
Abstract
The immunoglobulin receptor represents a central molecule in acquired immunity. The complete set of immunoglobulins present in an individual is known as immunological repertoire. The identification of this repertoire is particularly relevant in immunology and cancer research and diagnostics. In a seminal work we provided a proof of concept of the novel ARTISAN-PCR amplification method, we adapted this technology for sequencing using Nanopore technology. This approach may represent a faster, more portable and cost-effective alternative to current methods. In this study we present the pipeline for the analysis of immunological repertoires obtained by this approach. This paper shows the performance of immune repertoires sequenced by Nanopore technology, using measures of error, coverage and gene usage identification.
In the bioinformatic methodology used in this study, first, Albacore Base calling software, was used to translate the electrical signal of Nanopore to DNA bases. Subsequently, the sequons, introduced during amplification, were aligned using bl2seq from Blast. Finally, selected reads were mapped using IMGT/HighV-QUEST and IgBlast.
Our results demonstrate the feasibility of immune repertoire sequencing by Nanopore technology, obtaining higher depth than PacBio sequencing and better coverage than pair-end based technologies. However, the high rate of systematic errors indicates the need of improvements in the analysis pipeline, sequencing chemistry and/or molecular amplification.
Similar content being viewed by others
Keywords
1 Introduction
Immunoglobulins are central proteins of the adaptive immune system since they fulfill a fundamental role of defense against foreign agents. These proteins are synthesized for B Cells and are constituted by two identical heavy chains and two identical light chains, joined by disulfide bridges. Immunoglobulins that can be found forming part of their membrane or being secreted outside of the cell [1, 2]. Its formation is the result of two main phenomena: recombination of 3 gene segments (known as V (variable), D (diversity) and J (junction)) and point mutations (Somatic hypermutation) [3]. Both phenomena generate differences between each rearrangement of immunoglobulin producing a repertoire close to 1011 per individual [4].
The sequencing and identification of the nucleic acids that codify for variable region of these proteins has diverse applications in research, diagnosis and treatment of cancer and other diseases of the immune system [5, 6].
However, the sequencing and analysis of the immune repertoire represents a unique challenge for molecular biology and bioinformatics due to the intrinsic high variability attained to recombinant gene segments and somatic hypermutation. Accurate repertoire measurements requires unbiased PCR amplification, high depth, and full read coverage. We have previously overcame the primer binding bias of standard multiplex PCR [7] by a novel ARTISAN-PCR strategy (Anchoring reverse transcription immunoglobulin sequence and amplification by nested PCR) [8]. After amplification repertoire are subjected to high throughput sequencing. Whereas PacBio sequencing provides the required coverage (app. 1 Kb) the depth remains limited (10 K–100 K reads). On the other hand, Illumina pair-end sequencing provides excellent depth, albeit with insufficient coverage (app. 600 bp for Miseq 300 PE) [9]. Nanopore sequencing technology potentially provides high coverage and depth at a lower cost, therefore we tested the feasibility of immune repertoire sequencing of indexed amplicons generated by ARTISAN-PCR. These long-read technologies are very promising, but their error rates are higher than other current sequencing methods and require computational-based corrections and/or additional bioinformatics preprocessing before they can be valuable [10].
This work presents a pipeline for analysis of immune repertoire sequenced by Nanopore technology, showing performance measurements as error rate, percentage of substitutions and indels, coverage and identification of V(D)J genes of immunoglobulin.
2 Materials and Methods
2.1 Immunoglobulin Amplification, Library Preparation and Sequencing
In this study, 5 samples from healthy individuals were analyzed. All volunteers provided informed consent and the study as it was approved by the ethics review board of the University of Magallanes (registry 1180882).
RNA was extracted from peripheral blood mononuclear cells obtained by gradient centrifugation. Anchored cDNA was synthesized and amplified by ARTISAN-PCR as previously described [8].
A total of 5 amplicons representing each antibody chain: IgA, IgG, IgM, IgKappa and IgLambda were individually indexed and the library was processed using the 1D2 Sequencing Kit (R9.5) following the manufacturer protocol.
