of target compounds increased with an increase in liquid/solid ratio from 1:1 to 3:1 (200:200 to 600:200 μL/mg). From 3:1–5:1 (600:200–1000:200 μL/mg), no significant improvements were observed. A liquid/solid ratio of 600:200 μL/mg was thus sufficient to extract target compounds in the tested sample. Therefore, a liquid/solid ratio of 600:200 μL/mg was used for the rest of the study.
Effect of Time, Temperature, and Stirring Speed
In the next step, a rotational CCD was employed to further optimize the three significant factors. CCD is one of the most commonly used response surface designs and is constructed by superimposing several designs. The ranges of studied factors were the following: extraction temperature (X 1), 11–79 °C; extraction time (X 2), 3–74 min; and stirring speed (X 3), 136–1400 rpm. Table 3 shows the design matrix which involved 18 randomized experiments and experimental responses (as extraction recovery). Measurement precision was estimated from the four replicates of the central point (relative standard deviations (%RSD) for all the total responses were less than 4.6%).
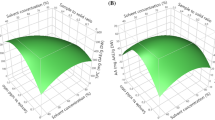
Figure 3 displays the response surface plots for the recovery of target analytes estimated from the CCD for each pair of independent factors. Figure 3A shows the response surface developed for extraction temperature and time, while maintaining a stirring speed of 768 rpm; Fig. 3B shows the response surface obtained by plotting extraction temperature against stirring speed with the extraction time fixed at 39 min; and Fig. 3C shows the response surface obtained as a function of time and speed, for a temperature of 45 °C.

Response surface plots of (A) extraction time (X 1) and temperature (X 2) at a constant stirring speed (X 3) of 768 rpm, (B) extraction time (X 1) and stirring speed (X 3) at a constant extraction temperature (X 2) of 45 °C, and (C) extraction temperature (X 2) and stirring speed (X 3) at a constant time (X 1) of 39 min (for (1) rutin, (2) naringin, (3) hesperidin, (4) neohesperidin, (5) quercetin, (6) naringenin, (7) hesperetin, and (8) chrysin)
Increasing temperature is the easiest method for enhancing desorption and dissolution of secondary metabolites in extraction solvents. In addition, at higher temperatures, solvent viscosity decreases and diffusivity increases, accelerating release of secondary metabolites from the sample matrix to the solvent. Temperature was therefore evaluated for extraction. As seen in Fig. 3A, extraction efficiencies of compounds significantly changed with an increase in temperature. Extraction efficiencies markedly increased when temperature was raised from 40 to 60 °C. However, extraction efficiencies slightly decreased with an increase in temperature from 60 to 79 °C, possibly due to decomposition at high temperatures. Thus, the extraction temperature (X 1) was set at 60 °C.
It is necessary to select the proper extraction time to guarantee completion of extraction. As confirmed in Fig. 3A, C, longer extraction times tended to favor extraction of polyphenols.
In our study, stirring was used to increase extraction efficiency. The extraction efficiency stabilized when stirring speed (X 3) was 1400 rpm.
The significance of the factors was evaluated by analysis of variance (ANOVA). Table 4 shows the results of the full quadratic model analysis, which includes all the terms. It shows the main and quadratic terms of X 1 and X 2 were important for recovery rates of RUT, NARG, HSD, NHSD, QUE, NAR, and HST, while interaction effects were not significant. In contrast, interaction effects were significant for recovery rates of CHS. Based on the experimental data, the following optimal conditions were selected: an extraction time of 45 min, an extraction temperature of 60 °C, and a stirring speed of 1400 rpm. The maximum spiked recovery estimated under the optimized conditions was in the range of 70.3 to 93.7%.
