
Abstract
Esthesioneuroblastoma, also often called olfactory neuroblastoma, is a rare tumor thought to arise from the olfactory neuroepithelium. It may occur at any age and accounts for 1–5% of intranasal tumors with an estimated incidence of 0.4/million population (Broich et al. 1997; Ferlito et al. 2003; Thompson 2009).
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Esthesioneuroblastoma, also often called olfactory neuroblastoma, is a rare tumor thought to arise from the olfactory neuroepithelium. It may occur at any age and accounts for 1–5% of intranasal tumors with an estimated incidence of 0.4/million population (Broich et al. 1997; Ferlito et al. 2003; Thompson 2009). No sex predisposition has been reported. Recent reports favor a unimodal age distribution with the majority of patients diagnosed in the fourth and fifth decades of life (Resto et al. 2000; Ferlito et al. 2003; Jethanamest et al. 2007), but previously an additional peak of incidence in the second decade has been claimed (Elkon et al. 1979).
In children esthesioneuroblastoma is rare with an estimated incidence of 0.1/100,000 children up to 15 years, but it is the most frequent cancer of the nasal cavity in this age group, representing 28% of cases registered in a series of 47 patients below 19 years with nasal cavity tumors in the SEER database from 1973 to 2002 (Benoit et al. 2008). Only single cases have been reported in young children below 10 years of age, the youngest reported case being as young as 2 years (Woerner et al. 1986; Perkkio et al. 1991; Bobele et al. 1994; Kumar et al. 2002; Eich et al. 2005; Jethanamest et al. 2007).
The clinical and histological diagnosis of esthesioneuroblastoma is confusing, and misdiagnosis has been reported in quite substantial parts of patients, where histology was reassessed according to current immunohistochemical criteria (Hirose et al. 1995; Resto et al. 2000; Cohen et al. 2002; Eich et al. 2005). Therefore, clinical features reported in the literature should be interpreted with cautiousness if not proved in recently reported series.

Esthesioneuroblastoma, histology
1 Pathology
Esthesioneuroblastoma is a tumor with small, round, blue tumor cells arranged in a lobular architecture in neurofibrillary stroma (Fig. 18.1). Rosettes and pseudorosettes as well as calcifications may be found. Based on assessment of lobular tumor architecture, mitotic activity, nuclear pleomorphism, rosettes, and tumor necrosis, Hyams et al. proposed a grading system, which is correlated to prognosis (Hyams et al. 1988). Immunohistochemically, esthesioneuroblastoma may stain positive for synaptophysin, chromogranin, CD56, neuron-specific enolase, NFP, and S-100 protein, but negative for desmin, myogenin, leukocyte common antigen, and CD99 (reviewed in Faragalla and Weinreb 2009; Thompson 2009).
Due to the rareness of the disease, histological evaluation by a second experienced pathologist should be aimed for. Other small round cell tumors as rhabdomyosarcoma, tumors of the Ewing tumor family, neuroblastoma, lymphoma, and, less common in childhood, neuroendocrine carcinoma, squamous cell carcinoma, and sinonasal undifferentiated carcinoma, have to be ruled out. Although previously controversially discussed (Sorensen et al. 1996), it has meanwhile been shown that esthesioneuroblastoma does not belong to the Ewing tumor family, as CD99/MIC staining and the typical translocations are lacking (Nelson et al. 1995; Argani et al. 1998; Mezzelani et al. 1999). From the histopathological point of view, metastatic neuroblastoma would present with identical findings as esthesioneuroblastoma. Amplification of the MYCN oncogen, which is found in many aggressive neuroblastomas, has so far not been reported in esthesioneuroblastoma. Thus, in single cases, molecular assessment of the tumor specimen, showing Ewing tumor family typic translocations or MYCN amplification, may be helpful to rule out esthesioneuroblastoma.
2 Staging
The origin of esthesioneuroblastoma is confined to the olfactory mucosa involving the superior turbinate, cribriform plate, and the superior one third of the nasal cavity. It may spread into the paranasal sinuses, the orbits, and – through the lamina cribriformis – into the cranial cavity. Although dystopic sites of origin in the nasopharynx, in the maxillary sinuses, and intracranially have been reported (Seccia et al. 2010; Banerjee et al. 1992; Jugie et al. 1992; Sharma et al. 2002; Mariani et al. 2004; Wormald et al. 2011), a diagnosis of esthesioneuroblastoma outside the nasal cavity should only be made with great cautiousness (Mills 2002).
