
Abstract
Agrobacterium radiobacter is the only known non-phytopathogenic species in Agrobacterium genus. In this study, the whole-genome sequence of A. radiobacter type strain DSM 30147T was described and compared to the other available Agrobacterium genomes. This bacterium has a genome size of 7,122,065 bp distributed in 612 contigs, including 6,834 protein-coding genes and 41 RNA genes. It harbors a circular chromosome and a linear chromosome but not a tumor-inducing (Ti) plasmid. To the best of our knowledge, this is the first report of a genome from the A. radiobacter species. In addition, an emended description of A. radiobacter is described. This study reveals information that enhances the current understanding of its non-phytopathogenicity and its phylogenetic position within Agrobacterium genus.
Similar content being viewed by others


Introduction
Agrobacterium radiobacter DSM 30147T (= ATCC 19358T) was first isolated from saprobic soil in 1902 as Bacillus radiobacter [1] and obtained its current name until Agrobacterium genus established by Conn in 1942 [2]. Based on phytopathogenic properties, Conn divided Agrobacterium into 3 species, A. radiobacter, A. tumefaciens and A. rhizogenes [2]. Subsequently, A. rubi, A. vitis and A. larrymoorei were also identified within the Agrobacterium genus [3–6]. Recently, A. rhizogenes was transferred to Rhizobium genus, as Rhizobium rhizogenes, based on multilocus sequence analysis (MLSA) using several housekeeping genes (rrs, atpD and recA) [7,8]. In addition, Young et al. proposed that A. radiobacter should have priority over A. tumefaciens, and A. tumefaciens may not officially represent a species [8,9]. Thus, currently, the genus Agrobacterium contains four validly named species, A. radiobacter, A. vitis, A. rubi and A. larrymoorei [7–9].
A taxonomic classification that relies on the phytopathogenic phenotypes may not accurately reflect the actual phylogenetic relationships of strains within Agrobacterium [10]. Accordingly, an alternative classification method was applied which divided most Agrobacterium strains into 3 biovariants (Biovars I, II and III) [10]. Among the 3 biovariants, Biovar I is the most complex group and includes several members (genomovars), designated as genomovar G1 through G9 and G13 [8,11]. At present, two strains in Biovar I have been completely sequenced: Agrobacterium sp. H13-3 (G1) and A. tumefaciens C58 (G8). The genome sequencing revealed that these strains contained two chromosomes and different numbers of plasmids. A. radiobacter DSM 30147T also belongs to Biovar I (it is classified as a member of genomovar G4), which indicates its close relationship to A. tumefaciens C58 and Agrobacterium sp. H13-3 [12].
Most strains in the genus Agrobacterium are phytopathogens and induce crown gall tumors or hairy root diseases in their host plants [2]. However, A. radiobacter is an exception because it does not have the tumor-inducing (Ti) plasmid that contributes to the pathogenicity [13–16]. A. radiobacter members have been widely found in soil, in the rhizosphere of plants and in clinical specimens [17]. A strain of A. radiobacter was reported to enhance soil arsenic phytoremediation, indicating a potential application in bioremediation [18]. However, some members have been identified as opportunistic human pathogens [19]. So far, a total of 11 Agrobacterium genomes (3 finished and 8 draft genomes, listed in Table 1) have been sequenced but no genome of A. radiobacter has been reported. Considering its essential biological feature and important phylogenetic position in the genus Agrobacterium, we present the genome sequence of A. radiobacter DSM 30147T, the first sequenced strain in this species.
The descriptions of A. radiobacter have been reported in 1902 [1], 1942 [2], 1980 [21] and 1993 [22]. After that, fatty acids and utilization of more carbon and nitrogen sources have been tested and showed that the major fatty acids (> 5%) are 16:0, 19:0 cyclo ω8c, summed feature 2 (one or more of 12:0 aldehyde, iso-16:1 I and 14:0 3-OH) and summed feature 8 (18:1ω7c and/or 18:1ω6c) [23]. The strain can utilize adonitol, D-fructose, D-galactose, D-mannitol, lactose and raffinose as sole carbon sources and L-ornithine, L-proline and L-serine as sole nitrogen sources [23]. Citrate utilization, nitrate reduction and urease are all positive [23]. In this study, we performed more physiological/biochemical analysis and present the emended description of A. radiobacter.
