
Abstract
Histone acetyltransferase and histone deacetylase are enzymes responsible for histone acetylation and deacetylation, respectively, in which the histones are acetylated and deacetylated on lysine residues in the N-terminal tail and on the surface of the nucleosome core. These processes are considered the most important epigenetic mechanisms for remodeling the chromatin structure and controlling the gene expression. Histone acetylation is associated with gene activation. Sodium phenylbutyrate is a histone deacetylase inhibitor that has been approved for treatement of urea cycle disorders and is under investigation in cancer, hemoglobinopathies, motor neuron diseases, and cystic fibrosis clinical trials. Due to its characteristics, not only of histone deacetylase inhibitor, but also of ammonia sink and chemical chaperone, the interest towards this molecule is growing worldwide. This review aims to update the current literature, involving the use of sodium phenylbutyrate in experimental studies and clinical trials.
Similar content being viewed by others


1. Introduction
The developmental biologist Conrad H. Waddington (1905–75) is considered to have coined the term ‘epigenetics’ in 1942[1] when he defined it as “the branch of biology which studies the causal interactions between genes and their products which bring the phenotype into being.”[2] The word ‘epigenetics’ derives from epi- (Greek: ɛπί- over, above) and -genetics (Greek: γɛνɛτικóς genetikos, ‘genitive’ and that from γε´νɛσις genesis, ‘origin’). Nowadays DNA methylation and histone modifications, i.e. acetylation, methylation, and phosphorylation, can be defined as epigenetic mechanisms important to determine when and where a gene will be expressed. Several inhibitors and activators of enzymes that catalyze DNA or histone modifications are considered useful as therapeutic agents, since the imbalance of epigenetic networks is known to cause several pathologic conditions.[3] The human genome is packaged so as to form the chromatin, which is made up of repetitive units called nucleosomes. A single nucleosome core structure can be described as a fragment of DNA, wrapped around a histone octamer formed by four histone partners (one H3-H4 tetramer and two H2A-H2B dimers).[4] Histone lysine and arginine residues are subject to post-translational modifications — i.e. methylation, citrullination, acetylation, ubiquitination, and sumoylation — and the combinatorial action of these modifications regulates the critical DNA processes, including replication, repair, and transcription. In addition, the enzymes that modify histone lysine and arginine residues have been connected to a variety of human diseases, including arthritis, cancer, heart disease, diabetes mellitus, and neurodegenerative disorders.[5] In particular, histone acetyl transferases (HATs) and histone deacetylases (HDACs) are responsible for the histone being acetylated or deacetylated (removal of an acetyl group from the ɛ-amino groups of the lysine side-chains) and have been related to transcription, cell cycle progression, gene silencing, lymphocyte and muscle differentiation, regulation of neuronal phenotype, DNA replication, and the response to DNA damage.[6] HATs acetylate the specific lysine residues in the amino terminals of histones, which are thought to neutralize the positive charge generating a more open DNA conformation, thus connecting to nucleosome remodeling and transcription regulation. The change of the charge induces a reduction in the affinity between the histones and DNA generates a more open DNA conformation. The histone deacetylation by HDACs is thought to restore the positive charges on histones by removing the acetyl groups and is responsible for transcriptional repression through chromatin condensation (hyperacetylated histones are linked to transcriptionally active domains, while hypoacetylated histones are generally associated with transcriptionally silent loci).[4,7–9] Histone deacetylase inhibitors (HDACIs) have been classified in different classes: short-chain fatty acid (as sodium butyrate, valproic acid, etc.), epoxides (as depudecin and trapoxin), cyclic peptides, benzamides, and hydroxamic acids (as trichostatin A [TSA], suberoylanilide hydroxamic acid [vorinostat; SAHA], and LAQ824).[10] Apart from these classic HDACIs, today, novel compounds, such as hydroxamic acids and benzamides, are also available.