Each amplicon has the following structure:
In the amplicons structure the Index identifies patient chain. The Constant Chain identifies Immunoglobulin chain (IgA, IgG, IgM, IgKappa and IgLambda).
The library with 25 indexed amplicons was then sequenced during 24 h in a MinION Nanopore Sequencing Device.
2.2 Basecalling and Qscore Stratification
The first bioinformatics step was to obtain the reads from the Nanopore electrical signal by ONT Albacore Sequencing Pipeline Software (version 2.3.3). In this step it was used the AXON server of the Universidad Católica del Maule, operating system CentOS Linux 7.4.1708 (core). This server has a hard disk of 1.8 Tera Bytes. 32 Gigabytes of Memory and has 2 processors Intel (R) Xeon (R) CPU E5-2623 v4 @ 2.60 GHZ with 4 cores each processor. This step took 70 h to work out the calculation.
Then sequencing quality was assessed by the internal Qscore (Qs) provided by Albacore and the reads were classified according to this score.
2.3 Demultiplexing of Indexed Amplicons
The second step of data processing corresponds to aligning the reads generated by Minion Nanopore to their respective sample indexes (Index, Anchor, Constant Chain, and End Adaptor) by using a custom script based on a Smith-Waterman heuristic algorithm using Bl2seq de BLAST software (Basic Local Alignment Search Tool) [11].
Consecutively, matching analysis was applied using the Levenshtein distance written in the C programming language. In this case the characters or bases of DNA that change are the product of insertions, deletions or substitutions of these. It is considered as a match when two sequences are identical, the mismatch is considered when at least one insertion, deletion or substitution. The reads that have up to 6 mismatches (6 differences between bases) contain motifs that align to their respective sample indexes. Alignments to indexes with a greater difference were discarded for subsequent analysis steps.
The general script for the demultiplexing of indexed amplicons was written in the R programming language [12].
2.4 Error Rate and Coverage
Each amplicon contains adapters and extensions located in 5’ and 3’ (Index, Anchor, Constant Chain, and End adaptor) that contain known DNA sequences, which are used to estimate sequencing error rate, substitution profiles, indel profiles, and coverage. The error rate constitutes the percentage of mismatched bases in the alignment and that can be subdivided into substitution, insertion and deletion rates, Eq. (1) [13].
The errors correspond to the total sum between substitutions, insertions and deletions in the motives. The alignment length is the sum of the number of eligible sequences (sequences that have up to 6 mismatch for the motif) multiplied by the length of the motif.
The error rate was estimated according to the criteria of stage 1.3 demultiplexing of indexed amplicons by using all motifs of the detected reads. The error rate was calculated as Eq. (2).
Reads were selected according to their internal Qscore (Qscore greater than 6) for subsequent analysis. Then, the reads were separated in forward and reverse complement to analysis of substitutions and indels. For these analysis, the sample indexes that are recognized in each read were considered. The united motifs of a read form a construct that eliminates the first 4 theoretical bases of Index and the last 4 bases of End Adapter (these amplicons have low quality in their end, according to preliminary analysis using FastQC tools [14]).
The profiles of each substitution analyzed were: C > A, C > G, C > T, T > A, T > C, T > G, G > T, G > C, G > A, A > T, A > G and A > C. In addition, all substitution were examined by incorporating information from the contiguous bases 5’ and 3’ for each mutated base generating 192 possible substitutions types (12 types of substitution \( * \) 4 types of 5’ base \( * \) 4 types of 3’ base). For each of the 192 possible substitutions types the percentage is obtained with respect to the total of substitutions. The procedure described above is similar for calculating the percentages of the indels.
Then, the coverage of the reads was analyzed respect to the structure of the amplicons (Fig. 1). The amplicons coverage is shown as the number of motifs that cover an area of the amplicons (does not correspond to coverage of known reference bases). In this case the presence of the motifs was quantified (Index, Anchor, Constant Chain, and End Adapter). Which are the motives of the reads is quantified, based on the criteria of stage 1.3 demultiplexing of indexed amplicons. In this stage, the presence of the V(D)J rearrangement motif was also quantified using Igblast software and Change-o tools for gene mapping [15].

