
Abstract
One of the important problems in functional genomics is how to select the disease genes. In this regard, the paper presents a new similarity measure to compute the functional similarity between two genes. It is based on the information of protein-protein interaction networks. A new gene selection algorithm is introduced to identify disease genes, integrating judiciously the information of gene expression profiles and protein-protein interaction networks. The proposed algorithm selects a set of genes from microarray data as disease genes by maximizing the relevance and functional similarity of the selected genes. The performance of the proposed algorithm, along with a comparison with other related methods, is demonstrated on colorectal cancer data set.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
- Disease Gene
- Functional Similarity
- Propose Similarity Measure
- Gene Selection Algorithm
- Genome Wide Association Study Data
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Genetic diseases such as Alzheimer’s disease, breast cancer, leukemia, colorectal cancer, down syndrome, and heart disease are caused by abnormalities in genes or chromosomes. A genetic disease may be heritable disorder or may not be. While some genetic diseases are passed down from the parent’s genes, others are frequently caused by new mutations or changes to the DNA. In other instances, the same disease, for example, some forms of cancer, may stem from an inherited genetic condition in some people, from new mutations in some people, and from non-genetic causes in other people. As the term genetic disease suggests, these diseases are caused by the dysfunction of some genes. Therefore, such genes are better known as disease genes [1].
Recent advancement and wide use of high-throughput biotechnologies have been producing an explosion in using gene expression phenotype for understanding the function of disease genes [4, 8]. Analyzing the difference of gene expression levels in particular cell types may provide an idea about the propensity of a disease. Specifically, if a set of genes shows a consistent pattern of different expression levels in sick subjects and a control group, then that gene set is likely a strong candidate of playing a pathogenic role. Differences in expression levels can be detected primarily by microarray studies. In this background, microarray gene expression data has been widely used for identification of disease genes using different feature selection algorithms [5, 16, 18, 22].
In [2, 6], it has been shown that the genes associated with the same disorder tend to share common functional features, reflecting that their protein products have a tendency to interact with each other. Hence, another indicative trait of a disease gene is that its protein product is strongly linked to other disease-gene proteins. In this background, the protein-protein interaction (PPI) data have been used in various studies to identify disease genes [13, 20]. Individually microarray data or the PPI network data can be used to identify potential disease genes, although there is a limited chance of finding novel disease genes from such an analysis. In this regard, data integration methods have been developed to identify pleiotropic genes involved in the physiological cellular processes of many diseases.
The integrated approaches assume that the protein products of disease genes tend to be close to differentially expressed genes in the protein interaction network. Chao et al. [24] developed a method by integrating gene expression data and the PPI network data to prioritize cancer-associated genes. Zhao et al. [25] also proposed an approach by integrating gene expression data and the PPI network data to select disease genes. Jia et al. [10] developed a dense module searching method to identify disease genes for complex diseases by integrating the association signal from genome wide association studies data sets into the human PPI network. Li and Li [17] developed another approach to identify candidate disease genes, where heterogeneous genomic and phenotype data sets are used. In this method, separate gene networks are first developed using different types of data sets. The various genomic networks are then merged into a single graph, and disease genes are identified using random walk. In [16], minimum redundancy-maximum relevance (mRMR) [5] approach has been used to select a set of genes from expression data, while maximum relevance-maximum significance (MRMS) criterion [18] has been used in [22]. The selected gene set is then used for identification of intermediate genes between a pair of selected genes using the PPI network data. However, all the methods reported earlier consider gene expression and PPI data separately while selecting candidate genes.
In this regard, this paper presents a new gene selection algorithm to identify disease genes. It selects a set of disease genes by maximizing the relevance and functional similarity of the selected genes. A new similarity measure is introduced to compute the functional similarity between two genes. The proposed algorithm judiciously integrates the information of gene expression profiles and PPI networks. The mutual information is employed to compute the relevance of the genes with respect to class labels based on gene expression profiles, while the PPI data is used to calculate the functional similarity between two genes. The mutual information is used to select differentially expressed genes as disease genes using gene expression profiles, on the other hand, the functional protein association network is used to study the mechanism of diseases. The performance of the proposed algorithm, along with a comparison with other related methods, is demonstrated on colorectal cancer data set. An important finding is that the proposed algorithm is shown to be effective for selecting relevant and functionally similar genes from microarray data, and the identified genes are significantly linked with colorectal cancer. Extensive experimental study on colorectal cancer establishes the fact that the genes identified by the proposed method have more colorectal cancer genes than those identified by existing methods and using the gene expression profiles alone, irrespective of any gene selection algorithm. All the results indicate that the proposed method is quite promising and may become a useful tool for identifying disease genes.
