
Abstract
Purpose
Inflammatory myofibroblastic tumor (IMT) is a rare benign neoplasm. The purpose of this study was to review the clinical characteristics, imaging and pathological features, and outcomes of children with IMTs from a single center in China.
Methods
A retrospective file review was conducted involving 23 cases of pathologically confirmed IMTs treated at the Children’s Hospital between April 2003 and April 2014.
Results
The tumor locations included multiple anatomic sites, as follows: abdomen or pelvis (n = 17); lungs (n = 2); head and neck (n = 1); trunk (n = 1); and extremities (n = 2). The tumors were associated with various clinical presentations. The predominant symptoms included an anemic appearance, fevers, and an asymptomatic mass. Computed tomography scanning showed solid, heterogeneous, well-demarcated masses; the appearance of enhancement was variable. MRI appeared hypointense on T1-weighted images and hypointense or hyperintense on T2-weighted images. Immunohistochemical staining revealed anaplastic lymphoma kinase was negative in 11 of 13 cases tested. One patient quit treatment for the unresectable mass after biopsy and died 2 years later, and another patient with incompletely resection is alive at 30 months following chemotherapy. The remaining 21 cases had complete resections; one patient died due to a recurrence, and the other 20 patients survived and were tumor free. The follow-up ranged from 7 to 141 months, with a mean of 56 months. The 3-year OS was 88 % (95 % CI, 57–97 %).
Conclusions
IMT is a benign neoplasm that rarely presents with malignant features. Complete resection is curative in most patients. ALK+ is variable for diagnosis. Close follow-up is necessary for patients who undergo surgical resection.
Similar content being viewed by others


Introduction
Inflammatory myofibroblastic tumors (IMTs) are neoplasms with intermediate biologic potential that frequently recur and rarely metastasize [1]. IMTs often occur primarily in children and young adults [1]. IMTs can be found in various tissues and organs in the body. Originally described in the lung, IMTs have been occasionally reported in various extrapulmonary sites, including the liver, skin, orbit, gingiva, breast, thyroid, thymus, spleen, lymph nodes, and salivary glands [2]. Most patients with IMTs are young (average = 10 years) and present with a mass, with or without constitutional manifestations, including fever, weight loss, and various clinical laboratory abnormalities [3]. Surgical resection is the principal treatment [4]. In the present study, to examine disease features, imaging characteristics, treatment approach, and outcomes for children with IMTs, a retrospective analysis was performed to describe a single institution’s experience with the management of children with IMTs.
Methods
Twenty-three patients with a diagnosis of IMT attending the Children’s Hospital between April 2003 and April 2014 were identified, and the clinical charts were reviewed retrospectively to document presentation, treatment, and follow-up subsequent to obtaining approval from the local Research Ethics Committee of the hospital and written informed consent from the parents of each patient. Overall survival (OS) was estimated using the Kaplan–Meier method.
Results
Clinical analysis
The participants included 13 male and ten female patients. The ages of the patients at the time of diagnosis ranged between 4 months and 13 years, with a mean age of 4.5 years. The tumor locations included multiple anatomic sites, as follows: lungs (n = 2); head and neck (n = 1); trunk (n = 1); extremities (n = 2); and abdomen or pelvis (n = 17) including the stomach (n = 5),mesentery (n = 3), bladder (n = 2), appendix (n = 1), omentum (n = 1), ileocecus (n = 1), rectum (n = 1), retroperitoneum (n = 1), pancreas (n = 1), liver (n = 1), and common bile duct (n = 1). One patient had an IMT in two anatomic sites. The presenting symptoms varied according to the location of the tumor. The predominant symptoms included an asymptomatic mass (n = 9), an anemic appearance (n = 8), and fevers (n = 7). In addition, variable presentations included abdominal pain (n = 3), constipation (n = 1), bloody stools (n = 1), cough (n = 1), jaundice (n = 3), and vomiting (n = 1) in select patients.
Laboratory investigations
A complete blood cell count revealed anemia in eight patients; the hemoglobin ranged from 36 to 90 g/L, with a mean of 58 g/L. Seven IMTs were gastrointestinal, especially gastric IMTs (n = 4). The serum AFP and CEA levels were normal in 17 patients. The tumor locations in three patients with jaundice included the pancreas (n = 1), liver (n = 1), and common bile duct (n = 1).