Symptoms are related to the site of origin and the local invasion and may present as long as several months prior to definite diagnosis (Dulguerov and Calcaterra 1992). Unilateral nasal obstruction, recurrent epistaxis, and – less common – anosmia are observed as well as ophthalmic manifestations like periorbital pain, excessive tearing, visual disturbance, or ptosis (Rakes et al. 1985). Occasionally headache, nerve palsies due to involvement of cranial nerves, or hormone excess syndromes such as Cushing syndrome or inappropriate antidiuretic hormone secretion have been reported (Osterman et al. 1986; Arnesen et al. 1994; Myers et al. 1994).
In 1976, Kadish et al. (1976) proposed a staging system based on the pattern of spread (Table 18.1). Kadish A tumors are confined to the nasal cavity, while Kadish B tumors infiltrate the paranasal cavities. Kadish C tumors extend beyond the nasal and paranasal cavities (Kadish et al. 1976). Later, the system has been modified by adding the category Kadish D for tumors with metastases. The Kadish system correlates to prognosis and is still in use, although other systems based on the TMN classification have been proposed (Biller et al. 1990; Dulguerov and Calcaterra 1992). Larger cohorts report about one third of the patients to present with Kadish stage C.
Distant metastases are thought to be very rare and to occur in less than 10% of patients. Metastases to lung, CNS, bone, liver, and bone marrow have been reported (Franklin et al. 1987; Koka et al. 1998; Chao et al. 2001; Argiris et al. 2003; Bradley et al. 2003; Eich et al. 2003; Bachar et al. 2008). It is assumed that, due to the difficult diagnosis, parts of those reports include misdiagnosed tumors and that the real proportion of esthesioneuroblastoma with distant metastases is even lower, as reflected in more recent published series (Diaz et al. 2005). However, locoregional spread to cervical lymph nodes seems to be encountered more often. Approximately 5–10% of all patients present with evidence of disease in the neck, while cervical lymph node involvement in the course of the disease will be found up to a quarter of patients (Dulguerov et al. 2001; Rinaldo et al. 2002; Bachar et al. 2008; Gore and Zanation 2009; Ozsahin et al. 2010).
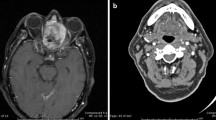
Investigations at initial work-up include CT scan which usually shows a homogenously enhancing lesion, with bone erosion, invasion of the adjacent structures, and often with calcifications. MRI images help to delineate the extent of the disease, which appear isointense or hypointense to brain on T1-weighted images and hyperintense on T2-weighted images with marked enhancement after gadolinium (reviewed in Thompson 2009) (Fig. 18.2).

Esthesioneuroblastoma, Kadish stage C, infiltrating the orbita, the nasal and paranasal cavities, and, through the lamina cribriformis, the brain (MRI)
Besides the imaging of the primary tumor site and the neck, the initial work-up should include the search for distant metastases in CNS, lung, liver, bone, and bone marrow. In bone marrow, multiple sites should be assessed as the diagnosis of neuroblastoma or rhabdomyosarcoma should be considered. Somatostatin receptor imaging and FDG-PET have been reported to give positive results in patients with esthesioneuroblastoma and might be helpful in assessing the extent of the disease at diagnosis and during treatment (Ramsay et al. 1996; Freeman et al. 2005; Nguyen et al. 2006; Rostomily et al. 2006). Only a single patient with esthesioneuroblastoma has been reported to show positive mIBG uptake (Kairemo et al. 1998), but mIBG scintigraphy might be helpful in those cases where a metastatic lesion of a neuroblastoma is discussed. However, these imaging techniques have not been systematically addressed in larger cohorts of patients with esthesioneuroblastoma so far.
3 Prognosis and Therapy
Due to the rareness of the disease, literature on prognosis is not reflected on large, homogenously treated cohorts. Nevertheless, it seems quite clear that grading, staging, the presence of metastases, and the treatment received influence prognosis. Kadish et al. reported patients with advanced stage (Kadish stage C) to be younger at diagnosis (median age 30.4 years) (Kadish et al. 1976). Vice versa, older age at diagnosis was correlated with better outcome in one series (<61 vs. >61 years) (Ozsahin et al. 2010), but this was not confirmed by others (<20 vs. >20 years (Eich et al. 2003); <50 vs. >50 years (Dulguerov and Calcaterra 1992)). In childhood, a high incidence of advanced stages is discussed (Lochrin 1989; Kumar et al. 2002), leading to the assumption that esthesioneuroblastoma in childhood shows an aggressive behavior; however this has never been proven in larger comparative studies.
