
Abstract.
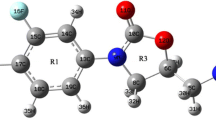
The Hatree-Fock method with 6-311G** split-valence molecular orbitals basis sets and the density function theory-B3LYP have been applied to geometrical optimizations and calculations of total electronic, zero point vibrational energies and proton affinities at 298 K for volatile organic compounds. Calculated values of proton affinities are compared with experimental data.
Similar content being viewed by others


References
R. Stewart, The Proton: Appellation to Organic Chemistry (Academic Press, New York, 1985)
F.A. Carrol, Perspectives on Structure and Mechanism in Organic Chemistry (Brooks-Cole, New York, 1998), p. 394
J. Zhao, R. Zhang, Atmos. Env. 38, 2177 (2004)
R.A. Kennedy, Ch.A. Mayhew, R. Thomas, P. Watts, Int. J. Mass Spectrom. 223–224, 627 (2003)
A. Hansel, N. Oberhofer, W. Lindinger, V.A. Zenevich, G.B. Billing, Int. J. Mass Spectr. 185, 186, 187, 559 (1999)
D.A. Dixon, S.G. Lias, Molecular Structure and Energetics, Vol. 2, Physical Measurements, edited by J.F. Liebman, A. Greenberg (VCH, Deereld Beach, FL, 1987)
M. Meot-Ner, J. Am. Chem. Soc. 101, 2396 (1979)
S.G. Lias, J.F. Liebman, R.D. Levin, J. Phys. Chem. Ref. Data 13, 695 (1984)
L.A. Curtiss, K. Raghavachari, P.A. Pople, J. Chem. Phys. 98, 1293 (1993)
J.E. Del Bene, J. Phys. Chem. 87, 367 (1983)
B.J. Smith, L. Radom, J. Phys. Chem. 99, 6468 (1995)
B.S. Jursic, J. Mol. Structure (Theochem.) 487, 193 (1999)
S. Hammerum, Chem. Phys. Lett. 300, 529 (1999)
J.L. Ozment, A.M. Schmiedekamp, Int. J. Quantum Chem. 43, 783 (1992)
D.A. McQuarrie, Statistical Mechanics (Harper & Row, New York, 1976)
R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, J. Chem. Phys. 72, 650 (1980)
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37, 785 (1988)
E.P.L. Hunter, S.G. Lias, J. Phys. Chem. Ref. Data 27, 413 (1998)
National Institute of Standards and Technology, http://webbook.nist.gov/chemistry/pa-ser.html
T. Wróblewski, L. Ziemczonek, K. Szerement, G.P. Karwasz, Czech. J. Phys. B 56, B1110 (2006)
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Wróblewski, T., Ziemczonek, L., Alhasan, A. et al. Ab initio and density functional theory calculations of proton affinities for volatile organic compounds. Eur. Phys. J. Spec. Top. 144, 191–195 (2007). https://doi.org/10.1140/epjst/e2007-00126-7
Issue Date:
DOI: https://doi.org/10.1140/epjst/e2007-00126-7