
Abstract
Context: Dietary management is the mainstay of effective treatment in PKU, but dietary restriction is difficult and additional treatment options are needed.
Objective: To systematically review evidence regarding sapropterin (BH4) use as an adjunct to dietary restriction in individuals with PKU.
Data Sources: Five databases including MEDLINE up to August 2011.
Study Selection: Two reviewers independently assessed studies against predetermined inclusion/exclusion criteria.
Data Extraction: Two reviewers independently extracted data regarding participant and intervention characteristics and outcomes and assigned overall quality and strength of evidence ratings based on predetermined criteria.
Results: BH4 research includes two randomized controlled trials (RCTs) and three uncontrolled open-label trials. Phenylalanine (Phe) levels were reduced by at least 30 % in up to half of treated participants (32–50 %). In one RCT comparing placebo on likelihood of a 30 % reduction in Phe, 9 % of those on placebo achieved this effect, compared with 44 % of the treated group after 6 weeks. Phe tolerance and variability were improved in treated participants in studies assessing those outcomes. No comparative studies assessed long-term outcomes including cognitive effects, nutritional status, or quality of life.
Conclusions: Adjuvant pharmacologic therapy has the potential to support individuals in achieving optimal Phe levels. BH4 has been shown to reduce Phe levels in some individuals, with significantly greater reductions seen in treated versus placebo groups. The strength of the evidence is moderate for short-term effects on reducing Phe in a subset of initially BH4-responsive individuals, moderate for a lack of significant harms, low for longer-term effects on cognition, and insufficient for all other outcomes.
Competing interests: none declared
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Approximately 1 in 13,500–19,000 infants in the United States is born with phenylketonuria (PKU) (Hegge et al. 2009; National Institutes of Health 2001). Individuals with PKU have defective phenylalanine hydroxylase activity, leading to a toxic accumulation of phenylalanine (Phe) in the blood and multiple tissues (Webster and Wildgoose 2010) and potentially to intellectual disability, delayed speech, seizures, and behavior abnormalities (Erbe and Levy 2002; Wilcox and Cederbaum 2002; Fisch and Stassart 2004).
The mainstay for treatment of PKU is a diet that restricts the intake of Phe to control the Phe concentration in the blood. In general, the usual treatment goal is a blood Phe level of 120 to 360 μmol/L (National Institutes of Health 2001; Poustie and Wildgoose 2010). In addition to the low-Phe diet, patients replace nutrients that are absent in their restricted diet with Phe-free medical foods (Giovannini et al. 2007). With adherence to a Phe-restricted diet, poor outcomes can be mitigated. While advances in dietary management are improving outcomes for individuals with PKU, management of PKU can be difficult and onerous, leading to interest in identifying new ways of managing this lifelong condition. (Demirkol et al. 2011; Harding and Blau 2010; Blau et al. 2010; Giovannini et al. 2012)
In 2007, the United States Food and Drug Administration (FDA) approved sapropterin dihydrochloride (Kuvan®, formerly known as Phenoptin), the first pharmacologic treatment for PKU, under the stipulation that additional studies be conducted on efficacy and long-term safety. The goal of treatment with sapropterin dihydrochloride (hereafter, BH4) is to control blood Phe concentrations. Although treatment with BH4 would potentially allow a relaxation of the low-Phe diet, it is not intended to serve as a complete substitute for dietary intervention (Somaraju and Merrin 2010).
The mechanism of action of BH4 is as a cofactor of the phenylalanine hydroxylase (PAH) enzyme, increasing the activity level of the enzyme and increasing the amount of Phe that can be converted to tyrosine. Hypothetically, it should be more effective in individuals with residual PAH activity than in individuals with negligible to no enzyme activity. Typically, a BH4 loading test is performed to identify potential candidates who are most likely to benefit from treatment. Loading tests used in practice and in research vary in terms of target reduction (e.g., 30 % reduction in blood Phe level) and timeframe, and none has been established as optimal for identifying those patients who respond to treatment.
As part of a systematic review of adjuvant therapies for individuals with PKU, we reviewed the literature on BH4 use in individuals with PKU. The full review is available at http://www.effectivehealthcare.ahrq.gov.