Data analysis gave a semi-empirical expression of extraction efficiency of flavonoids with the following equations:
The plot of experimental values for extraction efficiency fit those calculated from equations well, as presented in Fig. 4 (for the example of rutin). As a result of the full factorial design, a Pareto chart was drawn for each studied flavonoid to visualize the estimated effects of the main variables and their interactions. Figure 5 shows the Pareto graphical analysis for rutin. The Pareto chart gives a graphical presentation of these effects and allows an assessment of both the magnitude and the importance of an effect. In these Pareto charts, the bars (variables) that graphically exceed the significance line exert a statistically significant influence on the obtained results. Based on these results (Table 4), the following effects have a significant impact on extraction efficiency: for RUT: X 1 2, X 2, and X 2 2; for NARG: X 2 and X 2 2; for HSD: X 1 2, X 2, and X 2 2; for NHSD: X 2, X 2 2, X 1 X 2, and X 1 X 3; for QUE: X 2 and X 2 2; for NAR: X 2 and X 2 2; for HST: X 2 2; and for CHS: X 1 2, X 2, X 2 2, X 3, X 3 2, X 1 X 3, and X 2 X 3.

The experimental data versus predicted data for extraction efficiency of rutin

Pareto chart showing the values of effects from variables using the extraction efficiency of rutin
The Pareto charts show that interaction effects were considered to ensure that all the combinations of factor levels were explored in the full factorial design to avoid important effects going undetected.
Method Validation
The limit of detection (LOD), limit of quantification (LOQ), linearity, determination coefficient (R 2), intra- and inter-day precision and accuracy, and extraction recovery (ER%) were evaluated for the proposed method (Supplementary Material).
Application of the Developed Method to Real Samples
To show that the developed NADES-UHPLC-UV method is suitable for practical analyses, samples of fruits (cranberry, fruits of Lycium barbarum L., grape, plum, and orange peel), vegetables (onion and broccoli), and spices (mustard, rosemary, and black pepper) were analyzed (Table 5). Figure 6 shows an UHPLC-UV chromatogram of an extract of Lycium barbarum L. fruits. These chromatograms show that the peak of the analyte is free from interferences, demonstrating that the extraction procedure may be applicable to different samples. Additionally, the identity of the flavonoids in the samples was confirmed by UHPLC-MS/MS in MRM mode (Fig. 7).

Representative chromatograms obtained for an extract of Lycium barbarum L. fruits using the proposed SLE-NADES-UHPLC-UV method

Representative MRM chromatograms obtained for an extract of Lycium barbarum L. fruits after SLE-NADES using UHPLC-MS/MS
All target flavonoids were also observed in the Lycium barbarum L. fruits, with rutin and naringenin present in the highest amounts. The highest total content of naringin was found in the orange peel. Naringin was not found in mustard. Quercetin was detected in all samples, but in onion, it was found in the highest levels. In our study, naringenin was detected in all samples, with a high amount found in orange peel and a small amount found in mustard. The lowest concentrations of all target flavonoids were found in mustard and black pepper. Hesperidin, neohesperidin, and hesperetin were not quantified in the analyzed extracts of black pepper, while naringin, hesperidin, neohesperidin, and hesperetin were not quantified in the extract of mustard (<LOQ). The concentration of analytes in rosemary was slightly higher than in other spice samples.
Koh et al. (2009) reported a flavonoid content (quercetin and kaempferol) ranging from 1.0 to 6.0 mg/100 g of dry wt in commercial broccoli samples. Hertog et al. (1992) analyzed six food samples: cranberry and onion had quercetin at levels of 1485 mg/kg of dry wt and 5076 mg/kg of dry wt, respectively. Wang and Zuo (2011) reported quercetin concentrations of 1399 μg/g for cranberry samples (fruit), 194 μg/mL for cranberry juice, and 689 μg/g for fresh cranberry sauces. Inbaraj et al. (2010) reported hesperidin and rutin in L. barbarum fruits. Anagnostopoulou et al. (2005) found quercetin concentrations from 0.12 to 0.56 g/100 g dry orange peel, and Wang et al. (2008) found naringin, hesperidin, and neohesperidin from 0.21 to 29.8 mg/g, from 0.10 to 29.5 mg/g, and from 0.02 to 0.34 mg/g, respectively. In a study by Peng et al. (2005), hesperetin was found in rosemary (0.36 mg/g). Novak et al. (2008) reported rutin concentrations from 29.1 to 214 μg/g in the skin of four red grape varieties. Kim et al. (2003b) analyzed 11 cultivars of plums and found levels of rutin from 2.8 to 7.7 mg/100 g of fresh weight.