Classification and features
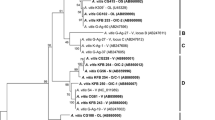
Genome sequences and 16S rRNA genes were used for phylogenetic analysis. In view of the close evolutionary relationship and the inconsistent phylogeny between Agrobacterium and Rhizobium [12], we pre-analyzed all sequenced strains in these two genera and found that two “Rhizobium” members were very closely related to the 12 Agrobacterium members (including strain DSM 30147T). Thus, all of the 12 Agrobacterium members with sequenced genomes, two Rhizobium strains [R. lupini HPC(L) and Rhizobium sp. PDO1-076] (Table 1) and an out-group strain R. rhizogenes K84 [7,8], were included in the phylogenetic analysis. A comparison of the 15 genomes revealed a total of 370 proteins that were shared across these genomes. A rooted neighbor-jointing (NJ) phylogenetic tree was constructed based on the shared amino acid sequences. As shown in Figure 1a, A. radiobacter DSM 30147T was in the same cluster as the Biovar I members Agrobacterium sp. H13-3 (G1) and A. tumefaciens C58 (G8), and showed the closest relationship with A. tumefaciens str. Cherry 2E-2-2. A NJ phylogenetic tree was also constructed based on the 16S rRNA genes (Figure 1b). When comparing the trees generated by the core protein sequences with those generated by 16S rRNA gene sequences, small topological differences in topology were found between them. In comparison to the tree generated using the 370 conserved proteins, some strains could not be distinguished with a high degree of clarity using the 16S rRNA genes. Therefore, phylogenomic analysis was considered a more robust approach than that using the 16S rRNA genes to infer the phylogeny, especially for closely related strains [21,25,26].

Phylogenetic trees highlighting the relationships among A. radiobacter DSM 30147T and other closely related sequenced strains. (a) A tree was built based on 370 conserved proteins shared among the 15 genomes (12 Agrobacterium strains, 2 Rhizobium strains very closely related to Agrobacterium and one out-group strain, R. rhizogenes K84); (b) A tree inferred from the 16S rRNA genes of the same strains. The phylogenies were inferred by MEGA 5.05 using the neighbor-joining algorithm [20,24], and 1,000 bootstrap repetitions were computed to estimate the reliability of the branching order. The genome accession numbers of the strains used in the phylogenetic reconstructions: A. albertimagni AOL15, ALJF00000000; Rhizobium sp. PDO1-076, AHZC00000000; A. vitis S4, A. radiobacter, ASXY01000000; GCA_000016285; Agrobacterium sp. H13-3, GCA_000192635; Agrobacterium sp. 10MFCol1.1, ARLJ00000000; A. tumefaciens 5A, AGVZ00000000; A. tumefaciens F2, AFSD00000000; A. tumefaciens C58, GCA_000092025; Agrobacterium sp. ATCC 31749, AECL00000000; R. lupini HPC(L), AMQQ00000000; A. tumefaciens str. Cherry 2E-2-2, APCC00000000; Agrobacterium sp. 224MFTsu3.1, ARQL00000000; A. tumefaciens CCNWGS0286, AGSM00000000 and R. rhizogenes K84 GCA_000016265.
Strain DSM 30147T is rod-shaped (0.6–0.8 × 1.5–1.8 µm) (Figure 2). The enzyme activities and carbon sources utilization of strain DSM 30147T were tested using API ZYM, API 20 NE and API ID 32 GN systems and the results are shown in Table 2 and in the emended description of A. radiobacter.

A transmission micrograph of A. radiobacter DSM 30147T, using 200 kV transmission electron microscopy FEI Tecnai G2 20 TWIN (USA). The scale bar represents 1 µm.
Genome sequencing and annotation
Genome project history
To make a comprehensive genomic comparison for the Agrobacterium genomes, the whole genome sequence of A. radiobacter DSM 30147T was determined. This draft genome sequence has been deposited at DDBJ/EMBL/GenBank under accession number ASXY00000000. The version described in this study is the first version, ASXY01000000. The project information is summarized in Table 3.
Growth condition and DNA isolation
A. radiobacter DSM 30147T was grown aerobically in LB medium [38] at 28 °C for 24 h. The DNA was extracted, concentrated and purified using the QiAamp kit according to the manufacturer’s instruction (Qiagen, Germany).
Genome sequencing and assembly
Illumina Hiseq2000 with the Paired-End library strategy (300 bp insert size) was used to determine the whole-genome sequence of A. radiobacter DSM 30147T and obtained a total of 15,140,909 reads (1.41 Gb data). The detailed methods of library construction and sequencing can be found at Illumina’s official website [39]. Using SOAPdenovo v1.05 [40], these reads were assembled into 612 contigs (> 200 bp) with a genome size of 7,122,065 bp and an average coverage of 196.3 ×.