Methods
Data Sources and Search Strategy
We searched the MEDLINE® (via the PubMed interface), PsycINFO, EMBASE, Cumulative Index of Nursing and Allied Health Literature, and the National Agricultural Library (AGRICOLA) databases. Our search strategies used a combination of subject heading terms appropriate for each database and key words relevant to PKU (e.g., phenylketonuria, pharmaceutical preparations, phenylalanine). We limited searches to the English language but did not set a date limit. We also manually searched the reference lists of included studies and of recent narrative and systematic reviews and meta-analyses addressing PKU.
Study Selection
We developed study inclusion and exclusion criteria in consultation with an expert panel of clinicians and researchers involved in treating or studying PKU. We included all study designs excepting individual case reports and required that studies include at least 10 individuals with PKU receiving BH4 for therapy after having shown positive results on a loading test.
Data Extraction and Synthesis
Using standardized forms, two investigators independently extracted data regarding study design; study populations; intervention and comparison groups; and baseline and outcome data, as well as data about harms or adverse effects. Principal outcomes of interest included cognitive and nutritional outcomes, quality of life, and ability to liberalize the diet while maintaining acceptable Phe levels. We also extracted data on the intermediate outcomes of Phe level, Phe tolerance (total Phe intake by weight an individual can tolerate while maintaining acceptable blood Phe concentration), and blood Phe variability. We present a descriptive analysis of the data given the low number of studies identified and variability in outcomes assessed, which precluded meta-analysis.
Quality Assessment
Two investigators independently assessed each study using quality assessment questions as appropriate for each study design. We adapted questions from quality tools including the Cochrane Risk of Bias tool. We evaluated the overall strength of the evidence (degree of confidence that the observed effect of an intervention is unlikely to change) for the primary outcomes. We used the approach to determine strength of evidence as described in the Agency for Healthcare Research and Quality Effective Health Care Program’s Methods Guide for Effectiveness and Comparative Effectiveness Reviews (Agency for Healthcare Research and Quality 2008). We assessed the strength of evidence for key outcomes identified as the most clinically important: cognitive outcomes including IQ and executive function, nutritional outcomes, quality of life, and liberalization of diet. Secondary outcomes included changes in blood Phe levels, Phe variability, and Phe tolerance.
Assessments were based on consideration of the domains of risk of bias, consistency in direction of the effect, directness in measuring intended outcomes, and precision of effect (Table 1). We determined the strength of evidence separately for major intervention-outcome pairs using an approach described in more detail in the full review.
Results
Overview of Study Characteristics
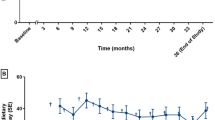
Ten studies evaluated the effects of BH4 in participants with PKU (Table 2, Fig. 1) (Burton et al. 2010, 2011; Levy et al. 2007; Lee et al. 2008; Trefz et al. 2009, 2010; Vernon et al. 2010; Burlina and Blau 2009; Lambruschini et al. 2005; Humphrey et al. 2011). Although study populations overlap, the studies were conducted as separate studies and thus are presented as such in our analysis. Four of the studies are linked by common participants: two are multisite placebo-controlled randomized trials (RCTs) that contributed to FDA approval of BH4 (Levy et al. 2007; Trefz et al. 2009). One additional uncontrolled open-label trial was conducted separately from the family of studies (Vernon et al. 2010) as were one prospective cohort study (Humphrey et al. 2011), two retrospective case series (Burlina and Blau 2009; Burton et al. 2010), and two prospective case series (Lambruschini et al. 2005; Trefz et al. 2010).

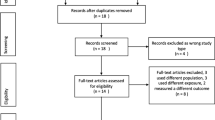
Flow of studies identified for the review. *The total number of articles in the exclusion categories exceeds the number of articles excluded because most of the articles fit into multiple exclusion categories. BH4 sapropterin, N number
The studies included up to 80 participants in the treatment arm, and the total number of individuals in all studies was 284, after accounting for duplication in participants across studies. The studies were performed in the United States, Canada, Australia, and Europe. Participants ranged in age from 3 to 58 years in the five trials and from birth to 34 years in the observational studies. Most participants had demonstrated responsiveness to BH4 in a loading study before being assessed for effectiveness response; however, the approach to assessing responsiveness varied by study, and the base populations tested for initial responsiveness were not consistent (Table 3).