Our results are in close agreement with those reported in the literature. To the best of our knowledge, no previous results for application of NADES to the extraction of these flavonoids in these products have been reported in the literature.
In summary, the recommended methods have short analysis times, simple extraction procedures, and wide linear dynamic ranges. Moreover, the proposed NADES extraction coupled with UHPLC analysis is superior to other methods in terms of ease and speed. The present method uses a smaller amount of solvent for extraction of flavonoids compared with all existing methods. In addition, the obtained recovery of analytes is either similar or higher than those obtained by other methods.
Conclusions
In the present study, a green NADES extraction method was described for comprehensive extraction of flavonoids from plant material. NADES was used as a green microextraction system and an alternative to traditional volatile organic solvents. Subsequent selection for flavonoid extraction indicated that the NADES composition, concentration of water in NADES, and solid-to-liquid ratio were key parameters affecting extraction yields.
The validated results demonstrated that the accuracy, precision, and sensitivity of the proposed UHPLC method were satisfactory for the comprehensive analysis of flavonoids in selected fruits, vegetables, and spices.
The developed method provides advantages including simplicity of experimental steps, relatively high speed of sample preparation, and use of inexpensive and low toxicity extraction solvents, since most NADES can be prepared from readily accessible chemicals, for example NADES derived from acetylcholine chloride and renewable chemicals.
The application of the method to real samples indicates that it could be an alternative for the extraction and quantitative assessment of active components in plant materials.
References
Abbott AP, Capper GD, Davies L, Rasheed RK, Tambyrajah V (2003) Novel solvent properties of choline chloride/urea mixtures. Chem Commun 7:70–71
Anagnostopoulou MA, Kefalas P, Kokkalou E, Assimopoulou AN, Papageorgiou VP (2005) Analysis of antioxidant compounds in sweet orange peel by HPLC–diode array detection–electrospray ionization mass spectrometry. Biomed Chromatogr 19(2):138–148. https://doi.org/10.1002/bmc.430
Bajkacz S, Adamek J (2017) Evaluation of new natural deep eutectic solvents for the extraction of isoflavones from soy products. Talanta 168:329–335. https://doi.org/10.1016/j.talanta.2017.02.065
Bakirtzi C, Triantafyllidou K, Makris DP (2016) Novel lactic acid-based natural deep eutectic solvents: efficiency in the ultrasound-assisted extraction of antioxidant polyphenols from common native Greek medicinal plants. JARMAP 3:120–127
Choi YH, van Spronsen J, Dai Y, Verberne M, Hollmann F, Arends IWCE, Witkamp GJ, Verpoorte R (2011) Are natural deep eutectic solvents the missing link in understanding cellular metabolism and physiology? Plant Physiol 156(4):1701–1705. https://doi.org/10.1104/pp.111.178426
Dai Y, Rozema E, Verpoorte R, Choi YH (2016) Application of natural deep eutectic solvents to the extraction ofanthocyanins from Catharanthus roseus with high extractability and stability replacing conventional organic solvents. J Chromatogr A 1434:50–56. https://doi.org/10.1016/j.chroma.2016.01.037
Dai Y, Spronsen J, Witkamp GJ, Verpoorte R, Choi YH (2013) Natural deep eutectic solvents as new potential media for green technology. Anal Chim Acta 766:61–68. https://doi.org/10.1016/j.aca.2012.12.019
Dai W, Witkamp GJ, Verpoorte R, Choi YH (2015) Tailoring properties of natural deep eutectic solvents with water to facilitate their applications. Food Chem 187:14–19