2 A New Gene-Gene Similarity Measure
In general, the genes, which are associated with the same disorder, tend to share common functional features. The protein products of these genes also have a tendency to interact with each other [2, 6]. Hence, an important characteristic of a disease gene is that its protein product is strongly linked to other disease-gene proteins. It has also been observed that proteins with short distances to each other in the network are more likely to involve in common biological functions [3, 14, 21], and that interactive neighbors are more likely to have identical biological function than non-interactive ones [11, 15]. This is because the query protein and its interactive proteins may form a protein complex to perform a particular function or be involved in a same pathway. Accordingly, a quantitative measure is required that can efficiently compute the similarity between two genes. In this paper, the information of PPI networks is used to calculate the functional similarity.
The PPI networks are commonly represented as graphs (Fig. 1), with nodes corresponding to proteins and edges representing PPIs. The weight of the edge in graph depends on experimental as well as predicted interaction information. Let \(\mathcal {N}_i\) be the set of interactive neighbors or successor genes of a candidate gene \({\mathcal A}_i\) and \(\omega _{ij} \in [0,1]\) is the weight value of the edge between gene \({\mathcal A}_j \in \mathcal {N}_i\) and candidate gene \({\mathcal A}_i\). The set of successors \(\mathcal {N}_i\) of gene \({\mathcal A}_i\) and corresponding weight value \(\omega _{ij}\) can be obtained from the information of PPI network. Let \(\mathcal {N}_{ik}\) be the set of genes, which are successors of both genes \({\mathcal A}_i\) and \({\mathcal A}_k\), that is, \(\mathcal {N}_{ik}=\mathcal {N}_i \cap \mathcal {N}_k\). Define \(\tilde{\mathcal {N}}_i=\mathcal {N}_i \setminus \mathcal {N}_{ik}\) as the set of genes those are successors of gene \({\mathcal A}_i\) but not of gene \({\mathcal A}_k\). The functional similarity between two genes \({\mathcal A}_i\) and \({\mathcal A}_k\), having sets of successor genes \(\mathcal {N}_i\) and \(\mathcal {N}_k\), respectively, is as follows:

An example of protein-protein interaction network
Hence, if the interactive neighbors and the corresponding edge weights of two genes are same, then the functional similarity between these two genes is high. On the other hand, two genes are functionally dissimilar if they have no common interactive neighbors. The following properties can be stated about the measure:
-
1.
\(0 \le \mathcal {S}({\mathcal A}_i,{\mathcal A}_k) \le 1\).
-
2.
\(\mathcal {S}({\mathcal A}_i,{\mathcal A}_k)=1\) if and only if two sets \({\mathcal N}_i\) and \({\mathcal N}_k\) contain exactly same set of successor genes, that is, \(\mathcal {N}_{ik}=\mathcal {N}_i=\mathcal {N}_k\), and weight value \(\omega _{ij}=\omega _{kj}, \forall {\mathcal A}_j \in {\mathcal N}_{ik}\).
-
3.
\(\mathcal {S}({\mathcal A}_i,{\mathcal A}_k)=0\) if and only if \({\mathcal N}_{ik}=\emptyset \).
-
4.
\(\mathcal {S}({\mathcal A}_i,{\mathcal A}_k)=\mathcal {S}({\mathcal A}_k,{\mathcal A}_i)\) (symmetric).
In this regard, it should be noted that if the weight value \(\omega _{ij} \in \{0,1\}\), then the proposed similarity measure reduces to
which is Jaccard index \(J({\mathcal A}_i,{\mathcal A}_k)\).