Amplicons structure.
2.5 Immunoglobulin Identification
Reads with Qscore greater than 6 and demultiplexing according to stage 1.3 were subsequently aligned using IMGT HighV-Quest and IgBlast. Then, sequences alignment using IMGT HighV-Quest were analyzed with the BcRep R package [16]. In case of sequences alignment using IgBlast were then analyzed with Change-o tools [15].
3 Results
3.1 Sequencing Yield, Quality, and Error Rate
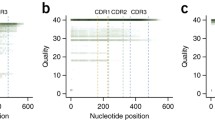
From the library of 25 indexed amplicons 1,450,507 reads were identified. The frequency distribution of read quality and error rate is showed in Table 1.
The proportion of reads with a Qscore equal or greater than 7, considered with acceptable quality, was 30.9% (n = 447,693) and the percentage of lower quality reads was 69.1% (n = 1,002,814). Due to the high proportion of low quality reads we performed a stratified analysis in order to establish whether lower quality reads contain useful sequencing data. With this information it is decided to analyze reads with a Qscore equal or greater than 6 (n = 914,069). It is observed that the reads with a Qscore greater than 7 have an error rate of up to 10%, in the case of reads with higher Qscore the error decreases by one percentage point.
In Fig. 2 is possible to see the reads that were demultiplexing for each type of immunoglobulin chain (IgA, IgG, IgM, IgKappa and IgLambda) and patient chain (DCR, JGP, LAH, MAN and MVC). Reads with Qscore between 5 and 9 were analyzed. 251,044 were identified with immunoglobulin chain and 317,395 reads were detected with patient chain. Besides, the amount of identified sequences show the depth and coverage of sequencing, allowing this technology as a greater platform in comparison to others commonly used for the sequencing of immunoglobulins [9].

Immunoglobulins (Ig) and Patient Chains identified. Sequences with a Qscore between 5 and 9 were analyzed.
The reads with a quality over 6, demultiplexing, were separated as forward and reverse complement. The forward reads was 45% (n = 273,215) and the percentage of reverse complement reads was 55% (n = 331,998). The substitutions, insertions and deletions of the reads were calculated separately for each type of sequence. Figure 3A and B shows the most important substitutions for forward and reverse complement reads. In this case the two most important substitutions G > A are shown (where G is the real base, being replaced by A) and A > G (where A is the real base, being replaced by G). It should be mentioned that in this case it is true that both for reads forward and reverse have as a maximum percentage the substitutions G > A and A > G. In the case of substitutions in the forward reads (Fig. 3A), it is observed that the profile CGC with the highest percentage (more than 20% of the substitutions in the forward reads), where G is replaced by A, and between two equal bases (between two C) Nanopore errors occur as expected. The second profile of the forward reads that has the highest percentage is AAC (more than 10%), where the central A is replaced by G, it is observed that when there are two equal bases, in this case A, the second base cannot recognize it and it is replaced.

Substitutions and indels. Bar graphs show the most important percentages of substitutions and indels.
In the case of substitutions of the reverse complement reads (Fig. 3B) it is observed that the highest substitution profile is TAC (greater than 15%), where A is replaced by G. The second most important profile corresponds to AGC with almost 15% of the substitutions, where G is replaced by A which is contiguous to the base that we wish to infer.
In summary, it is observed that in the case of forwards reads there are greater errors when two bases are equal in the profile. In the case of the reverse complement reads the substitutions change the base resembling a contiguous base.
The other substitution profiles are not shown in this document because of their space and also they have very low percentages, all of which are less than 5%.