To some extent, the amount of therapy needed is discussed controversially in the literature. Table 18.1 sets our proposal for the treatment in children in relation to the survival estimates as reported from a large cohort of 261 patients with esthesioneuroblastoma (Jethanamest et al. 2007).
Complete resection of the primary has been correlated to prognosis and is considered the backbone of all treatment strategies (Goldsweig and Sundaresan 1990; Devaiah and Andreoli 2009). Planning of the surgery may involve different subdisciplines (head and neck surgeons, neurosurgeons, ophthalmologists). Although most often open surgeries have been performed, endoscopic resections seem not to be inferior, as long as oncologic principles with clearance of margins are maintained (Lund et al. 2010; Snyderman et al. 2008; Folbe et al. 2009). To gain resectability in locally extended tumors, preoperative chemotherapy or radiotherapy proved efficient (Foote et al. 1993; Eich et al. 2003; Eich et al. 2005).
Kadish stage A tumors, especially when presenting with low-grade histology, seem to be sufficiently treated by surgery alone (with or without radiotherapy), but account only for a minor part of patients. In higher stages, the addition of radiotherapy with doses ranging from 55 to 65 Gy is claimed (Foote et al. 1993; Chao et al. 2001; Dulguerov et al. 2001; Eich et al. 2001). The planning of the radiotherapy may be hampered by adjacent endangered structures as eye and CNS and may require modern radiation techniques as intensity-modulated radiotherapy, proton irradiation, or stereotactic radiosurgery (Bhattacharyya et al. 1997; Walch et al. 2000; Zabel et al. 2002a, b; Tselis et al. 2008; Sterzing et al. 2009).
Although some authors abrogate the positive influence of chemotherapy, most authors support the need for a multimodal treatment strategy including surgery, radiotherapy, and chemotherapy in tumors of Kadish stage C (with or without metastases), however, without reaching a consensus on kind and amount of chemotherapy needed (Goldsweig and Sundaresan 1990; McElroy et al. 1998; Oskouian et al. 2002; Eich et al. 2003, 2005; Loy et al. 2006; McLean et al. 2007; Kiyota et al. 2008; Nichols et al. 2008; Porter et al. 2008). In children, chemotherapy regimen of soft tissue sarcoma or neuroblastoma protocols have often been used; in adults, treatment with platinum-based regimens and others have been reported.
If cervical lymph nodes are involved, neck dissection and postoperative radiation therapy are discussed (Zanation et al. 2010), while cervical treatment seems not required in N0 patients. However, to discover late-evolving lymph nodes, adequate imaging of the neck should always be included in the follow-up of the patients (Gore and Zanation 2009).
Relapses and metastases usually develop within the first 2–3 years after diagnosis, but late relapses more than 5 years after therapy have been reported (Morita et al. 1993; Eden et al. 1994; Loy et al. 2006), indicating the need for a long follow-up, which, in a childhood population, should always include the care for therapy-related sequelae. The need for local control including expanded surgical procedures and high-dose radiotherapy poses specific problems in the pediatric age. Long-term sequelae in children include damage to craniofacial growth and permanent dentition, endocrine dysfunctions, and loss of sense of smell.