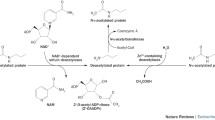
Sodium phenylbutyrate (figure 1), a HDACI, is an aromatic fatty acid that is converted/oxidized in vivo into phenylacetate (PAA) by β-oxidation.[11] In humans the so formed PAA is eliminated by conjugation with glutamine to form phenacetylglutamine, which is excreted in the urine. This metabolic pathway is the mechanism by which phenylbutyrate acts as an ammonia scavenger in patients with urea cycle disorders (UCDs) and hyperammonemia.[12] It has been shown that 1 g of phenylbutyrate would allow the removal of the equivalent of 1 g of protein.[13] Plasma PAA and urinary phenacetylglutamine are also endpoints of normal tyrosine metabolism. In vitro, phenylbutyrate shows the ability to induce differentiation by various mechanisms,[14] and the US FDA has approved its clinical use in patients with hyperammonemia. Phenylbutyrate therapy for infants, children, and adults with UCD, which leads to nitrogen accumulation in the form of ammonia, must be undertaken daily and lifelong, and it is generally well tolerated and associated with improvements in ammonia and liver function.[15] Phenylbutyrate is relatively stable, with a 0.8–1 hour half-life in human serum.[16] It has been shown that phenylbutyrate is able to lower very-long-chain fatty acid levels in the brain of mice with x-linked adrenoleukodystrophy, suggesting that phenylbutyrate can cross the blood-brain barrier.[17] An experimental study has shown its penetration into the cerebrospinal fluid after intravenous administration in nonhuman primates.[18] In the US it has been proposed as an alternative pathway for waste nitrogen excretion in female patients with ornithine transcarbamylase (OTC) deficiency, which is the most common UCD.[19] Phenylbutyrate has also been proposed during pregnancy of heterozygote OTC-deficient females, without leading to deleterious consequences for the fetus.[20] The ability of phenylbutyrate to inhibit HDACs makes it a good choice as an anti-tumor agent able to entice cellular differentiation through modification of chromatin and reprogramming the gene expression.[21] It has been reported that phenylbutyrate exerts a potent anti-tumor effect in vitro, causing growth inhibition and differentiation in various human cancer cells such as colon carcinoma, Burkitt lymphoma, primary acute myeloid leukemia, retinoblastoma, prostate cancer, U138 MG, T98G, U373 MG and A-172 glioma cells, medulloblastoma, and hepatocellular carcinoma. Phenylbutyrate has been safely used in phase I and II clinical trials for solid tumors and high-grade astrocytoma, and has shown efficacy in the treatment of different kinds of tumors, including hormone refractory prostate cancer, hematologic malignancy, and high grade astrocytoma.[22] Moreover, phenylbutyrate has reduced the neuroinflammation and overall disease process in multiple sclerosis cases,[23] has attenuated cerebral infarction and neuronal apoptosis, and improved the neurologic status in a mouse model featuring cerebral hypoxia-ischemia.[24] Its brain permeability remains a major limitation for drug treatment of CNS disorders, and it has been demonstrated that several HDACIs, such as vorinostat, sodium butyrate, phenylbutyrate, MS275 and valporic acid, are able to cross the blood-brain barrier. That is why they are considered good candidates for treating brain and spinal cord disorders.[25] Interestingly, a study, concerning PAA and phenylbutyrate intravenous administration in three Rhesus monkeys, has shown high cerebrospinal fluid penetration of both drugs, suggesting that their activity in brain tumors, and especially in meningeal malignancy, should be investigated; phenylbutyrate seems to bring some advantages, since it results in exposure in plasma and cerebrospinal fluid to both active compounds.[18]

Chemical structure of sodium phenylbutyrate (sodium 4-phenylbutanoate).
An experimental in vitro study has been conducted to test sodium butyrate, phenylbutyrate and TSA in relation to fragile X syndrome, the most common form of inherited mental retardation. It is characterized by an abnormal expansion of the repeated trinucleotide sequence CGG, which is contained in the regulatory region of the FMR1 gene and causes transcriptional inactivation.[26] These compounds lead to a consistent, but modest (1–2% of wild-type levels), reactivation of the FMR1 gene in fragile X lymphoblasts using reverse transcription polymerase chain reaction.[27]
2. Review Criteria
We searched the literature (all articles listed in PubMed Central up to June 2011) for both clinical trials and experimental studies in vitro and in vivo, involving the use of sodium phenylbutyrate and derivative compounds, using the keywords ‘triButyrate’, ‘sodium phenylbutyrate’, and ‘4-phenylbutyric acid sodium salt’. The results have been divided into different categories, based on the way in which these compounds are able to interact with different pathologic conditions.