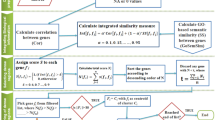
3 Proposed Disease Gene Selection Algorithm
Recent advancement and wide use of high-throughput biotechnologies have been producing huge amount of gene expression profiles data, which have been widely used in different studies to understand the function of disease genes. If a set of genes shows a consistent pattern of different expression levels in sick subjects and a control group, then that gene set is likely a strong candidate of playing a pathogenic role. The difference of gene expression levels in particular cell types can be studied to get an idea about the propensity of a disease. On the other hand, the genes associated with the same disease tend to share common functional features. Also, the protein products of disease genes have a tendency to interact with other disease-gene proteins.
In this regard, the paper presents a new gene selection algorithm, integrating judiciously the gene expression and PPI data, to identify pleiotropic genes involved in the physiological cellular processes of the disease. The proposed method assumes that the protein products of disease genes tend to be close to differentially expressed genes in the protein interaction network. Hence, the proposed gene selection algorithm selects a set \(\mathbb {S}\) of disease genes from the whole gene set \(\mathbb {C}\) of the given microarray gene expression data set by maximizing both relevance and functional similarity of genes present in \(\mathbb {S}\). Let \({\mathbb C}=\{{\mathcal A}_1,\cdots , {\mathcal A}_i,\cdots ,{\mathcal A}_j,\cdots ,{\mathcal A}_m\}\) be the set of m genes of a given microarray gene expression data set and \({\mathbb S}\) is the set of selected genes. Define \(\gamma _{{\mathcal A}_i} ({\mathbb D})\) as the relevance of the gene \({\mathcal A}_i\) with respect to the class labels \({\mathbb D}\) while \(\mathcal {S}({\mathcal A}_i,{\mathcal A}_j)\) as the functional similarity between two genes \({\mathcal A}_i\) and \({\mathcal A}_j\). Hence, the total relevance of all selected genes is
while the total functional similarity among the selected genes is
Hence, the problem of selecting a set \({\mathbb S}\) of relevant and functionally similar genes from the whole set \({\mathbb C}\) of m genes is equivalent to maximizing both \({\mathcal J}_\mathrm{relevance}\) and \({\mathcal J}_\mathrm{similarity}\), that is, to maximize the objective function
where a is a weight parameter. To solve the above problem, following greedy algorithm is used in the current study:
-
1.
Initialize \({\mathbb C} \leftarrow \{{\mathcal A}_1,\cdots , {\mathcal A}_i,\cdots ,{\mathcal A}_j,\cdots ,{\mathcal A}_m\}, {\mathbb S} \leftarrow \emptyset \).
-
2.
Calculate the relevance \(\gamma _{{\mathcal A}_i} ({\mathbb D})\) of each gene \({\mathcal A}_i \in {\mathbb C}\).
-
3.
Select the gene \({\mathcal A}_i\) as the most relevant gene that has the highest relevance value \(\gamma _{{\mathcal A}_i} ({\mathbb D})\). In effect, \({\mathcal A}_i \in {\mathbb S}\) and \({\mathbb C}={\mathbb C} \setminus {\mathcal A}_i\).
-
4.
Repeat the following two steps until the desired number of genes is selected.
-
5.
Calculate the functional similarity between each of the remaining genes of \({\mathbb C}\) with respect to the selected genes of \({\mathbb S}\) and remove it from \({\mathbb C}\) if it has zero functional similarity value with respect to any one of the selected genes.
-
6.
From the remaining genes of \({\mathbb C}\), select gene \({\mathcal A}_j\) that maximizes the following condition:
$$\begin{aligned} a \gamma _{{\mathcal A}_j} ({\mathbb D}) + \frac{(1-a)}{|{\mathbb S}|} \sum _{{\mathcal A}_i \in {\mathbb S}} \mathcal {S}({\mathcal A}_i,{\mathcal A}_j). \end{aligned}$$(6)As a result of that, \({\mathcal A}_j \in {\mathbb S}\) and \({\mathbb C}={\mathbb C} \setminus {\mathcal A}_j\).
-
7.
Stop.