References
Argani P, Perez-Ordonez B et al (1998) Olfactory neuroblastoma is not related to the Ewing family of tumors: absence of EWS/FLI1 gene fusion and MIC2 expression. Am J Surg Pathol 22(4):391–398
Argiris A, Dutra J et al (2003) Esthesioneuroblastoma: the Northwestern University experience. Laryngoscope 113(1):155–160
Arnesen MA, Scheithauer BW et al (1994) Cushing’s syndrome secondary to olfactory neuroblastoma. Ultrastruct Pathol 18(1–2):61–68
Bachar G, Goldstein DP et al (2008) Esthesioneuroblastoma: the Princess Margaret Hospital experience. Head Neck 30(12):1607–1614
Banerjee AK, Sharma BS et al (1992) Intracranial olfactory neuroblastoma: evidence for olfactory epithelial origin. J Clin Pathol 45(4):299–302
Benoit MM, Bhattacharyya N et al (2008) Cancer of the nasal cavity in the pediatric population. Pediatrics 121(1):e141–e145
Bhattacharyya N, Thornton AF et al (1997) Successful treatment of esthesioneuroblastoma and neuroendocrine carcinoma with combined chemotherapy and proton radiation. Results in 9 cases. Arch Otolaryngol Head Neck Surg 123(1):34–40
Biller HF, Lawson W et al (1990) Esthesioneuroblastoma: surgical treatment without radiation. Laryngoscope 100(11):1199–1201
Bobele GB, Sexauer C et al (1994) Esthesioneuroblastoma presenting as an orbital mass in a young child. Med Pediatr Oncol 22(4):269–273
Bradley PJ, Jones NS et al (2003) Diagnosis and management of esthesioneuroblastoma. Curr Opin Otolaryngol Head Neck Surg 11(2):112–118
Broich G, Pagliari A et al (1997) Esthesioneuroblastoma: a general review of the cases published since the discovery of the tumour in 1924. Anticancer Res 17(4A):2683–2706
Chao KS, Kaplan C et al (2001) Esthesioneuroblastoma: the impact of treatment modality. Head Neck 23(9):749–757
Cohen ZR, Marmor E et al (2002) Misdiagnosis of olfactory neuroblastoma. Neurosurg Focus 12(5):e3
Devaiah AK, Andreoli MT (2009) Treatment of esthesioneuroblastoma: a 16-year meta-analysis of 361 patients. Laryngoscope 119(7):1412–1416
Diaz EM Jr, Johnigan RH 3rd et al (2005) Olfactory neuroblastoma: the 22-year experience at one comprehensive cancer center. Head Neck 27(2):138–149
Dulguerov P, Calcaterra T (1992) Esthesioneuroblastoma: the UCLA experience 1970–1990. Laryngoscope 102(8):843–849
Dulguerov P, Allal AS et al (2001) Esthesioneuroblastoma: a meta-analysis and review. Lancet Oncol 2(11):683–690
Eden BV, Debo RF et al (1994) Esthesioneuroblastoma. Long-term outcome and patterns of failure–the University of Virginia experience. Cancer 73(10):2556–2562
Eich HT, Staar S et al (2001) Radiotherapy of esthesioneuroblastoma. Int J Radiat Oncol Biol Phys 49(1):155–160
Eich HT, Hero B et al (2003) Multimodality therapy including radiotherapy and chemotherapy improves event-free survival in stage C esthesioneuroblastoma. Strahlenther Onkol 179(4):233–240
Eich HT, Muller RP et al (2005) Esthesioneuroblastoma in childhood and adolescence. Better prognosis with multimodal treatment? Strahlenther Onkol 181(6):378–384
Elkon D, Hightower SI et al (1979) Esthesioneuroblastoma. Cancer 44(3):1087–1094
Faragalla H, Weinreb I (2009) Olfactory neuroblastoma: a review and update. Adv Anat Pathol 16(5):322–331
Ferlito A, Rinaldo A et al (2003) Contemporary clinical commentary: esthesioneuroblastoma: an update on management of the neck. Laryngoscope 113(11):1935–1938
Folbe A, Herzallah I et al (2009) Endoscopic endonasal resection of esthesioneuroblastoma: a multicenter study. Am J Rhinol Allergy 23(1):91–94
Foote RL, Morita A et al (1993) Esthesioneuroblastoma: the role of adjuvant radiation therapy. Int J Radiat Oncol Biol Phys 27(4):835–842
Franklin D, Miller RH et al (1987) Esthesioneuroblastoma metastatic to the trachea. Head Neck Surg 10(2):102–106
Freeman SR, Mitra S et al (2005) Expression of somatostatin receptors in arginine vasopressin hormone-secreting olfactory neuroblastoma–report of two cases. Rhinology 43(1):61–65