3. Phenylbutyrate and Cancer
Histone acetylation is associated with gene activation, while deacetylation, mediated by HDACs, is associated with gene silencing, explaining why HDACs are considered a powerful drug target.[28] The inhibitors of HDAC modulate the chromatin structure, which results in loosening the chromatin and changing transcription factor loading to the DNA,[29] modulating the expression pattern of various tumor-relevant genes for the control of the cell cycle or apoptosis,[30,31] and causing the inhibition of cell growth and differentiation.[32–34] These inhibitors are currently under clinical investigation in patients with different types of malignancies,[35] and phenylbutyrate is one of some HDACIs already tested in clinical trials in treatment of recurrent malignant gliomas or myelodysplastic syndrome (MDS).[36,37]
The following multiple activities have been assigned to the ability of phenylbutyrate to protect normal tissues: (i) the activity of an HDACI;[38] (ii) the activity of a chemical chaperone;[39–41] and (iii) the activity of an ammonia sink.[42,43] HDACIs, such as phenylbutyrate, have shown a wide range of abilities: (i) they are able to cause cell cycle arrest in the G1 and/or G2 phase; (ii) they induce differentiation and/or apoptosis in a variety of cell types;[44] (iii) they induce the transcriptional activation of certain genes, such as the one for cyclin-dependent kinase inhibitor p21, which has been related to tumor suppression growth and prevention of cell cycle progression;[45,46] (iv) they inhibit tumor growth in animal models with little toxicity in non-tumor cells;[47] and (v) they enhance the expression of a wide variety of transiently transfected transgenes in tumors, both in vitro and in vivo, through their effect on the acetylation of histones.[46,48] The combination of p53 gene therapy with an HDACI, for example, FR901228 (romedipsin/depsipeptide) or phenylbutyrate, has shown enhanced therapeutic efficacy in vitro[46] and in vivo.[49]
Phenylbutyrate can upregulate the transcription of epigenetically silenced genes and could be therapeutically useful for treatment of certain types of cancer conditions.[50] Differentiating agents in cancer therapy may potentially alter tumor growth and progression, slow or inhibit metastases, and/or effect response to other forms of therapy. Among them, phenylbutyrate has shown, in experimental tumor model systems, to be able to exert broad effects on tumor cytostasis and tumor differentiation, altering the gene expression for tumor growth, invasion, angiogenesis, and immunogenicity through mechanisms of action, such as inhibition of histone deacetylase, hypomethylation, modification of lipid metabolism, and activation of peroxisome proliferation activator receptor.[16] The effect of phenylbutyrate on tumor growth inhibition has been related to several mechanisms, including differentiation and apoptosis induction,[51] expression of silenced genes via histone deacetylase inhibition,[16] and induction of transforming growth factor-α secretion.[52] Particularly, the activation of c-Jun N-terminal kinase and extracellular signal-regulated kinase, in the mitogen-activated protein kinase pathway, seems to mediate the pro-apoptotic effects of phenylbutyrate.[53] Moreover, it has been reported that a differentiation therapy for epithelial malignancies may potentially alter tumor growth and progression, slow or inhibit metastases, inhibit angiogenesis, and/or effect response to other forms of therapy.[54] The ability of phenylbutyrate to be a potent cellular differentiating and cytostatic agent has been related to DNA methylation, a strong association with the peroxisome proliferator-activated receptor-γ and systemic glutamine depletion.[55] Differentiating agents have been used as therapy for MDS since the bone marrow, in this condition, is hypercellular, with aberrant differentiation and concomitant bone marrow failure. It has been reported that these agents have three potential roles in the treatment of myeloid neoplasms: (i) terminal differentiation of a malignant clone to clonal extinction, as in retinoic acid induction of acute promyelocytic leukemia; (ii) enforced clonal differentiation leading to functional even though clonal hematopoiesis; and (iii) prolongation of remission duration in patients with acute myelogenous leukemia (AML) or MDS with residual disease after chemotherapy through suppression of proliferation of the malignant clone.[56]