The mutual information [22] can be used to calculate the relevance of a gene with respect to class labels, while the proposed similarity measure, based on the information of PPI data, can be used for computing functional similarity between two genes. However, in microarray gene expression data sets, the class labels of samples are represented by discrete symbols, while the expression values of genes are continuous. Hence, to measure the gene-class relevance of a gene with respect to class labels using mutual information, the continuous expression values of a gene are divided into several discrete partitions. The a prior (marginal) probabilities and their joint probabilities are then calculated to compute the gene-class relevance using the definitions for discrete cases. In this paper, the discretization method reported in [5, 22] is employed to discretize the continuous gene expression values. The expression values of a gene are discretized using mean \(\mu \) and standard deviation \(\sigma \) computed over n expression values of that gene: any value larger than \((\mu + \sigma /2)\) is transformed to state 1; any value between \((\mu - \sigma /2)\) and \((\mu + \sigma /2)\) is transformed to state 0; any value smaller than \((\mu - \sigma /2)\) is transformed to state -1. These three states correspond to the over-expression, baseline, and under-expression of genes. On the other hand, the STRING (Search Tool for the Retrieval of Interacting Genes) is an online database resource that provides both experimental as well as predicted PPI information, along with a confidence score. In the current work, STRING is used for computing functional similarity between two genes considering confidence score as the weight value.
4 Experimental Results and Discussions
This section presents the performance of the proposed maximum relevance-maximum functional similarity (MRMFS) criterion based proposed gene selection algorithm, along with a comparison with other related methods. The algorithms compared are t-test, MR (maximum relevance), mRMR [5], MRMS [18], MR+PPIN [22], mRMR+PPIN [16], and MRMS+PPIN [22]. The mutual information is used to compute the relevance, redundancy, and significance of the genes. The value of a in (6) is set to 0.5.
In this study, the gene expression data from the colorectal cancer study of Hinoue et al. [7] is used. The gene expression profiling of 26 colorectal tumors and matched histologically normal adjacent colonic tissue samples were retrieve from the NCBI Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) with the accession number of GSE25070. The number of genes and samples in this data set are 24526 and 52, respectively. The data set is pre-processed by standardizing each sample to zero mean and unit variance.
The performance of different methods is compared with respect to the degree of overlapping with three gene lists, namely, LIST-1, LIST-2, and LIST-3. The LIST-1 contains 742 cancer related genes, which are collected from the Cancer Gene Census of the Sanger Centre, Atlas of Genetics and Cytogenetic in Oncology [9], and Human Protein Reference Database [12]. On the other hand, both LIST-2 and LIST-3 consist of colorectal cancer related genes. While the LIST-2 is retrieved from the study of Sabatas-Bellver et al. [23], the LIST-3 is prepared from the work of Nagaraj and Reverter [19]. While LIST-2 contains 438 colorectal cancer genes, LIST-3 consists of 134 colorectal cancer genes.
4.1 Performance of Different Gene Selection Algorithms
This section presents the comparative performance analysis of different gene selection algorithms with respect to the degree of overlapping with the three gene lists. The algorithms compared are t-test, MR, mRMR [5], MRMS [18], and the proposed MRMFS. Results are reported for first twenty genes selected by different algorithms.
Table 1 presents the lists of genes selected by different gene selection algorithms, along with their degree of overlapping with any one of the three cancer gene lists. From the results reported in Table 1, it can be seen that the proposed method provides better results than that of other methods with respect to degree of overlapping with known gene lists. Out of 20 selected genes, 14 genes selected by the proposed algorithm overlap with known disease genes, while t-test, MR, mRMR, and MRMS algorithms can identify 10, 10, 7, and 11 disease genes.
4.2 Performance of Different Disease Gene Identification Methods
Finally, the performance of the proposed algorithm is compared with two algorithms, namely, MR+PPIN [22] and mRMR+PPIN [16], which combine gene expression and PPIN data for selection of disease genes. The results are reported in Table 2 considering 41 genes as both MR+PPIN and mRMR+PPIN methods consider 41 genes for their analysis. Table 2 also presents the statistical significance test of the gene sets selected by the MR+PPIN, mRMR+PPIN, and proposed methods with respect to the genes of LIST-1, LIST-2, and LIST-3. Using the Fisher’s exact test, statistical analysis of the overlapped genes is performed.