Goldsweig HG, Sundaresan N (1990) Chemotherapy of recurrent esthesioneuroblastoma. Case report and review of the literature. Am J Clin Oncol 13(2):139–143
Gore MR, Zanation AM (2009) Salvage treatment of late neck metastasis in esthesioneuroblastoma: a meta-analysis. Arch Otolaryngol Head Neck Surg 135(10):1030–1034
Hirose T, Scheithauer BW et al (1995) Olfactory neuroblastoma. An immunohistochemical, ultrastructural, and flow cytometric study. Cancer 76(1):4–19
Hyams V, Batsakis JG, Michaels L, Armed Forces Institute of Pathology Fascicles (1988) Tumors of the upper respiratory tract and ear, vol 2. American Registry of Pathology Press, Washington D.C
Jethanamest D, Morris LG et al (2007) Esthesioneuroblastoma: a population-based analysis of survival and prognostic factors. Arch Otolaryngol Head Neck Surg 133(3):276–280
Jugie M, Marsot-Dupuch K et al (1992) Unusual localization of an olfactory esthesioneuroma. Ann Radiol (Paris) 35(6):477–482
Kadish S, Goodman M et al (1976) Olfactory neuroblastoma. A clinical analysis of 17 cases. Cancer 37(3):1571–1576
Kairemo KJ, Jekunen AP et al (1998) Imaging of olfactory neuroblastoma–an analysis of 17 cases. Auris Nasus Larynx 25(2):173–179
Kiyota N, Tahara M et al (2008) Nonplatinum-based chemotherapy with irinotecan plus docetaxel for advanced or metastatic olfactory neuroblastoma: a retrospective analysis of 12 cases. Cancer 112(4):885–891
Koka VN, Julieron M et al (1998) Aesthesioneuroblastoma. J Laryngol Otol 112(7):628–633
Kumar M, Fallon RJ et al (2002) Esthesioneuroblastoma in children. J Pediatr Hematol Oncol 24(6):482–487
Lochrin C (1989) Esthesioneuroblastoma. Med Pediatr Oncol 17(5):433–438
Loy AH, Reibel JF et al (2006) Esthesioneuroblastoma: continued follow-up of a single institution’s experience. Arch Otolaryngol Head Neck Surg 132(2):134–138
Lund VJ, Stammberger H, et al (2010) European position paper on endoscopic management of tumours of the nose, paranasal sinuses and skull base. Rhinol Suppl (22):1–143
Mariani L, Schaller B et al (2004) Esthesioneuroblastoma of the pituitary gland: a clinicopathological entity? Case report and review of the literature. J Neurosurg 101(6):1049–1052
McElroy EA Jr, Buckner JC et al (1998) Chemotherapy for advanced esthesioneuroblastoma: the Mayo Clinic experience. Neurosurgery 42(5):1023–1027, discussion 1027–1028
McLean JN, Nunley SR et al (2007) Combined modality therapy of esthesioneuroblastoma. Otolaryngol Head Neck Surg 136(6):998–1002
Mezzelani A, Tornielli S et al (1999) Esthesioneuroblastoma is not a member of the primitive peripheral neuroectodermal tumour-Ewing’s group. Br J Cancer 81(4):586–591
Mills SE (2002) Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinomas. Mod Pathol 15(3):264–278
Morita A, Ebersold MJ et al (1993) Esthesioneuroblastoma: prognosis and management. Neurosurgery 32(5):706–714, discussion 714–705
Myers SL, Hardy DA et al (1994) Olfactory neuroblastoma invading the oral cavity in a patient with inappropriate antidiuretic hormone secretion. Oral Surg Oral Med Oral Pathol 77(6):645–650
Nelson RS, Perlman EJ et al (1995) Is esthesioneuroblastoma a peripheral neuroectodermal tumor? Hum Pathol 26(6):639–641
Nguyen BD, Roarke MC et al (2006) F-18 FDG PET/CT staging and posttherapeutic assessment of esthesioneuroblastoma. Clin Nucl Med 31(3):172–174
Nichols AC, Chan AW et al (2008) Esthesioneuroblastoma: the Massachusetts eye and ear infirmary and Massachusetts general hospital experience with craniofacial resection, proton beam radiation, and chemotherapy. Skull Base 18(5):327–337
Oskouian RJ Jr, Jane JA Sr et al (2002) Esthesioneuroblastoma: clinical presentation, radiological, and pathological features, treatment, review of the literature, and the University of Virginia experience. Neurosurg Focus 12(5):e4