Figure 3C and D, shows the most important deletion profiles to forward and reverse complement reads. It is observed that the deletion of A is the most important for the two types of reads. The most important deletions for the forward reads are those of A and G. The most important deletion for the forward reads is A> - with a percentage of almost 40% of the total deletions for this type of reads. The profile corresponds to CAA, where the A base is not inferred by the neural network that uses the Nanopore Albacore software’s neural network. The second deletion corresponds to the AGT profile (with a percentage greater than 15%), where G is not inferred.
The most important deletion for reverse complement reads is A> - with a percentage of more than 45% of all deletions for this type of reads. The profile of the deletion corresponds to GAG, where A is not inferred. The second most important deletion profile is GGA, where the bold type G is not recognized.
Based on the results it is observed that the most important deletions occur in profiles that have two equal bases. The deletions of C and T are not shown given that the percentages of each are less than 5% for all.
In Fig. 3E, it is shown that the most important forward reads insertions correspond to C and G. The profile with the highest percentage is T-C (greater than 25%), where a C is inserted into the middle of the profile (mistakenly inferring a TCC profile). The second most important profile corresponds to G-A where a G is inserted into the middle of the profile (erroneously inferring a GGA profile). It is observed based on these results that in the insertions an adjacent base is duplicated. The inserts of T and A, as the inserts of the reverse complement reads have profiles with percentages less than 10%.
The coverage of the reads was analyzed (Fig. 4). We found that all sequences have V(D)J Rearrangement (400 to 880 bp) when analyzing the results of IgBlast used MakeDb.py of Change-o (using the partial option: which allows to include incomplete alignments to V(D)J). However, very few complete amplicons were detected 6,682. It is observed that a large number of reads that has the Anchor and Index motif.

Read coverage. The number of reads that have the patterns is marked in color. (Color figure online)
3.2 Nanopore Sequencing Allows V(D)J Gene Usage Identification
Demultiplexed reads were aligned to the immunoglobulin databases HighV-Quest and IgBlast. The results we have obtained using IgBlast can be better recognized incomplete immunoglobulins compared to HighV-Quest, however HighV-Quest has more tools used in the analysis of the sequence as BcRep R package that are presented below.
The overall V, D, and J gene segments usage for heavy and light chains is depicted in Fig. 5. The frequency distribution is consistent with the previously reported usage in repertoires obtained in healthy individuals.

Percentage of variable region from V, D, and J segments associated with families. Bar graphs in upper panels correspond to immunoglobulin heavy chains (blue bars) and graphs in the lower section to light chains (orange bar). (Color figure online)
Figure 6 represents the combinatorial gene segments usage according to each individual index for demultiplexed samples. As expected for healthy individuals the gene segments usage and combinations correlates.

Heavy chain gene segments usage and V-D recombination in demultiplexed samples. Each panel represents one individual.
4 Conclusions
This work corresponds to the first description for feasibility to amplifiying immune repertoire by ARTISAN-PCR protocol sequenced by nanopores.
The bioinformatic analysis obtained from the pipeline previously described allow us the use of gene information for the assembly of the immunoglobulin variable region, providing relevant information, for example, for the prognosis of various pathologies. This method provides full rearrangement coverage and sequencing depth that could potentially reach 106 sequences per flow cell.
In our previous experiments using PacBio we were able to identify an average of 50,000 reads, whereas with this novel approach we rescued more than 300,000 reads with adequate quality.
Nevertheless, this technology still faces important challenges such as high error rate [17, 18]. In our experiments error rate was as high as 10% for reads with a Qscore between 7 and 8, selection of reads with higher Qscore only decreased this error rate by 1 percentual point. Although this error rates allows accurate identification of V(D)J family usage, it is not sufficient for other analysis such as precise hypermutation rate measurement. However, the principal disadvantage of this platform is the accuracy, apparently driven by systematic errors [19]. Since most of the error rate could be associated to predictable patterns, it is envisioned that bioinformatics tools could be readily developed [10].