It has been shown that in rats injected with methylnitrosourea to induce mammary cancers, the efficacy of anti-inflammatory agents such as phenylbutyrate, vitamin C, and diindoylmethane to prevent mammary carcinogenesis and to inhibit the growth of established tumors, has been compared with their potential to inhibit the proliferation index and increase the apoptotic index in tumor cells showing that it is not significantly effective (reducing tumor multiplicity by <35%).[57]
Adriamycin is a potent anticancer drug that is used for treating both hematologic and solid tumors, although severe cardiomyopathy and heart failure have been observed in adriamycin-treated cancer patients due to the potent oxidative stress induced by this drug. In wild-type male mice of inbred strain C57BL/6 it has been observed that (i) phenylbutyrate (intraperitoneal 400 mg/kg/day injected 1 day before and daily after the adriamycin injection for 2 days) significantly decreases the adriamycin (intraperitoneal injection [20 mg/kg])-associated elevation of serum lactase dehydrogenase activities; (ii) it diminishes adriamycin-induced ultrastructural damage of cardiac tissue by >70%; (iii) it completely rescues adriamycin-caused reduction of cardiac function exemplified by ejection fraction and fraction shortening; and (iv) it increases the cardiac manganese superoxide dismutase protein and activity, suggesting that a combination therapy of HDACIs and adriamycin could be beneficial for treatment of cancer and simultaneously decrease adriamycin-induced cardiotoxicity.[58]
In vitro studies have shown that phenylbutyrate can (i) induce differentiation and inhibit proliferation of AML cell lines and primary leukemic cells;[59,60] (ii) inhibit CFU-L production from bone marrow specimens from patients with MDS;[60] and (iii) induce differentiation associated with induction of p21WAF1/CIP1 expression, hypophosphorylation of retinoblastoma protein, and G1 arrest in the ML-1 myeloid leukemia cell line.[59] At least some of the pharmacodynamic effects of phenylbutyrate seem to result because of its ability to inhibit HDACs,[61] considering that histone acetylation contributes to the regulation of the gene transcription.[62]
The in vitro effects of decitabine, phenylbutyrate and TSA on clonogenic AML human cells have been investigated. It has been observed that decitabine and HDACIs increase the expression of the stem cell marker CD117 for intermediate and erythroid colonies. Decitabine and HDACIs decrease the expression of the other stem cell marker CD34 and of CD64. These effects are common to different colony subsets. The two HDACIs TSA and S4PB differ in their effects on the expression of some membrane molecules, and this fact is especially seen in CD33 and CD11b. All three drugs modulate erythroid differentiation. Both decitabine and HDACIs decrease CD71 expression of non-erythroid colonies and increase its expression for mature erythroid colonies. The authors conclude that decitabine and the two HDACIs alter AML cell expression of differentiation markers, but the drugs do not have any major influence on cell cycle distribution.[63]
Burkitt and Ljungman,[64] after demonstrating that a subset of head and neck cancer cell lines have a defective Fanconi anemia DNA damage response pathway, which correlates with cisplatin sensitivity, have showed that phenylbutyrate sensitizes human cells to cisplatin (cisplatin is a widely used chemotherapeutic agent used against many different types of tumors, but unfortunately tumors initially responsive to it later acquire resistance). The same authors observe that phenylbutyrate (sodium phenylbutyrate 2 mmol/L for 48 hours) sensitizes cisplatin-resistant head and neck cancer cell lines UM-SCC-1, -6, -25 to cisplatin by inhibiting the Fanconi Anemia (FA)/Breast Cancer (BRCA) pathway through the downregulation of BRCA1 as well as by an FA/BRCA-independent mechanism. Therefore, they suggest that the cisplatin-sensitizing effect of phenylbutyrate is related to its role in targeting the expression of the apoptosis-antagonist B-cell lymphoma — extra large (Bcl-XL).[64]