Genome annotation
The draft genome of A. radiobacter DSM 30147T was annotated using the National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline (PGAP) [41], which combines the gene caller GeneMarkS+ [42] with the similarity-based gene detection approach. Protein function classification was performed by searching all the predicted coding sequences of strain DSM 30147T against the Clusters of Orthologous Groups (COGs) protein database [43] using Blastp algorithm with E-value cutoff 1-e10.
Genome properties
The whole genome of A. radiobacter DSM 30147T is 7,122,065 bp in length, with an average GC content of 59.9%, and distributed in 612 contigs. Compared to the complete reference genome A. tumefaciens C58 [44] (also belonging to Biovar I, Figure 1), the whole genome of strain DSM 30147T could clearly be divided into 2 replicons, a circular chromosome and a linear chromosome (Figure 3). In accordance with its non-phytopathogenicity phenotype, strain DSM 30147T did not contain a Ti plasmid. Of the 6,894 genes predicted, 6,853 were protein-coding genes (CDSs), and 41 RNA genes. A total of 5,320 CDSs (77.85%) were assigned with putative functions, and the remaining proteins were annotated as the hypothetical proteins. The genome properties and statistics are summarized in Table 4 and Figure 3. The distribution of the genes into COG functional categories is shown in Table 5.

The circular representation of the A. radiobacter DSM 30147T circular chromosome (left) and linear chromosome (right). From outside to center, ring 1, 4 show protein-coding genes colored by COG categories on forward/reverse strand; ring 2, 3 denote genes on forward/reverse strand; ring 5 shows G+C% content plot, and the innermost ring shows GC skew.
Comparative genome analysis of A. radiobacter DSM 30147T with the other related genomes
Strain DSM 30147T has the largest genome size of the 12 Agrobacterium strains sequenced to date and is larger than the 2 very closely related Rhizobium strain genomes as well (Table 1). OrthoMCL [45] was used to perform orthologs clustering analysis for the 14 genomes (Table 1). The results indicate that A. radiobacter DSM 30147T shares 1,636 genes with the other 13 strains and contains 548 strain-specific genes (Table 1), which potentially encode products that contribute to species-specific features differentiating A. radiobacter from other Agrobacterium species [46]. In addition, on average, only 31% core genes were shared among the 14 genomes, which reveals a high-degree of diversity within Agrobacterium genus.
Emended description of Agrobacterium radiobacter (Beijerinck and van Delden 1902) Conn 1942 (Approved Lists 1980) emend. Sawada et al. 1993
This emended description is based on that given by Beijerinck and van Delden 1902, Conn 1942 (Approved Lists 1980) and Sawada et al. 1993 with the following changes. Positive results are observed for acid phosphatase, α-glucosidase, alkaline phosphatase, arginine dihydrolase, β-glucosidase, citrate utilization, esterase (C4), leucine arylamidase, N-acetyl-β-glucosaminidase, naphthol-AS-BI-phosphohydrolase, nitrate reduction, urease and valine arylamidase, but negative results for α-galactosidase, α-mannosidase, β-fucosidase, β-galactosidase, β-glucuronidase, chymotrypsin, cystine arylamidase, esterase lipase (C8), lipase (C14) and trypsin. Arabinose, D-glucose, D-melibiose, D-ribose, D-sorbitol, gluconates, histidine, 4-hydroxybenzoate, 3-hydroxybutyrate, inositol, 2-ketogluconate, L-alanine, L-fucose, L-lactate, L-rhamnose, malate, maltose, mannose, N-acetyl glucosamine, propionate, salicin, sodium acetate and sucrose source while cannot assimilate adipate, caprate, 3-hydroxy-benzoate, itaconic acid, glycogen, 5-ketogluconate, phenylacetate, potassium, sodium malonate, suberate and valerate are utilized as the sole carbon sources. L-ornithine, L-proline and L-serine are utilized as nitrogen sources. The major fatty acids (> 5%) are 16:0, 19:0 cyclo ω8c, summed feature 2 (one or more of 12:0 aldehyde, iso-16:1 I and 14:0 3-OH) and summed feature 8 (18:1ω7c and/or 18:1ω6c). The members of this species are nonphytopathogenic, but in individual cases, some members of this species are detected as possible human pathogens.
References
Beijerinck MW, van Delden A. Über die Assimilation des freien Stickstoffs durch Bakterien. Zbl. Bakterbl. Parasitenkd. Infektionskr. Hyg. Abt. 1902; 11:3–43.