BH4 was studied in doses ranging from 5 to 20 mg/kg/day. The follow-up period for the two RCTs was 10 weeks, and some participants in multiple studies (including the extension studies) were exposed to the drug for up to 2.6 years. One case series followed participants up to 7 years (Lambruschini et al. 2005), with an average follow-up of 3.5 years. The mean treatment duration among participants in another case series (Trefz et al. 2010) was 4 years and 8 months (range = 24–110 months). The degree to which participants were adherent to a restricted diet varied. One RCT (Levy et al. 2007) and its follow-on trial (Lee et al. 2008) included participants with PKU who were at least 8 years old with a mean age of 20 years, consuming a relaxed or unrestricted diet, and had baseline blood Phe level of >450/umol/L. The second RCT (Trefz et al. 2009) examined the effect of 20 mg/kg/day for 10 weeks in children ages 4–12 who were on a Phe-restricted diet with baseline blood Phe levels <480 umol/L. One uncontrolled open-label study examined a differential effect in those who maintained a restricted diet versus those who did not (Vernon et al. 2010).
All randomized trials (Table 4) and three case series evaluated reduction in blood Phe levels. Two trials and three case series reported on Phe tolerance (Trefz et al. 2009; Vernon et al. 2010; Burlina and Blau 2009; Lambruschini et al. 2005; Trefz et al. 2010), and one cohort study (Humphrey et al. 2011) and one case series reported on Phe variability (Burton et al. 2010). Only one case series (Lambruschini et al. 2005) assessed longer-term outcomes, including cognition and nutritional status. No study evaluated quality of life. BioMarin, the pharmaceutical company that holds the patent for sapropterin dihydrochloride, sponsored five studies (Burton et al. 2011; Levy et al. 2007; Lee et al. 2008; Trefz et al. 2009; Burton et al. 2010), including both RCTs (Levy et al. 2007; Trefz et al. 2009).
Key Study Results
One good-quality RCT (Levy et al. 2007) randomized 89 participants with PKU to receive either 10 mg/kg of BH4 (N = 42) or placebo (N = 47) once daily for 6 weeks. The primary outcome was the change in blood Phe from baseline to week 6. Participants’ mean age was 21.5 years in the treatment group and 19.5 years in the placebo group. Adherence to treatment during the 6-week trial was reported as high, with 82 % of participants taking all doses correctly.
After 6 weeks of treatment, participants in the BH4 group had a significant decrease in mean blood Phe levels of −235.9 ± 257 μmol/L from baseline (843 μmol/L) compared with a 2.9 ± 239.5 μmol/L increase in mean Phe levels from baseline (888 μmol/L) in the control group (p < 0.0001). The mean blood Phe decreased in the BH4 group at 1 week and remained at that lower level until the 6-week end point, when the mean Phe level was 607 μmol/L. The estimated difference between treatment and placebo groups in the mean change in blood Phe at 6 weeks compared with baseline was −245 (p < 0.0002). A significantly higher proportion of participants receiving BH4 (44 %) had a 30 % or greater reduction in blood Phe levels compared with controls (9 %).
Almost all participants (16 of 17) for whom genotyping was performed had at least one mutation known to be associated with residual enzymatic activity. Responsiveness was not consistently linked to specific mutations. Despite enrolling only those participants who had at least a 30 % reduction in blood Phe while taking BH4 in a 1-week loading test, not all participants were responsive to BH4 in the trial.
A 22-week uncontrolled, open-label trial (Lee et al. 2008) of good quality followed this RCT (Levy et al. 2007) and included three phases: dose titration, dose analysis, and a fixed dose phase. All participants enrolled in the previous RCT were eligible if they had taken at least 80 % of their scheduled dose in the trial and were willing to continue their current diet. The primary end point was mean plasma Phe levels at week 22 and mean changes from week 0. Of 87 participants who completed the previous RCT (Levy et al. 2007), 80 were enrolled in the extension trial (Lee et al. 2008), of whom 39 had previously received BH4 and 41 placebo. Participants’ mean age was 20.4 years. High doses of BH4 (10 or 20 mg/kg/day were more effective at lowering Phe levels in the dose titration phase. By the end of the dose analysis phase with 10 weeks at 10 mg/kg/day, 46 % of participants had a decrease in plasma Phe of at least 30 % compared with week 0. During the fixed dose phase, most participants (92 %) received either 10 (46 %) or 20 mg/kg/day (46 %). By week 22, plasma Phe was reduced by 190.5 μmol/L compared to week 0. Mean plasma Phe decreased from 844 μmol/L at baseline to 645 μmol/L at week 10 and was maintained at a mean of 652 μmol/L at week 22. At week 22, 46 % of participants had achieved a 30 % reduction in plasma Phe concentration compared with week 0. The corresponding reductions for those receiving 5, 10, and 20 mg/kg/day were 50 %, 49 %, and 42 %, respectively.