Out of total 41 genes selected by the proposed method, 16 and 8 genes are related to colorectal cancer with respect to the LIST-2 and LIST-3, respectively, while only 7 and 5 genes obtained using MR+PPIN are colon cancer related genes. On the other hand, only 4 and 3 genes selected using mRMR+PPIN are related to colon cancer with respect to two lists. Hence, the Fisher’s exact test for the proposed method generates lower p-values for both LIST-2 and LIST-3, which are significantly better than the p-values obtained by other two methods. However, the degree of overlapping by the proposed algorithm with cancer related genes of LIST-1 is lower than that by existing methods. The last column of Table 2 depicts the degree of overlapping with respect to the two colorectal cancer gene lists. While the proposed method can identify 19 colorectal cancer related genes, only 10 and 5 disease genes are identified by the MR+PPIN and mRMR+PPIN methods.
4.3 KEGG Enrichment Analysis
The hundred genes selected by the proposed method are further analyzed using the functional annotation tool of David. The enriched p-value was corrected to control family-wide false discovery rate under certain threshold (for example, \(<\)0.05) with Benjamin multiple testing correction method. Table 3 represents the KEGG pathway enrichment analysis of the gene set obtained by the proposed algorithm. From the table, it is seen that most of the networks are associated with cancer. Various processes, those are associated with colon cancer like p53 signaling pathway and colorectal cancer, are also observed in the result. Moreover, the gene set is found to be highly associated with colorectal cancer disease according to the OMIM disease database as analyzed by the functional annotation tool of David.
5 Conclusion
The main contribution of the paper is to present a new gene selection algorithm to identify disease genes. The proposed algorithm integrates judiciously the information of gene expression profiles and protein-protein interaction networks. It selects a set of genes from microarray data as disease genes by maximizing the relevance and functional similarity of the selected genes. A new similarity measure is introduced to compute the functional similarity between two genes. It is based on the information of protein-protein interaction networks. The performance of the proposed algorithm, along with a comparison with other related methods, is demonstrated on colorectal cancer data set. Extensive experimental study on colorectal cancer establishes the fact that the genes identified by the proposed method have more colorectal cancer genes than the genes identified by the existing gene selection algorithms. All these results indicate that the proposed method is quite promising and may become a useful tool for identifying disease genes.
References
Altshuler, D., Daly, M.J., Lander, E.S.: Genetic mapping in human disease. Science 322(5903), 881–888 (2008)
Barrenas, F., Chavali, S., Holme, P., Mobini, R., Benson, M.: Network properties of complex human disease genes identified through genome-wide association studies. PLoS ONE 4(11), e8090 (2009)
Bogdanov, P., Singh, A.K.: Molecular function prediction using neighborhood features. IEEE/ACM Trans. Comput. Biol. Bioinform. 7(2), 208–217 (2010)
Cai, Yu-Dong, Huang, T., Feng, K.-Y., Hu, L., Xie, L.: A unified 35-gene signature for both subtype classification and survival prediction in diffuse large B-cell lymphomas. PLoS ONE 5(9), e12726 (2010)
Ding, C., Peng, H.: Minimum redundancy feature selection from microarray gene expression data. J. Bioinf. Comput. Biol. 3(2), 185–205 (2005)
Goh, K.-I., Cusick, M.E., Valle, D., Childs, B., Vidal, M., Barabsi, A.-L.: The human disease network. Proc. National Acad. Sci. USA 104(21), 8685–8690 (2007)
Hinoue, T., Weisenberger, D.J., Lange, C.P.E., Shen, H., Byun, H.M., Van Den Berg, D., Malik, S., Pan, F., Noushmehr, H., van Dijk, C.M., Tollenaar, R.A.E.M., Laird, P.W.: Genome-scale analysis of aberrant dna methylation in colorectal cancer. Genome Res. 22(2), 271–282 (2012)
Huang, T., Chen, L., Cai, Y.-D., Chou, K.-C.: Classification and analysis of regulatory pathways using graph property, biochemical and physicochemical property, and functional property. PLoS ONE 6(9), e25297 (2011)