Conn HJ. Validity of the Genus Alcaligenes. J Bacteriol 1942; 44:353–360. PubMed
Bouzar H, Jones JB. Agrobacterium larrymoorei sp. nov., a pathogen isolated from aerial tumours of Ficus benjamina. Int J Syst Evol Microbiol 2001; 51:1023–1026. PubMed http://dx.doi.org/10.1099/00207713-51-3-1023
Ophel K, Kerr A. Agrobacterium vitis sp. nov. for strains of Agrobacterium biovar 3 from grapevines. Int J Syst Evol Microbiol 1990; 40:236–241.
Hildebrand EM. Cane gall of brambles caused by Phytomonas rubi n.sp. J Agric Res 1940; 61:685–696.
Starr MP, Weiss JE. Growth of phytopathogenic bacteria in a synthetic asparagin medium. Phytopathology 1943; 33:314–318.
Velázquez E, Palomo JL, Rivas R, Guerra H, Peix A, Trujillo ME, Garcia-Benavides P, Mateos PF, Wabiko H, Martinez-Molina E. Analysis of core genes supports the reclassification of strains Agrobacterium radiobacter K84 and Agrobacterium tumefaciens AKE10 into the species Rhizobium rhizogenes. Syst Appl Microbiol 2010; 33:247–251. PubMed http://dx.doi.org/10.1016/j.svapm.2010.04.004
Lindström K, Young JP. International Committee on Systematics of Prokaryotes Subcommittee on the taxonomy of Agrobacterium and Rhizobium: minutes of the meeting, 7 September 2010, Geneva, Switzerland. Int J Syst Evol Microbiol 2011; 61:3089–3093. PubMed http://dx.doi.org/10.1099/ijs.0.036913-0
Young JM, Pennycook SR, Watson DR. Proposal that Agrobacterium radiobacter has priority over Agrobacterium tumefaciens. Request for an opinion. Int J Syst Evol Microbiol 2006; 56:491–493. PubMed http://dx.doi.org/10.1099/ijs.0.64030-0
Kersters K, de Ley J. Genus III. Agrobacterium Cohn 1942. in Bergey’s manual of systematic bacteriology, eds Krieg NR, Holt JG. (The Williams & Wilkins Co. Baltimore, Md), Volume 1, 1984; p. 244–254.
Costechareyre D, Rhouma A, Lavire C, Portier P, Chapulliot D, Bertolla F, Boubaker A, Dessaux Y, Nesme X. Rapid and efficient identification of Agrobacterium species by recA allele analysis: Agrobacterium recA diversity. Microb Ecol 2010; 60:862–872. PubMed http://dx.doi.org/10.1007/s00248-010-9685-7
Slater SC, Goldman BS, Goodner B, Setubal JC, Farrand SK, Nester EW, Burr TJ, Banta L, Dickerman AW, Paulsen I, et al. Genome sequences of three Agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J Bacteriol 2009; 191:2501–2511. PubMed http://dx.doi.org/10.1128/JB.01779-08
Moore L, Warren G, Strobel G. Involvement of a plasmid in the hairy root disease of plants caused by Agrobacterium rhizogenes. Plasmid 1979; 2:617–626. PubMed http://dx.doi.org/10.1016/0147-619X(79)90059-3
Sigee DC. Bacterial Plant Pathology: Cell and Molecular Aspects. Cambridge University Press, 1993.
Kerr A. Transfer of virulence between isolates of Agrobacterium. Nature 1969; 223:1175–1176. http://dx.doi.org/10.1038/2231175a0
Kerr A. Acquisition of virulence by nonpathogenic isolates of Agrobacterium radiobacter. Physiol Plant Pathol 1971;1:241–246. http://dx.doi.org/10.1016/0048-4059(71)90045-2
Freney J, Gruer LD, Bornstein N, Kiredjian M, Guilvout I, Letouzey MN, Combe C, Fleurette J. Septicemia caused by Agrobacterium sp. J Clin Microbiol 1985; 22:683–685. PubMed
Wang Q, Xiong D, Zhao P, Yu X, Tu B, Wang G. Effect of applying an arsenic-resistant and plant growth-promoting rhizobacterium to enhance soil arsenic phytoremediation by Populus deltoides LH05-17. J Appl Microbiol 2011; 111:1065–1074. PubMed http://dx.doi.org/10.1111/j.1365-2672.2011.05142.x
Detrait M, D’Hondt L, Andre M, Lonchay C, Holemans X, Maton JP, Canon JL. Agrobacterium radiobacter bacteremia in oncologic and geriatric patients: presentation of two cases and review of the literature. Int J Infect Dis 2008; 12:e7–e10. PubMed http://dx.doi.org/10.1016/j.ijid.2008.03.010
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 2008; 9:75. PubMed http://dx.doi.org/10.1186/1471-2164-9-75
Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Evol Microbiol 1980; 30:225–420.