Another RCT (Trefz et al. 2009) of fair quality enrolled children with PKU between 4 and 12 years of age who were on a Phe-restricted diet, had maintained blood Phe control (blood Phe level ≤480 μmol/L), and had an estimated Phe tolerance of ≤1,000 mg/day. The objective was to determine the safety and efficacy of BH4 at 20 mg/kg/day for 10 weeks in increasing Phe tolerance (defined as cumulative increase or decrease in Phe supplement at the last visit for which blood Phe level was ≤360 μmol/L) while maintaining blood Phe control. Participants maintained a stable, Phe-restricted diet, monitored by food diaries. Starting at the third week, a dietary Phe supplement was added or removed every 2 weeks based on Phe levels. Children with a blood Phe level of ≥1200 μmol/L in two consecutive weeks were withdrawn from study drug treatment and received dietary counseling.
Thirty-three children were randomized to 20 mg/kg/day of BH4 for 10 weeks, and 12 children received a placebo. After 10 weeks of treatment, the total mean ± SD of Phe supplement tolerated by participants on BH4 increased significantly from 0 mg/kg/day at baseline to 20.9 ± 15.4 mg/kg/day. In contrast, the placebo group tolerated an increase of 2.9 mg/kg/day of Phe supplement. The adjusted mean difference between the groups in Phe tolerance was 17.7 ± 4.5 mg/kg/day (p < 0.001). Total Phe intake (dietary Phe intake plus total Phe supplement) also increased significantly from baseline in the BH4 group, approximately doubling to 43.8 mg/kg/day at 10 weeks compared to a slight increase in total Phe intake in the placebo group (16.3 mg/kg/day at baseline to 23.5 ± 12.6 at 10 weeks). Mean blood Phe levels decreased significantly in the BH4 group between baseline and the beginning of supplementation in week 3 (decrease of 148.5 ± 134.2 μmol/L). Some participants in the BH4 group had transient low blood Phe levels (<26 μmol/L) corrected with increased Phe supplementation.
An uncontrolled, open-label trial (Vernon et al. 2010) of good quality included participants with classic or variant PKU with any Phe level or diet. Eligible subjects received 7 days of open-label BH4 at 10 mg/kg/day with plasma Phe measurement on day 8 and weekly during a dietary modification period with dosage increased to 20 mg/kg/day until day 30 for nonresponders. Responders who were on a Phe-restricted diet underwent gradual liberalization of their diet to the maximum tolerated natural protein intake while still maintaining plasma levels in the range of 120 to 360 μmol/L.
Of the 29 individuals completing the study, 59 % were on some form of protein-restricted diet and had a mean baseline blood Phe of 587 μmol/L. Forty-one percent were not following protein-restricted diets and had a mean baseline blood Phe level of 1,372 μmol/L. Overall, 62 % were determined to be responders, with variable doses required for response. Four (27 %) of the classic PKU participants (defined as off-diet plasma Phe of >1,200 μmol/L) were responders, and 100 % of the variant PKU participants (>400 and <1,200 μmol/L) were responders. Of the 12 participants who were not on a Phe-restricted diet, 33 % were responders with a significantly decreased mean blood Phe level (554 μmol/L) compared with baseline (1049 μmol/L). Of the 17 participants who were on a Phe-restricted diet, 82 % were responders with significantly reduced mean blood Phe level of 226 μmol/L compared with baseline (485 μmol/L). Among individuals who were responders and on a Phe-restricted diet, the average Phe tolerance increased from 21 to 41 mg/kg/day. However, responders’ Phe tolerance varied widely from an increase of 20 to 22 mg/kg/day to a non-protein-restricted diet in two participants.