Bandres et al.,[65] after identifying five microRNAs (miRNAs) downregulated in patients with colorectal cancer (CRC) and located around/on a cytosine-phosphate-guanine (CpG) island, have observed that treatment with a DNA methyltransferase inhibitor (5-aza-2′-deoxycytidine) and phenylbutyrate have restored the expression of the three miRNAs hsa-miR-9, hsa-miR-129, and hsa-miR-137 in three CRC cell lines. The expression of hsa-miR-9 is inversely correlated with the methylation of their promoter regions as measured by methylation-specific polymerase chain reaction (PCR).[MSP] and bisulfate sequencing. Moreover, the methylation of the hsa-miR-9-1, hsa-miR-129-2, and hsa-miR-137 CpG islands is frequently observed in CRC cell lines and in primary CRC tumors, but not in normal colonic mucosa. Finally, the methylation of hsa-miR-9-1 is associated with the presence of a lymph node metastasis. This study shows that miRNA-specific hypermethylation in CRC and histone-deacetylation could be an important molecular mechanism, as it causes the global downregulation of miRNAs. miRNA gene methylation could be considered as a useful tumor marker considering the high frequency of miRNA hypermethylation found in CRC cancer.[65]
Hurtubise et al.[66] have observed that phenylbutyrate, among other HDACIs (MS-275, trichostatin-A, LAQ824, and depsipeptide), has enhanced the antineoplastic action of 5-aza-2′-deoxycytidine (5AZA-CdR; a potent inhibitor of DNA methylation that has been approved for treatment of hematologic malignancies and is able to reactivate the expression of silenced tumor suppressor genes [TSGs]) on Ewing sarcoma cells.
Connexin (Cx) protein deregulation is believed to be involved in the carcinogenesis process and to play a key role in gap junction intercellular communication,[67] and it is essential for both proliferation and activation of differentiation pathways.[68] It has been observed that highly proliferating malignant cells have less gap junctions than the most differentiated ones.[69] In particular, connexin 43 (Cx43) is a tumor suppressor,[69] and its expression is reduced in various tumors.[70,71] The forced expression of the Cx43 gene in several Cx43-deficient tumor cell lines attenuates their malignant phenotype,[72,73] although the mechanisms by which the Cx43 gene inhibits the tumor growth remain unclear. The role of Cx43 alone and combined with an HDACI in tumor growth inhibition has shown that (i) trasfecting Cx43 plasmid DNA (pCMV-Cx43) into human nasopharyngeal cancer KB cells, using folate-linked nanoparticles, induces inhibition of cell growth (Cx43 induced a tumor suppressive effect via a gap junctional intercellular communication-independent mechanism); and (ii) the transfection of pCMV-Cx43 along with phenylbutyrate greatly enhances Cx43 expression in vitro, and significantly inhibits the tumor growth of KB cells and xenografts compared with that of pCMV-Cx43 alone. Phenylbutyrate induces an increased expression of genes of DNA damage checkpoints and of apoptosis via the downregulation of anti-apoptotic β-cell lymphoma (bcl-2) messenger RNA (mRNA) expression and upregulation of the activity of the apoptosis-associated enzyme caspase-3/7.[74]
The combined treatment 50-azacytidine/phenylbutyrate results in a partial but efficient demethylation of the miR-203 upstream region both in the two T-cell leukemia cell lines, KARPAS-45 and PEER, and in the two Ph-positive chronic myelogenous leukemia (CML) cell lines, K562 and KCL-22, restoring miR-203 expression (a tumor suppressor miRNA controlling the expression levels of ABL1, a classic oncogene extensively characterized in hematopoietic malignancies, and the expression levels of the BCR-ABL1 translocation protein, produced by the Philadelphia chromosome, which is a hallmark of CML and some B-cell leukemias in children). The re-expression of this miRNA dramatically reduces the proliferation of tumor cells and strongly correlates with a significant reduction in both ABL1 and BCRABL1 protein levels. This fact suggests that epigenetic drugs, in addition to re-expressing other methylated complementary DNAs, can result in oncogene downregulation mediated by the chemical restoration of miRNA function.[75]