Diuzheva A, Carradori S, Andruch V, Locatelli M, De Luca E, Tiecco M, Germani R, Menghini L, Nocentini A, Gratteri P, Campestre C (2017) Use of innovative (micro)extraction techniques to characterize Harpagophytum procumbens root and its commercial food supplements. Phytochem Anal doi: https://doi.org/10.1002/pca.2737, in press
Duan L, Dou LL, Guo L, Li P, Liu EH (2016) Comprehensive evaluation of deep eutectic solvents in extraction of bioactive natural products. ACS Sustain Chem Eng 4(4):2405–2411. https://doi.org/10.1021/acssuschemeng.6b00091
Faggian M, Sut S, Perissutti B, Baldan V, Grabnar I, Acqua SD (2006) Natural deep eutectic solvents (NADES) as a tool for bioavailability improvement: pharmacokinetics of rutin dissolved in proline/glycine after oral administration in rats: possible application in nutraceuticals. Molecules 21:1–11
Francisco M, Bruinhorst A, Kroon MC (2013) Low-transition-temperature mixtures (LTTMs): a new generation of designer solvents. Angew Chem Int Edit 52(11):3074–3085. https://doi.org/10.1002/anie.201207548
Ghasemzadeh A, Ghasemzadeh N (2011) Flavonoids and phenolic acids: role and biochemical activity in plants and human. J Med Plants Res 5:6697–6703
Hertog MGL, Hollman PCH, Venema DP (1992) Optimization of a quantitative HPLC determination of potentially anticarcinogenic flavonoids in vegetables and fruits. J Agric Food Chem 40(9):1591–1598. https://doi.org/10.1021/jf00021a023
Huie CW (2002) A review of modern sample-preparation techniques for the extraction and analysis of medicinal plants. Anal Bioanal Chem 373(1-2):23–30. https://doi.org/10.1007/s00216-002-1265-3
Huang Y, Feng F, Jiang J, Qiao Y, Wu T, Voglmeir J, Chen Z (2017) Green and efficient extraction of rutin from tartary buckwheat hull by using natural deep eutectic solvents. Food Chem 221:1400–1405. https://doi.org/10.1016/j.foodchem.2016.11.013
Inbaraj BS, Lu H, Kao TH, Chen BH (2010) Simultaneous determination of phenolic acids and flavonoids in Lycium barbarum Linnaeus by HPLC-DAD-ESI-MS. J Pharm Biomed Anal 51(3):549–556. https://doi.org/10.1016/j.jpba.2009.09.006
Kerton FM (2009) Room Temperature Ionic Liquids and Eutectic Mixtures. Alternative Solvents for Green Chemistry, RSC Publishing, Oxford, UK, pp. 118–142. (Chapter 6)
Kim DO, Chun OK, Kim YJ, Moon HY, Lee CY (2003a) Quantification of polyphenolics and their antioxidant capacity in fresh plums. J Agric Food Chem 51(22):6509–6515. https://doi.org/10.1021/jf0343074
Kim DO, Jeong SW, Lee CY (2003b) Antioxidant capacity of phenolic phytochemicals from various cultivars of plums. Food Chem 81(3):321–326. https://doi.org/10.1016/S0308-8146(02)00423-5
Koh E, Wimalasiri KMS, Chassy AW, Mitchell AE (2009) Content of ascorbic acid, quercetin, kaempferol and total phenolics in commercial broccoli. J Food Compos Anal 22(7-8):637–643. https://doi.org/10.1016/j.jfca.2009.01.019
Lobo V, Patil A, Phatak A, Chandra N (2010) Free radicals, antioxidants and functional foods: impact on human health. Phcog Rev 4(8):118–126. https://doi.org/10.4103/0973-7847.70902
Locatelli M, Melucci D, Carlucci G, Locatelli C (2012) Recent HPLC strategies to improve sensitivity and selectivity for the analysis of complex matrices. ISAT 40:112–137
Magiera S, Baranowska I, Kusa J (2012) Development and validation of UHPLC-ESI-MS/MS method for the determination of selected cardiovascular drugs, polyphenols and their metabolites in human urine. Talanta 89:47–56. https://doi.org/10.1016/j.talanta.2011.11.055
Mulia K, Krisanti E, Terahadi F, Putri S (2015) Selected natural deep eutectic solvents for the extraction of α-mangostin from mangosteen (Garcinia mangostana L.) pericarp. Int. J Technol 7:1211–1220