One poor-quality prospective cohort study (Humphrey et al. 2011) assessed variability in blood Phe. Participants included nine children who were responsive to a 20 mg/kg BH4 loading test and 25 who were nonresponsive. Among those who were BH4 responsive, two were treated with BH4 alone and the rest also needed dietary modifications. Overall, there were no significant differences in mean and median blood Phe levels between the groups; however, above blood Phe levels of 600 μmol/L, confidence intervals around the mean were wider among BH4 nonresponsive participants. The authors equate these differences with variability in response.
Four poor-quality case series (Burlina and Blau 2009; Lambruschini et al. 2005; Burton et al. 2010; Trefz et al. 2010) evaluated dosages of BH4 ranging from 5 to 26 mg/kg/day for durations of 6 months to up to 9 years among BH4-responsive participants. All reported positive outcomes in terms of reduction in blood Phe and increased Phe tolerance. One case series (Lambruschini et al. 2005) also examined longer-term functional outcomes, including IQ and developmental quotient after 1 year of treatment, reporting no adverse effects as participants’ Phe tolerance increased and the diet was liberalized. Nutritional status was unchanged with the exception of increases in selenium. In another case series (Burlina and Blau 2009), 12 participants were studied for up to 7 years on a dosage of 10 mg/kg twice a day. In this group, ranging in age from 2 to 16 years old, all participants eventually stopped Phe supplementation and relaxed dietary restrictions.
Another longer-term case series (Trefz et al. 2010) assessed Phe levels and increase in Phe tolerance in 16 individuals receiving BH4 for between 24 months to 9 years (mean = 56 months). The mean blood Phe level in responders (n = 14) was 321 ± 236 μmol/L, and the mean decrease in blood Phe was 54.6 % (range 28.4–85.6 %). Seven patients had stable Phe control without any dietary restriction. Of the remaining seven patients who were on dietary restrictions, six increased their Phe intake from a baseline of 200–300 mg/day to 800–1,000 mg/day. Psychomotor development among children 5–6 years of age was reported to be within normal range; however, results were not presented. One case series (Burton et al. 2010) provided data on Phe variability by measuring blood Phe at least six times before and after treatment initiation. Individual variability in Phe levels was lessened after treatment.
Harms Reported in Studies of BH4
Of the ten studies examining the effectiveness of BH4, four studies with overlapping participants (Burton et al. 2011; Levy et al. 2007; Lee et al. 2008; Trefz et al. 2009) reported any type of harm related to the intervention drug. Three studies (Burlina and Blau 2009; Lambruschini et al. 2005; Trefz et al. 2010) reported that no adverse events were observed during intervention, one study reported that BH4 was well tolerated with mild diarrhea occurring rarely (Humphrey et al. 2011), and there was no mention of harms in two studies (Vernon et al. 2010; Burton et al. 2010). The most common side effects reported during BH4 trials were headache, throat pain, upper respiratory infection, diarrhea, abdominal pain, and nausea and vomiting. Harms probably or possibly related to study treatment occurred at similar rates in both BH4 and placebo (23 vs. 20 % (Levy et al. 2007), 27 vs. 25 % (Trefz et al. 2009)) arms in RCTs.
Strength of Evidence
We rated the strength of the evidence (confidence that the current effect estimate will not change with future research) for the effects of BH4 on reducing blood Phe levels to clinically acceptable levels among BH4 responders in the short term (12 weeks or less) as moderate based on few studies. The strength of evidence for the indirect relationship of BH4 on IQ is low, based on current evidence and lack of direct measurement. Harms were noted to be rare and mild, and the strength of evidence for this observation is moderate. The strength of the evidence is insufficient for the direct effect of BH4 on improving all other outcomes (Phe tolerance and the ability to liberalize the diet, Phe variability, quality of life, and cognitive and nutritional outcomes).
Discussion
The treatment for PKU with dietary restriction of Phe in natural protein and use of Phe-free medical foods has been critical in reducing the incidence of irreversible neurocognitive impairment in individuals with PKU, and together with newborn screening has been a tremendous success story. However, families and patients report that, especially with the onset of adolescence and adulthood, dietary adherence and supplement use can be challenging. Little is known about rates of adherence to diet, and clinicians, patients, and families have lacked therapeutic options other than a lifetime of strict dietary management. Importantly, the ability to liberalize the diet has the potential to affect the quality of life of individuals with PKU who must be constantly vigilant about what they consume. Ideally, a therapeutic adjunct to dietary management would increase Phe tolerance, allowing for increased intake of dietary protein and micronutrients, and reducing (but likely not eliminating) the necessity for Phe-free medical foods.