Huret, J.L., Dessen, P., Bernheim, A.: Atlas of genetics and cytogenetics in oncology and haematology. Nucleic Acids Res. 31(1), 272–274 (2003)
Jia, P., Zheng, S., Long, J., Zheng, W., Zhao, Z.: dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics 27(1), 95–102 (2011)
Karaoz, U., Murali, T.M., Letovsky, S., Zheng, Y., Ding, C., Cantor, C.R., Kasif, S.: Whole-genome annotation by using evidence integration in functional-linkage networks. Proc. National Acad. Sci. USA 101(9), 2888–2893 (2004)
Keshava Prasad, T.S., Goel, R., Kandasamy, K., Keerthikumar, S., Kumar, S., Mathivanan, S., Telikicherla, D., Raju, R., Shafreen, B., Venugopal, A., Balakrishnan, L., Marimuthu, A., Banerjee, S., Somanathan, D.S., Sebastian, A., Rani, S., Ray, S., Harrys Kishore, C.J., Kanth, S., Ahmed, M., Kashyap, M.K., Mohmood, R., Ramachandra, Y.L., Krishna, V., Rahiman, B.A., Mohan, S., Ranganathan, P., Ramabadran, S., Chaerkady, R., Pandey, A.: Human protein reference database-2009 update. Nucleic Acids Res. 37(suppl 1), D767–D772 (2009)
Kohler, S., Bauer, S., Horn, D., Robinson, P.N.: Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Gen. 82(4), 949–958 (2008)
Kourmpetis, Y.A.I., van Dijk, A.D.J., Bink, M.C.A.M., van Ham, R.C.H.J., ter Braak, C.J.F.: Bayesian markov random field analysis for protein function prediction based on network data. PLoS ONE 5(2), e9293 (2010)
Letovsky, S., Kasif, S.: Predicting protein function from protein/protein interaction data: a probabilistic approach. Bioinformatics 19(suppl 1), i197–i204 (2003)
Li, B.-Q., Huang, T., Liu, L., Cai, Y.-D., Chou, K.-C.: Identification of colorectal cancer related genes with mrmr and shortest path in protein-protein interaction network. PLoS ONE 7(4), e33393 (2012)
Li, Y., Li, J.: Disease gene identification by random walk on multigraphs merging heterogeneous genomic and phenotype data. BMC Genomics 13(Suppl 7), S27 (2012)
Maji, P., Paul, S.: Rough set based maximum relevance-maximum significance criterion and gene selection from microarray data. Int. J. Approximate Reasoning 52(3), 408–426 (2011)
Nagaraj, S., Reverter, A.: A boolean-based systems biology approach to predict novel genes associated with cancer: application to colorectal cancer. BMC Syst. Biol. 5(1), 35 (2011)
Navlakha, S., Kingsford, C.: The power of protein interaction networks for associating genes with diseases. Bioinformatics 26(8), 1057–1063 (2010)
Ng, K.-L., Ciou, J.-S., Huang, C.-H.: Prediction of protein functions based on function-function correlation relations. Comput. Biol. Med. 40(3), 300–305 (2010)
Paul, S., Maji, P.: Gene expression and protein-protein interaction data for identification of colon cancer related genes using \(f\)-information measures. Natural Computing (2015). doi:10.1007/s11047-015-9485-6
Sabates-Bellver, J., Van der Flier, L.G., de Palo, M., Cattaneo, E., Maake, C., Rehrauer, H., Laczko, E., Kurowski, M.A., Bujnicki, J.M., Menigatti, M., Luz, J., Ranalli, T.V., Gomes, V., Pastorelli, A., Faggiani, R., Anti, M., Jiricny, J., Clevers, H., Marra, G.: Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 5(12), 1263–1275 (2007)
Chao, W., Zhu, J., Zhang, X.: Integrating gene expression and protein-protein interaction network to prioritize cancer-associated genes. BMC Bioinform. 13(1), 182 (2012)
Zhao, J., Yang, T.-H., Huang, H., Holme, P.: Ranking candidate disease genes from gene expression and protein interaction: a katz-centrality based approach. PLoS ONE 6(9), e24306 (2011)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this paper
Cite this paper
Maji, P., Shah, E., Paul, S. (2015). A New Similarity Measure for Identification of Disease Genes. In: Kryszkiewicz, M., Bandyopadhyay, S., Rybinski, H., Pal, S. (eds) Pattern Recognition and Machine Intelligence. PReMI 2015. Lecture Notes in Computer Science(), vol 9124. Springer, Cham. https://doi.org/10.1007/978-3-319-19941-2_43
Download citation
DOI: https://doi.org/10.1007/978-3-319-19941-2_43
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19940-5
Online ISBN: 978-3-319-19941-2
eBook Packages: Computer ScienceComputer Science (R0)