It has been observed that phenylbutyrate (i) induces up to 70% apoptosis in pancreatic carcinoma cell lines Panc 1, T4M-4, COLO 357, and BxPc3, while it leads to cell cycle arrest in only T3M-4 and Colo357 cells, which have been found to express p21 to a higher extent than Panc1 and BxPc3 cells; (ii) it increases gap junction communication between adjacent T3M-4 cells in a concentration-dependent manner and efficiently inhibits cellular export mechanisms in pancreatic carcinoma cell lines Panc 1, T4M-4, COLO 357, and BxPc3 cells; (iii) in combination with gemcitabine, phenylbutyrate shows an overadditive effect on induction of apoptosis in BxPc3 and T3M-4 cells (up to 4.5-fold compared with single drug treatment) with accompanied activation of caspase 8, BH3 interacting domain death agonist (Bid) and poly (ADP-ribose) polymerase family, member 1 (PARP) cleavage; (iv) in concentrations exceeding 2.0 mmol/L it sensitizes H6c7 cells (a pancreatic ductal epithelial cell line not of malignant origin, but characterized by immortality and a high proliferation rate), which suggests that phenylbutyrate does not act on malignant cells specifically, but also on other highly proliferating immortalized cells; and (v) it does not affect low proliferating primary human fibroblasts and peripheral blood mononuclear cells.[76]
A pilot study of five African patients with Burkitt lymphoma, has shown that phenylbutyrate, administered before cyclophosphamide, induced shrinkage of cyclophosphamide-resistant tumors in two patients, at least temporarily. A comparison of the responses of these two patients suggests that phenylbutyrate pretreatment for 6 days, before cyclophosphamide, is more effective than pretreatment for 2.5 days.[77]
It has been shown that phenylbutyrate significantly reduces the number of inclusions in glucose oxidase and acetyl-leucyl-leucyl-norleucinal-treated cells (human hepatoma cell line established from a hepatocellular carcinoma [Huh7 cells] and a highly differentiated immortalized human hepatocyte cell line [OUMS-29 cells]), indicating that endoplasmic reticulum (ER) dysfunction, induced by glucose oxidase and acetyl-leucyl-leucyl-norleucinal, influences the formation of inclusions. These phenomena are reversed by a co-treatment with phenylbutyrate, which, due to its properties of chemical chaperone, reduces the ER stress.[78]
The effect of a combination treatment with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-aza-CdR) and the HDACI phenylbutyrate on a set of CCD-1070SK (human normal fibroblasts) and cancer cell lines (T24 [bladder transitional carcinoma cells], CFPAC-1 [pancreatic carcinoma cells], CALU-1 [lung carcinoma cells], NCCIT [embryonal carcinoma cells]) has been studied and the results have shown that (i) low doses of the drug combination cause cell cycle arrest, whereas high doses induce apoptosis in T24 bladder carcinoma cells; (ii) both p16 (CDKN2A/INK4) and p21 (CIP1/SDI1/WAF1) expression is induced to similar levels in normal and cancer cells in a dose-dependent fashion after combination treatments; (iii) a distinct increase of histone H3 acetylation at lysine 9/14 near the transcription start sites, in both LD419 normal fibroblasts and T24 bladder carcinoma cells, has been observed, and instead the acetylation changes in the p21 locus are less apparent; (iv) the levels of trimethylation of histone H3 on lysine 9 do not change after drug treatments and there is evidence that the remethylation of the p16 promoter CpG island in T24 cells after 5-aza-CdR treatment cannot be halted by a subsequent continuous phenylbutyrate treatment; and (v) the p16 gene is resilenced with kinetics similar to 5-aza-CdR-only-treated cells, which is also marked by a localized loss of histone acetylation at the transcription start site.[79]
Cyclo-oxygenase-2 is frequently overexpressed in non-small cell lung cancer (NSCLC) and results in increased levels of prostaglandin E2, an important signaling molecule implicated in tumorigenesis, with levels that have increased following cyclo-oxygenase-2 overexpression in NSCLC. It exerts its effects through the E prostanoid (EP) receptors (EPs1-4). In NSCLC cell lines (Beas-2B [transformed normal human bronchoepithelial], H460 [large cell], H647 [adenosquamous carcinoma], A549 [adenocarcinoma] and SK-MES-1 [squamous cell carcinoma]), histone acetylation has been found to be a critical regulator of EP expression with TSA, phenylbutyrate, and suberoylanilide hydroxamic acid, as they induce increased expression of EPs2-4. In addition, direct chromatin remodeling has been demonstrated at the promoters for EPs2-4.[80]