All studies of BH4 evaluated intermediate outcomes (change in blood Phe levels and Phe tolerance). Almost no information is yet available, and none from RCTs studying longer-term outcomes, including cognitive impairment, quality of life, nutritional impact and status, and the ability to liberalize diet. In enriched populations (all participants had reduced Phe in initial loading tests) with varying levels of dietary and Phe control, up to half of the participants had Phe reductions of at least 30 % and the relationship of reductions in Phe to clinical outcomes was not assessed.
Further, a high proportion of treated participants who achieved a reduction in blood Phe of the study target of 30 % continued to have Phe levels above the clinical target. Nonetheless, an open-label extension trial demonstrated that reductions in Phe observed early in treatment could be maintained up to 22 weeks (Lee et al. 2008). On the other hand, the clinical goal for individuals maintaining dietary control could be to improve their quality of life by liberalizing their diet. In the trial targeting children with Phe <480 who were successfully maintaining a restricted diet, Phe tolerance was increased (Trefz et al. 2009). Even so, the impact on Phe tolerance was not uniform across the study population, and although many of the participants could modestly increase their protein intake, none could be on an unrestricted diet. Of note, a number of conditions may affect Phe tolerance, including illness, type of mutation, and degree of BH4 response among others; these are not assessed in the studies.
In all of the studies, compliance with BH4 was reported to be good over the short term. However, long-term sustainability of compliance with both BH4 and dietary therapy, especially given the variability in response, has not been evaluated, nor has durability of treatment effects. Authors from the uncontrolled open-label trial note that one responder reportedly discontinued BH4 after the trial as the small increases in Phe intake that BH4 allowed were not significant enough to warrant taking the medication. Certainly, observed increases in Phe tolerance were moderate at best in classic PKU in terms of allowing changes in diet, and the decision about trade-offs between reliance on medication and carefully titrating liberalization of the diet will need to be made by patients and their clinicians on an individual basis that balances available evidence with the individual’s context.
Even though studies reporting harms consistently indicate that BH4 is well tolerated and without serious side effects, not all studies assessed and reported harms, and data are based on a small number of individuals, so ongoing registries will be important for supplementing these data.
Future Directions
Research on the use of BH4 as an adjuvant therapy in PKU management is relatively new and consists of small, tightly controlled multisite efficacy studies. The greatest research need in this area is thus for larger studies that include adequate numbers of participants. Given the known difficulty of accruing large numbers of participants, however, researchers should also use existing data sets and a consortium and multisite approach to gathering data. Ideally, studies will be conducted in both tightly controlled and nonadherent populations, and among different age groups, with appropriate design and power for subgroup analyses. Research should continue to include RCTs, but prospective cohort studies that may have the potential to provide additional effectiveness data – including outside of a controlled clinical setting – adherence, and longer-term evidence would also be helpful to support understanding of the role of BH4 in clinical care. These studies should provide substantially more details on the range of benefits and harms associated with treatment.
Data are not currently available to understand potential modifiers of treatment effectiveness in order to select the best populations for targeting further research and treatment. Moreover, the significant variability in responsiveness to BH4 is unexplained, and subpopulations that have a unique response to this medication have not been well characterized. Causes of variability may be multifactorial and likely include individual patient and genotype differences, drug dose, and individual patient behavior such as dietary adherence. It is unclear, in particular, why a high proportion of individuals who have an initial response in loading studies do not have a durable response even over a few weeks in efficacy trials, even while those who do have a response demonstrate a significant effect. The degree to which this observed variation may be associated with suboptimal adherence should be assessed both in clinical trials and other types of studies.