Sawada H, Ieki H, Oyaizu H, Matsumoto S. Proposal for rejection of Agrobacterium tumefaciens and revised descriptions for the genus Agrobacterium and for Agrobacterium radiobacter and Agrobacterium rhizogenes. Int J Syst Bacteriol 1993; 43:694–702. PubMed http://dx.doi.org/10.1099/00207713-43-4-694
Kaur J, Verma M, Lal R. Rhizobium rosettiformans sp. nov., isolated from a hexachlorocyclohexane dump site, and reclassification of Blastobacter aggregatus Hirsch and Muller 1986 as Rhizobium aggregatum comb. nov. Int J Syst Evol Microbiol 2011; 61:1218–1225. PubMed http://dx.doi.org/10.1099/ijs.0.017491-0
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28:2731–2739. PubMed http://dx.doi.org/10.1093/molbev/msr121
Li X, Hu Y, Gong J, Zhang L, Wang G. Comparative genome characterization of Achromobacter members reveals potential genetic determinants facilitating the adaptation to a pathogenic lifestyle. Appl Microbiol Biotechnol 2013; 97:6413–6425. PubMed http://dx.doi.org/10.1007/s00253-013-5018-3
Pennisi E. Evolution. Building the tree of life, genome by genome. Science 2008; 320:1716–1717. PubMed http://dx.doi.org/10.1126/science.320.5884.1716
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1
Woese CR, Kandier O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1–6. PubMed http://dx.doi.org/10.1099/ijs.0.64188-0
Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1.
Kuykendall LD. Order VI. Rhizobiales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 324.
Conn HJ. Taxonomic relationships of certain nonsporeforming rods in soil. J Bacteriol 1938; 36:320–321.
Allen ON, Holding AJ. Genus II. Agrobacterium Conn 1942, 359; Nom. gen. cons. Opin. 33, Jud. Comm. 1970, 10. In: Buchanan RE, Gibbons NE (eds), Bergey’s Manual of Determinative Bacteriology, Eighth Edition, The Williams and Wilkins Co., Baltimore, 1974, p. 264–267.
Editorial Secretary (for the Judicial Commission of the International Committee on Nomenclature of Bacteria). OPINION 33: Conservation of the Generic Name Agrobacterium Conn 1942. Int J Syst Bacteriol 1970; 20:10. http://dx.doi.org/10.1099/00207713-20-1-10
BAuA. Classification of bacteria and archaea in risk groups. http://www.baua.de. TRBA 2010; 466:112.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Bertani G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia col. [pmid:14888646]. J Bacteriol 1951; 62:293–300. PubMed
Illumina. www.illumina.com.cn
SOAPdenovo v1.05. http://soap.genomics.org.cn/
Prokaryotic Genome Annotation Pipeline. http://www.ncbi.nlm.nih.gov/genome/annotationprok
Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 2001; 29:2607–2618. PubMed http://dx.doi.org/10.1093/nar/29.12.2607
Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 2001; 29:22–28. PubMed http://dx.doi.org/10.1093/nar/29.1.22
Wood DW, Setubal JC, Kaul R, Monks DE, Kitajima JP, Okura VK, Zhou Y, Chen L, Wood GE, Almeida NF, Jr., et al. The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science 2001; 294:2317–2323. PubMed http://dx.doi.org/10.1126/science.1066804
Li L, Stoeckert CJ, Jr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 2003; 13:2178–2189. PubMed http://dx.doi.org/10.1101/gr.1224503
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev 2005; 15:589–594. PubMed http://dx.doi.org/10.1016/j.gde.2005.09.006
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31010103903 and J1103510). The authors thank the German Collection of Microorganisms and Cell Cultures (DSMZ) for providing A. radiobacter DSM 30147T cultures.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors contributed equally.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhang, L., Li, X., Zhang, F. et al. Genomic analysis of Agrobacterium radiobacter DSM 30147T and emended description of A. radiobacter (Beijerinck and van Delden 1902) Conn 1942 (Approved Lists 1980) emend. Sawada et al. 1993. Stand in Genomic Sci 9, 574–584 (2014). https://doi.org/10.4056/sigs.4688352
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.4688352