Imaging analysis
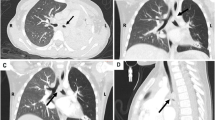
US revealed that IMTs were solid tumors, which had heterogeneity accompanied by hypoechoic areas and abundant blood flow signals with or without a capsule. CT scanning revealed low-density, solid, heterogeneous, well-demarcated (sometimes infiltrative) masses with necrotic or calcified components in several cases. The appearance of enhancement was variable. After contrast enhancement, peripheral enhancement may occur in tumors in the early phases of dynamic CT, while moderate or marked homogeneous or heterogeneous enhancement may be seen in solid tumors. MRI revealed varying images that appeared hypointense on T1-weighted images and hypointense or hyperintense on T2-weighted images. With intravenous contrast, there was peripheral enhancement; the capsule was enhanced, particularly on delayed phase images with increased enhancement of the central areas.
Pathologic analysis
The gross appearance was a circumscribed solitary or multi-nodular mass with a white-to-tan fleshy cut surface, ranging in size between 1 cm × 2 cm × 1.7 cm and 12 cm × 12 cm × 12 cm, with an average volume of 297.6 cm3, and the maximum diameter was 12 cm. Tumors in four patients showed infiltrative growth. The tumors in five patients were well circumscribed with capsules. The remaining tumors had no infiltrative growth or capsules. One case involved both the bladder and appendix. Necrosis and calcifications were noted in a few cases. Two histologic patterns were demonstrated in our study [a myxoid-vascular pattern (n = 1) and a compact spindle cell pattern (n = 22; Fig. 1a)]. On immunohistochemical staining (Table 1), spindle cells were positive for vimentin (VIM 20/20), muscle-specific actin (MSA, 8/11; Fig. 1b), and smooth muscle actin (SMA, 16/19; Fig. 1c), but negative for cluster of differentiation 34 [CD34 (15/16)], cluster of differentiation 117 [CD117 (11/14)], and anaplastic lymphoma kinase [ALK (11/13); Fig. 1d].

a Hematoxylin and eosin staining. The tumor comprised plump spindle cells with a background of an inflammatory infiltrate of plasma cells. b Immunohistochemical staining, negative for ALK. c Immunohistochemical staining, positive for SMA. d Immunohistochemical staining, positive for MSA
Diagnosis
The diagnosis, based on the clinical manifestations and imaging characteristics, had little specificity. Ten cases were initially misdiagnosed [10/23 (43 % and included lymphomas (n = 3), rhabdomyosarcomas (n = 2), a neuroblastoma (n = 1), a teratoma (n = 1; Fig. 2], a gastrointestinal stromal tumor (n = 1), a hemangioma (n = 1), and an appendicitis (n = 1). All patients had a pathologically confirmed diagnosis of IMT after surgical intervention.

Children with gastric IMTs. CT scan showed calcification (arrow), misdiagnosed as teratoma before surgery
Management and outcomes
All the patients had biopsies or surgical treatment. One patient with a rectal IMT quit treatment for the unresectable mass after biopsy and died 2 years later. A patient with a bladder IMT underwent incomplete resection in a local hospital; however, multiple local recurrences were noted in 1 year. After transfer to our hospital he received adjuvant chemotherapy and incomplete resection again and has survived with the tumor for 30 months. Complete resection was performed in the other 21 patients. A recurrence occurred extensively on the thoracic wall of a patient with a lung IMT after 7 months. This may have been a satellite lesion which was overlooked, originating from implanted metastatic lesions of the original tumor. Also, it was possible the original resection was incomplete and a neoplastic blood metastasis caused the reoccurrence. The patient discontinued treatment and died after 16 months. Recurrences in two patients were observed an average of 8.5 months after surgery. The other patients survived without recurrences. Two of the patients received post-operative chemotherapy. Both patients were treated with CYVADIC regimen (CTX, VCR, ADM, DTIC) and IVA regimen (IFO, VCR, ACTD), respectively. Both survived tumor free. In total, two patients died at an average of 23 months after discontinuing treatment; one survived with a tumor, and the other 20 patients currently have tumor-free survival. The follow-up ranged from 7 to 141 months, with a mean of 56 months. The 3-year OS was 88 % (95 % CI 57–97 %).
Discussion
IMT was previously described as an inflammatory pseudotumor, inflammatory myofibroblastoma, lymphoplasmacytic, histiocytoma, and fibrous pseudotumor; however, these lesions were synonymous and all shared the following key pathologic differentiation: spindle-shaped myofibroblastic cells; and an intense inflammatory process, mainly involving lymphocytes and plasma cells [5]. In 1994, IMTs were established as distinct low-grade malignancies by the World Health Organization classification [6, 7]. The exact etiology and pathogenesis of IMTs are unclear. In a review of the medical literature, it has been suggested that trauma, surgery, autoimmune etiologies, inflammation, and infection by the Epstein-Barr virus or human herpes virus could result in the development of IMTs [5, 8].