Another area of potential research that could be explored in combination with studies of BH4 is the use of adherence supports for both drug and diet to optimize potentially positive outcomes. Long-term efficacy outcomes beyond 22 weeks and safety outcomes beyond 3 years are currently unavailable, as are measures of behavioral change and cognition and patient-reported outcomes including quality of life. Better markers of treatment effectiveness are needed; in addition, responsiveness to BH4 with reduction in blood Phe and improvement in Phe tolerance may not translate into responsiveness in cognitive outcomes. The degree to which reductions in blood Phe are associated with measurable cognitive outcomes or even patient perception of increased mental clarity is unknown; foundational research should be done to identify target outcomes for additional studies. Furthermore, explicit assessment of the potential for liberalization of the diet, and the subsequent nutritional effects on cognition has yet to be conducted.
Larger studies are also necessary to determine whether pharmacologic intervention is more advantageous in certain age groups or among individuals of varying dietary control of Phe, genotype, or severity of disease. A number of studies are reportedly under way to address gaps in the current literature. These include a long-term study of the effect of BH4 on neurocognitive function in young children, a study of the effect in adolescent patients with attention deficit hyperactivity disorder, and a registry that includes pregnant women (PKUMOMS).
Conclusions
Dietary management remains the mainstay of treatment for PKU, and maintaining control over the lifetime is an appropriate goal. Nonetheless, there is potential for supporting patients in achieving their clinical goals and possibly liberalizing their diet with adjuvant therapy. BH4 has been shown in two RCTs and three open-label trials to reduce Phe levels in some patients, with significantly greater reductions seen in treated versus placebo groups. We do not yet have the ability to reliably predict which patients are most likely to be responders, as all participants in the trials were initially responsive in screening tests, but not necessarily so in the efficacy studies.
One RCT also demonstrated increased Phe tolerance using BH4 among children on restricted diets. Overall, harms associated with the drug were minor and did not occur more frequently in the treatment group than in placebo arms. To date, there are no data to directly establish the potential effects of BH4 on longer-term clinically important outcomes, including cognition, executive function, and quality of life. Significant gaps in the evidence remain, including effectiveness of the drug in a range of patients outside of the clinical trial setting. Thus, while the strength of evidence is moderate for a large, positive effect of BH4 on reducing Phe levels over the short term in groups of patients showing initial responsiveness, evidence for the effect of BH4 on longer-term clinical outcomes is low and based on indirect associations.
References
Agency for Healthcare Research and Quality (2008) Methods guide for effectiveness and comparative effectiveness reviews. AHRQ publication No. 10(12)-EHC063-EF. Agency for Healthcare Research and Quality, Rockville, MD, April 2012. Chapters available at: www.effectivehealthcare.ahrq.gov
Blau N, van Spronsen FJ, Levy HL (2010) Phenylketonuria. Lancet 376:1417–1427
Burlina A, Blau N (2009) Effect of BH(4) supplementation on phenylalanine tolerance. J Inherit Metab Dis 32:40–45
Burton BK, Grange DK, Milanowski A et al (2007) The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis 30:700–707
Burton BK, Bausell H, Katz R, Laduca H, Sullivan C (2010) Sapropterin therapy increases stability of blood phenylalanine levels in patients with BH4-responsive phenylketonuria (PKU). Mol Genet Metab 101:110–114
Burton BK, Nowacka M, Hennermann JB et al (2011) Safety of extended treatment with sapropterin dihydrochloride in patients with phenylketonuria: results of a phase 3b study. Mol Genet Metab 103:315–322
Demirkol M, Giżewska M, Giovannini M, Walter J (2011) Follow up of phenylketonuria patients. Mol Genet Metab 104:S31–S39
Erbe RW, Levy HL (2002) Neonatal screening. In: Rimoin L et al (eds) Emery and Rimoin’s principles and practice of medical genetics. Churchill Livingstone, New York, pp 826–841
Fisch RO, Stassart JP (2004) Normal infant by a gestational carrier for a phenylketonuria mother: alternative therapy. Mol Genet Metab 82:83–86
Giovannini M, Verduci E, Salvatici E, Fiori L, Riva E (2007) Phenylketonuria: dietary and therapeutic challenges. J Inherit Metab Dis 30(2):145–152