Novak I, Janeiro P, Seruga M, Oliveira-Brett AM (2008) Ultrasound extracted flavonoids from four varieties of Portuguese red grape skins determined by reverse-phase high-performance liquid chromatography with electrochemical detection. Anal Chim Acta 630(2):107–115. https://doi.org/10.1016/j.aca.2008.10.002
Paiva A, Craveiro R, Aroso I, Martins M, Reis RL, Duarte ARC (2014) Natural deep eutectic solvents—solvents for the 21st century. ACS Sustain Chem Eng 2(5):1063–1071. https://doi.org/10.1021/sc500096j
Pandey KB, Rizvi SI (2009) Plant polyphenols as dietary antioxidants in human health and disease. Oxidative Med Cell Longev 2(5):270–278. https://doi.org/10.4161/oxim.2.5.9498
Peng Y, Yuan J, Liu F, Ye J (2005) Determination of active components in rosemary by capillary electrophoresis with electrochemical detection. J Pharm Biomed Anal 39(3-4):431–437. https://doi.org/10.1016/j.jpba.2005.03.033
Radošević K, Ćurko N, Srček VG, Bubalo MC, Tomašević M, Ganić KK, Redovniković IR (2016) Natural deep eutectic solvents as beneficial extractants for enhancement of plant extracts bioactivity. LWT - Food Sci Technol 73:45–51. https://doi.org/10.1016/j.lwt.2016.05.037
Sasidharan S, Chen Y, Saravanan D, Sundram KM, Latha LY (2011) Extraction, isolation and characterization of bioactive compounds from plants’ extracts. AJTCAM 8(1):1–10
Saxena M, Saxena J, Pradhan A (2012) Flavonoids and phenolic acids as antioxidants in plants and human health. IJPSRR 16:130–134
Shishov A, Bulatov A, Locatelli M, Carradori S, Andruch V (2017) Application of deep eutectic solvents in analytical chemistry. A review. Microchem J 135:33–38. https://doi.org/10.1016/j.microc.2017.07.015
Wang YC, Chuang YC, Hsu HW (2008) The flavonoid, carotenoid and pectin content in peels of citrus cultivated in Taiwan. Food Chem 106(1):277–284. https://doi.org/10.1016/j.foodchem.2007.05.086
Wang C, Zuo Y (2011) Ultrasound-assisted hydrolysis and gas chromatography–mass spectrometric determination of phenolic compounds in cranberry products. Food Chem 128(2):562–568. https://doi.org/10.1016/j.foodchem.2011.03.066
Wei ZF, Wang XQ, Peng X, Wang W, Zhao ChJ ZYG, Fu YJ (2015a) Fast and green extraction and separation of main bioactive flavonoids from Radix Scutellaria. Ind Crop Prod 63:175–181. https://doi.org/10.1016/j.indcrop.2014.10.013
Wei Z, Qi X, Li T, Luo M, Wang W, Zu Y, Fu Y (2015b) Application of natural deep eutectic solvents for extraction and determination of phenolics in Cajanus cajan leaves by ultra performance liquid chromatography. Sep Purif Technol 149:237–244. https://doi.org/10.1016/j.seppur.2015.05.015
Zhuang B, Dou LL, Li P, Liu EH (2017) Deep eutectic solvents as green media for extraction of flavonoid glycosides and aglycones from Platycladi Cacumen. J Pharm Biomed Anal 134:214–219. https://doi.org/10.1016/j.jpba.2016.11.049
Funding
This project was supported by funds from the National Science Center within the framework of the project SONATA no. 2014/13/D/ST4/01863 for 2015–2018, Krakow, Poland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Sylwia Bajkacz declares that she has no conflict of interest. Jakub Adamek declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human or animal subjects.
Informed Consent
Informed consent was not applicable.
Electronic supplementary material
ESM 1
(DOC 108 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bajkacz, S., Adamek, J. Development of a Method Based on Natural Deep Eutectic Solvents for Extraction of Flavonoids from Food Samples. Food Anal. Methods 11, 1330–1344 (2018). https://doi.org/10.1007/s12161-017-1118-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-017-1118-5