IMTs have been reported in various sites of the body and are more commonly seen in the lungs than the abdomen [9]. In the present study, however, abdominal and pelvic IMTs (74 %) were more frequently observed, which was in agreement to the report by Coffin et al. [10]. A slight male preponderance (M:F = 1.3) was reported in previous studies [1, 5, 11]. The clinical presentation was determined by the site of origin and the effects of the mass. The non-specific symptoms can include an anemic appearance, fevers, abdominal pain, or a palpable abdominal mass with non-specific laboratory findings, such as iron deficiency anemia and jaundice. We noted that gastrointestinal IMTs, especially gastric IMTs [4/5 (80 %)] were usually accompanied by anemia. Some symptoms were related to the compression effect of the tumor, such as jaundice, when the tumor was located in the pancreas, liver, and common bile duct. With respect to imaging characteristics, on CT scan a solid heterogeneous mass was usually observed with a variable appearance of enhancement. MRI showed peripheral enhancement, which was believed to reflect the slow washout from the inflamed fibrous tissue [12]. In the absence of pathognomonic features on imaging, the diagnosis was usually obtained only by histopathologic examination of the specimens. The differential diagnosis was made with malignant tumors, such as teratomas, rhabdomyosarcomas, and lymphomas.
Three histologic appearances have been described, as follows: a myxoid-vascular pattern resembling nodular fasciitis; a compact spindle cell pattern resembling a variety of spindle cell neoplasms; and a hypocellular fibrous pattern resembling a scar or desmoid fibromatosis [3]. In the present study, the myxoid-vascular pattern accounted for 4 % (1/23) of all cases, the compact spindle cell pattern accounted for 96 % (22/23) of all cases, and no hypocellular fibrous pattern type was observed, which suggested that the compact spindle cell pattern was more common than the other patterns. These cells often express classical myogenic markers (SMA and MSA), which is still compatible with the diagnosis of IMT and supports the myofibroblastic nature. The expression of the ALK was recently suggested as a good marker for IMT, and the frequency of positive ALK in IMTs was reported to range from 50 to 70 % [3, 7, 10, 13]; however, ALK was a variable marker for IMT diagnosis in this study. ALK immunohistochemistry was negative in 11 of 13 cases (85 %). Though the reason for the low positive rate maybe we have not checked the ALK rearrangements by much more precise methods like FISH or RT-PCR, or it’s the genetic characteristics of this Chinese children population, which need further research. It was risky to diagnose IMT based mainly on ALK staining, especially when the histology was atypical.
IMTs share neoplastic characteristics of aggressive local tissue infiltration, recurrence after resection, and occasionally, distant metastases [10, 14]. Nevertheless, IMTs are generally considered benign tumors, for which the primary therapeutic approach is surgery if the anatomic location is amenable to a resection. In our group, the patient with a rectal IMT discontinued treatment for the unresectable mass, which had infiltrative local growth. The other three patients observed with infiltrative IMTs received complete resections and survived tumor free. Due to the possible multi-focality or metastases, a patient was described involving two anatomic sites (appendix and bladder), with complete resection and tumor-free survival. Chemotherapy, radiotherapy, immunomodulation with corticosteroids, and non-steroidal anti-inflammatory drugs have not been reported to be consistently effective against this aggressive tumor [9, 12, 15]. Consequently, radical resection is the curative treatment for IMTs. In patients with unresectable IMTs, treatment options are limited, and better recommendations have not been offered.
The role of chemotherapy is unclear based upon our limited experience. In our study, the patient with a bladder IMT and incomplete resection followed by subsequent rounds of chemotherapy with three varying chemotherapeutic regimens survived with tumor at 30 months of follow-up. The other two patients received post-operative chemotherapy. There is little evidence within the literature regarding chemotherapy for IMTs. The effect of chemotherapy is controversial; various chemotherapeutic drugs or regimens have been used with variable responses [6].
The recurrence rate has been reported to range from 25 to 40 %, and has been suggested to be more common if the lesions are extrapulmonary [5, 16]. Precise removal with adequate margins has great prognostic significance,and the recurrence rate is <10 % when complete resection is achieved [4, 17]. Our series demonstrates a recurrence rate of 5 % (1/21) with a primarily resective approach, which re-occurred after 7 months. The patient with a bladder IMT underwent incomplete resection. Residual tumor typically grows progressively over a follow-up period of 10 months [1/1 (100 %)]. Given this finding, we would suggest that radical surgical resection should be recommended for all lesions if not prohibited by anatomic location or morbidity. For the possibility of recurrence and metastases, careful clinical, biologic, and radiographic follow-up is required after surgical resection [18].
In conclusion, IMTs are benign neoplasms that rarely present with malignant features. Complete resection is recommended as the curative treatment for most patients with IMTs. The compact spindle cell pattern is the most common histopathologic type of IMT. ALK+ is a variable marker for diagnosis. The effect of chemotherapy for IMT is controversial; more experience is required. Close follow-up is necessary for patients with IMTs after surgical resection.