Giovannini M, Verduci E, Salvatici E, Paci S, Riva E (2012) Phenylketonuria: nutritional advances and challenges. Nutr Metab (Lond) 9:7
Harding CO, Blau N (2010) Advances and challenges in phenylketonuria. J Inherit Metab Dis 33:645–648
Hegge KA, Horning KK, Peitz GJ, Hegge K (2009) Sapropterin: a new therapeutic agent for phenylketonuria. Ann Pharmacother 43:1466–1473
Humphrey M, Nation J, Francis I, Boneh A (2011) Effect of tetrahydrobiopterin on Phe/Tyr ratios and variation in Phe levels in tetrahydrobiopterin responsive PKU patients. Mol Genet Metab 104:89–92
Lambruschini N, Perez-Duenas B, Vilaseca MA et al (2005) Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol Genet Metab 86(Suppl 1):S54–S60
Lee P, Treacy EP, Crombez E et al (2008) Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. Am J Med Genet A 146A:2851–2859
Levy HL, Milanowski A, Chakrapani A et al (2007) Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 370:504–510
Muntau AC, Roschinger W, Habich M et al (2002) Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med 347:2122–2132
National Institutes of Health (2001) National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16–18, 2000. Pediatrics 108:972–982
Poustie VJ, Wildgoose J (2010) Dietary interventions for phenylketonuria. Cochrane Database Syst Rev 20(1):CD001304
Somaraju UR, Merrin M (2010) Sapropterin dihydrochloride for phenylketonuria. Cochrane Database Syst Rev 16(6):CD008005
Trefz FK, Burton BK, Longo N et al (2009) Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr 154:700–707
Trefz FK, Scheible D, Frauendienst-Egger G (2010) Long-term follow-up of patients with phenylketonuria receiving tetrahydrobiopterin treatment. J Inherit Metab Dis
Vernon HJ, Koerner CB, Johnson MR, Bergner A, Hamosh A (2010) Introduction of sapropterin dihydrochloride as standard of care in patients with phenylketonuria. Mol Genet Metab 100:229–233
Webster D, Wildgoose J (2010) Tyrosine supplementation for phenylketonuria. Cochrane Database Syst Rev 4(8):CD001507
Wilcox W, Cederbaum S (2002) Amino acid metabolism. In: Rimpoin L et al (eds) Emery and Rimoin’s principles and practice of medical genetics. Churchill Livingstone, New York, pp 2405–2440
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Nenad Blau
Appendices
Disclaimer
This project was funded under Contract No. HHSA 290 2007 10065 I from the Agency for Healthcare Research and Quality, US Department of Health and Human Services. The authors of this report are responsible for its content. Statements in the report should not be construed as endorsement by the Agency for Healthcare Research and Quality or the US Department of Health and Human Services.
Synopsis
BH4 is effective in reducing blood phenylalanine levels in some individuals with PKU; however, understanding of who will benefit and effects on longer-term outcomes such as cognition and quality of life is lacking.
Contributors’ Statement
We acknowledge that all authors of this manuscript meet authorship criteria and have made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; have participated in drafting the article or revising it critically for important intellectual content; and have given final approval of the version to be published.
Article Guarantor
Mary Lou Lindegren, M.D., accepts full responsibility for the work/conduct of the study, had access to the data, and controlled the decision to publish.
Competing Interest Statement
None of the authors has any competing interests.
Financial Disclosure
This project was supported by the Agency for Healthcare Research and Quality (contract number: HHSA 290 2007 10065 I). The authors confirm independence from the sponsor; the content of the article has not been influenced by the sponsor.
Ethics Approval
Ethics approval was not required for this study.
Note: The material included in this manuscript has been published by the Agency for Healthcare Research and Quality. The full review can be accessed at http://effectivehealthcare.ahrq.gov/ehc/products/259/958/CER56_PKU_FinalReport.pdf
Rights and permissions
This chapter is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
Copyright information
© 2012 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Lindegren, M.L., Krishnaswami, S., Reimschisel, T., Fonnesbeck, C., Sathe, N.A., McPheeters, M.L. (2012). A Systematic Review of BH4 (Sapropterin) for the Adjuvant Treatment of Phenylketonuria. In: Zschocke, J., Gibson, K., Brown, G., Morava, E., Peters, V. (eds) JIMD Reports - Case and Research Reports, 2012/5. JIMD Reports, vol 8. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2012_168
Download citation
DOI: https://doi.org/10.1007/8904_2012_168
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-33432-0
Online ISBN: 978-3-642-33433-7
eBook Packages: MedicineMedicine (R0)