Osterman J, Calhoun A et al (1986) Chronic syndrome of inappropriate antidiuretic hormone secretion and hypertension in a patient with olfactory neuroblastoma. Evidence of ectopic production of arginine vasopressin by the tumor. Arch Intern Med 146(9):1731–1735
Ozsahin M, Gruber G et al (2010) Outcome and prognostic factors in olfactory neuroblastoma: a rare cancer network study. Int J Radiat Oncol Biol Phys 78(4):992–997
Perkkio M, Majuri S et al (1991) A child with esthesioneuroblastoma with metastases to the spinal cord and the bone marrow. Med Pediatr Oncol 19(1):66–69
Porter AB, Bernold DM et al (2008) Retrospective review of adjuvant chemotherapy for esthesioneuroblastoma. J Neurooncol 90(2):201–204
Rakes SM, Yeatts RP et al (1985) Ophthalmic manifestations of esthesioneuroblastoma. Ophthalmology 92(12):1749–1753
Ramsay HA, Kairemo KJ et al (1996) Somatostatin receptor imaging of olfactory neuroblastoma. J Laryngol Otol 110(12):1161–1163
Resto VA, Eisele DW et al (2000) Esthesioneuroblastoma: the Johns Hopkins experience. Head Neck 22(6):550–558
Rinaldo A, Ferlito A et al (2002) Esthesioneuroblastoma and cervical lymph node metastases: clinical and therapeutic implications. Acta Otolaryngol 122(2):215–221
Rostomily RC, Elias M et al (2006) Clinical utility of somatostatin receptor scintigraphic imaging (octreoscan) in esthesioneuroblastoma: a case study and survey of somatostatin receptor subtype expression. Head Neck 28(4):305–312
Seccia V, Lenzi R et al (2010) Ectopic olfactory neuroblastoma arising in the pterygopalatine fossa. Otolaryngol Head Neck Surg 142(3):460–461
Sharma SC, Reddy CE et al (2002) Isolated esthesioneuroblastoma of sphenoid sinus. Am J Otolaryngol 23(5):287–289
Snyderman CH, Carrau RL et al (2008) Endoscopic skull base surgery: principles of endonasal oncological surgery. J Surg Oncol 97(8):658–664
Sorensen PH, Wu JK et al (1996) Olfactory neuroblastoma is a peripheral primitive neuroectodermal tumor related to Ewing sarcoma. Proc Natl Acad Sci USA 93(3):1038–1043
Sterzing F, Stoiber EM et al (2009) Intensity Modulated Radiotherapy (IMRT) in the treatment of children and adolescents–a single institution’s experience and a review of the literature. Radiat Oncol 4:37
Thompson LD (2009) Olfactory neuroblastoma. Head Neck Pathol 3(3):252–259
Tselis N, Heyd R et al (2008) Interstitial high-dose-rate-brachytherapy in advanced esthesioneuroblastoma. Laryngoscope 118(11):2006–2010
Walch C, Stammberger H et al (2000) The minimally invasive approach to olfactory neuroblastoma: combined endoscopic and stereotactic treatment. Laryngoscope 110(4):635–640
Woerner SJ, Lazerson J et al (1986) Olfactory neuroblastoma (esthesioneuroblastoma) in a 2-year-old boy. Pediatr Hematol Oncol 3(2):167–174
Wormald R, Lennon P et al (2011) Ectopic olfactory neuroblastoma: report of four cases and a review of the literature. Eur Arch Otorhinolaryngol 268(4):555–560
Zabel A, Thilmann C et al (2002a) The role of stereotactically guided conformal radiotherapy for local tumor control of esthesioneuroblastoma. Strahlenther Onkol 178(4):187–191
Zabel A, Thilmann C et al (2002b) Comparison of forward planned conformal radiation therapy and inverse planned intensity modulated radiation therapy for esthesioneuroblastoma. Br J Radiol 75(892):356–361
Zanation AM, Ferlito A et al (2010) When, how and why to treat the neck in patients with esthesioneuroblastoma: a review. Eur Arch Otorhinolaryngol 267(11):1667–1671
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Hero, B., Bisogno, G. (2012). Esthesioneuroblastoma. In: Schneider, D., Brecht, I., Olson, T., Ferrari, A. (eds) Rare Tumors In Children and Adolescents. Pediatric Oncology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-04197-6_18
Download citation
DOI: https://doi.org/10.1007/978-3-642-04197-6_18
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-04196-9
Online ISBN: 978-3-642-04197-6
eBook Packages: MedicineMedicine (R0)