References
Mehta B, Mascarenhas L, Zhou S, Wang L, Venkatramani R (2013) Inflammatory myofibroblastic tumors in childhood. Pediatr Hematol Oncol 30:640–645
Nagarajan S, Jayabose S, McBride W, Prasadh I, Tanjavur V, Marvin MR, Rodriguez-Davalos MI (2013) Inflammatory myofibroblastic tumor of the liver in children. J Pediatr Gastroenterol Nutr 57:277–280
Cook JR, Dehner LP, Collins MH, Ma Z, Morris SW, Coffin CM, Hill DA (2001) Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol 25:1364–1371
Diop B, Konate I, Ka S, Sall I, Fall D, Dieng M, Wone Y (2011) Mesenteric myofibroblastic tumor: NSAID therapy after incomplete resection. J Visc Surg 148:e311–e314
Kovach SJ, Fischer AC, Katzman PJ, Salloum RM, Ettinghausen SE, Madeb R, Koniaris LG (2006) Inflammatory myofibroblastic tumors. J Surg Oncol 94:385–391
Mattei P, Barnaby K (2008) Rapid regression of duodenal inflammatory myofibroblastic tumor after intravenous ketorolac: case report and review of the literature. J Pediatr Surg 43:1196–1199
Fletcher CDM, Unni KK, Mertens F (2002) World Health Organization classification of tumors. Pathology and genetics of tumors of soft tissue and bone. IARC Press, Lyon, pp 48–106
Tao YL, Wang ZJ, Han JG, Wei P (2012) Inflammatory myofibroblastic tumor successfully treated with chemotherapy and nonsteroidals: a case report. World J Gastroenterol 18:7100–7103
Bertocchini A, Lo ZC, Callea F, Gennari F, Serra A, Monti L, de Ville DGJ (2011) Unresectable multifocal omental and peritoneal inflammatory myofibroblastic tumor in a child: revisiting the role of adjuvant therapy. J Pediatr Surg 46:e17–21
Coffin CM, Hornick JL, Fletcher CD (2007) Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 31:509–520
Karnak I, Senocak ME, Ciftci AO, Caglar M, Bingol-Kologlu M, Tanyel FC, Buyukpamukcu N (2001) Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 36:908–912
Yang X, Miao R, Yang H, Chi T, Jiang C, Wan X, Xu Y, Xu H, Du S, Lu X, Mao Y, Zhong S, Zhao H, Sang X (2014) A retrospective and comparative study of inflammatory myofibroblastic tumor of the liver. J Gastroenterol Hepatol 30:885–890
Cessna MH, Zhou H, Sanger WG, Perkins SL, Tripp S, Pickering D, Daines C, Coffin CM (2002) Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol 15:931–938
Dong A, Wang Y, Dong H, Gong J, Cheng C, Zuo C, Lu J (2014) Inflammatory myofibroblastic tumor: FDG PET/CT findings with pathologic correlation. Clin Nucl Med 39:113–121
Fernandez DLPG, Garcia AJ, Lechuga VM, Blanco MM (1999) Hepatic inflammatory pseudotumor: apropos a case with a response to steroid treatment. Gastroenterol Hepatol 22:14–17
Coffin CM, Watterson J, Priest JR, Dehner LP (1995) Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 19:859–872
Janik JS, Janik JP, Lovell MA, Hendrickson RJ, Bensard DD, Greffe BS (2003) Recurrent inflammatory pseudotumors in children. J Pediatr Surg 38:1491–1495
Lazure T, Ferlicot S, Gauthier F, Doz F, Couturier J, Fabre M, Bedossa P (2002) Gastric inflammatory myofibroblastic tumors in children: an unpredictable course. J Pediatr Gastroenterol Nutr 34:319–322
Acknowledgments
The authors are indebted to all participating patients and physicians for their participation in the study. The authors are also indebted to Dr. Lian Chen and Dr. Yangyang Ma who participated in our research and provided lots of suggestions to pathologic analysis.
This study received grants support from Shengkang Hospital Development Center (SHDC12012220 by Xiaolong Zhao, Department of Endocrinology, Huashan Hospital of Fudan University) and Shanghai Human Resources and Social Security (201342, by Kai LI, Department of Pediatric Surgery, Children’s Hospital of Fudan University).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts to disclose relative to this article.
Rights and permissions
About this article

Cite this article
Wang, Z., Zhao, X., Li, K. et al. Analysis of clinical features and outcomes for inflammatory myofibroblastic tumors in China: 11 years of experience at a single center. Pediatr Surg Int 32, 239–243 (2016). https://doi.org/10.1007/s00383-015-3840-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-015-3840-7