References
Kipnis, J.: Multifaceted interactions between adaptive immunity and the central nervous system. Science 353, 766–771 (2016)
Labrecque, N., Cermakian, N.: Circadian clocks in the immune system. J. Biol. Rhythms 30, 277–290 (2015)
Rajewsky, K.: Clonal selection and learning in the antibody system. Nature 381, 751–758 (1996)
Greiff, V., Miho, E., Menzel, U., Reddy, S.T.: Bioinformatic and statistical analysis of adaptive immune repertoires. Trends Immunol. 36, 738–749 (2015)
Navarrete, M.A., Bertinetti-Lapatki, C., Michelfelder, I., Veelken, H.: Usage of standardized antigen-presenting cells improves ELISpot performance for complex protein antigens. J. Immunol. Methods 391, 146–153 (2013)
van Bergen, C.A.M., et al.: Selective graft-versus-leukemia depends on magnitude and diversity of the alloreactive T cell response. J. Clin. Invest. 127, 517–529 (2017)
Koning, M.T., Nteleah, V., Veelken, H., Navarrete, M.A.: Template-switching anchored polymerase chain reaction reliably amplifies functional lambda light chain transcripts of malignant lymphoma. Leuk. Lymphoma 55, 1212–1214 (2014)
Koning, M.T., et al.: ARTISAN PCR: rapid identification of full-length immunoglobulin rearrangements without primer binding bias. Br. J. Haematol. 178, 983–986 (2017)
Chaudhary, N., Wesemann, D.R.: Analyzing immunoglobulin repertoires. Front. Immunol. 9(462), 5–6 (2018)
Warren, R.L., et al.: LINKS: scalable, alignment-free scaffolding of draft genomes with long reads. GigaScience 4, 35 (2015)
Tatusova, T.A., Madden, T.L.: BLAST 2 sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174, 247–250 (2006)
Salas, Christian: ¿Por qué comprar un programa estadístico si existe R? Ecol. Austral 18, 223–231 (2008)
Rang, F.J., Kloosterman, W.P., de Ridder, J.: From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 19, 90 (2018)
Brown, J., Pirrung, M., McCue, L.A.: FQC dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139 (2017)
Gupta, N.T., Vander Heiden, J.A., Uduman, M., Gadala-Maria, D., Yaari, G., Kleinstein, S.H.: Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358 (2015)
Bischof, J., Ibrahim, S.M.: bcRep: R package for comprehensive analysis of B cell receptor repertoire data. PLoS One 11, e0161569 (2016)
Deamer, D., Akeson, M., Branton, D.: Three decades of nanopore sequencing. Nat. Biotechnol. 34, 518–524 (2016)
Lu, H., Giordano, F., Ning, Z.: Oxford Nanopore MinION sequencing and genome assembly. Genomics Proteomics Bioinform. 14, 265–279 (2016)
Laehnemann, D., Borkhardt, A., McHardy, A.C.: Denoising DNA deep sequencing data—high-throughput sequencing errors and their correction. Brief. Bioinform. 17, 154–179 (2016)
Acknowledgments
This project was funded by grants ESR-MAG1895 to RUP and Fondecyt#1180882 to MAN.
Author information
Authors and Affiliations
Contributions
RAG performed bioinformatics analysis, JGP performed molecular biology experiments, DAS provided concept analysis and performed bioinformatics analysis, XLC, RB and RUP provided concept analysis, MAN designed and conceptualized the project and experiments. RAG, MAN, XLC and JGP wrote the paper.
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Ahumada-García, R. et al. (2019). Identification of Immunoglobulin Gene Usage in Immune Repertoires Sequenced by Nanopore Technology. In: Rojas, I., Valenzuela, O., Rojas, F., Ortuño, F. (eds) Bioinformatics and Biomedical Engineering. IWBBIO 2019. Lecture Notes in Computer Science(), vol 11465. Springer, Cham. https://doi.org/10.1007/978-3-030-17938-0_27
Download citation
DOI: https://doi.org/10.1007/978-3-030-17938-0_27
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-17937-3
Online ISBN: 978-3-030-17938-0
eBook Packages: Computer ScienceComputer Science (R0)