AML cells (cell lines K562, CMK, HL-60, KG-1a, and SK-Hep-1) have been cultured in the presence of phenylbutyrate, valproate, suberoylanilide hydroxamic acid, or TSA and analyzed for drug transporter expression and function as well as sensitivity to anticancer drugs. Phenylbutyrate has induced P-glycoprotein and breast cancer resistance protein expression and the efflux of drugs as determined with labeled substrates. KG-1a cells, treated with phenylbutyrate, have developed resistance to daunorubicin, mitoxantrone, etoposide, vinblastine, paclitaxel, topotecan, gemcitabine, and 5-fluorouracil, resulting in an impairment of drug-induced apoptosis.[81]
p21waf1/Cip1 (p21) is a tumor suppressor gene involved in apoptosis in many cancer cell types induced by different agents. In hepatocellular hepatocarcinoma, Hep3B cells transfected by EGFP-p21 anti-sense or sense plasmids, apoptosis induced by phenylbutyrate, and TSA has been assessed by terminal transferase dUTP nick end labeling (TUNEL) assay. The results have shown that the p21 anti-sense construct prevents apoptosis induced by HDACIs in Hep3B cells. The obtained results suggest an important role for p21 in mediating the apoptotic effect of HDACIs.[82]
An experimental study has investigated the antineoplastic effect of TSA and phenylbutyrate on the human glioblastoma cell lines GBM-29, U-343 MG, and U-343 MGa Cl. 2 : 6, giving the following results: (i) TSA and phenylbutyrate have induced apoptosis in the three cell lines in a dose- and time-dependent manner, and caspase-3 activation has been detected in all three cell lines; (ii) U-343 MG cells have been more sensitive to the apoptotic effect of HDACI compared with U-343 MGa Cl. 2 : 6; (iii) TSA and phenylbutyrate have induced differentiation in the three cell lines, each cell line developing unique phenotypic characteristics; (iv) during long-term treatment with a low dose of HDACI U-343 MGa Cl. 2 : 6 cells have developed an astrocytic morphology with expression of glial fibrillary acidic protein (GFAP); (v) GFAP-negative U-343 MG cells have changed their morphology in response to HDACI and downregulated their expression of vimentin; and (vi) the nestin and vimentin positive GBM-29 cells have also shown a morphologic differentiation, while the expression of the two malignancy markers has decreased.[22]
Treatment of tumor cells (human prostate [PC3, DU-145 (ATCC)] and colon carcinoma cell lines [HCT116,HCT116 p21¡/¡, HCT116 p53¡/¡]) with phenylbutyrate and 13-cis-retinoic acid (CRA) and with paclitaxel and doxorubicin has been studied with the following results: (i) inhibition of tumor cell growth has been greatly enhanced when compared with phenylbutyrate + CRA, paclitaxel or doxorubicin alone, with >90% growth inhibition; (ii) when the cells have been pretreated with phenylbutyrate + CRA, followed by paclitaxel or doxorubicin, the enhanced inhibition is abolished; (iii) treatment with phenylbutyrate + CRA restores sensitivity to doxorubicin in the PC-3 human prostate cancer cell line; (iv) phenylbutyrate + CRA induces p21 expression and cell cycle arrest in the G1 phase, while paclitaxel and doxorubicin induce G2/M arrest; (v) p21- and p53-deficient colon carcinoma cell lines are more sensitive to the effect of phenylbutyrate + CRA and paclitaxel as single agents and in combination, compared with the wild-type cells; (vi) when the p21-deficient cells are pretreated with phenylbutyrate + CRA, followed by paclitaxel, the protective effect is still observed; (vii) treatment of tumor cells with a combination of these drugs induces cell cycle delay at multiple mitotic checkpoints before undergoing apoptosis; (viii) the tumor growth is significantly inhibited and delayed in male athymic nude or female BALBc mice treated with either paclitaxel or concomitantly with paclitaxel and phenylbutyrate + CRA compared with control; and (ix) animals, treated with all three agents, demonstrate further growth inhibition or delay compared with the paclitaxel alone or phenylbutyrate + CRA arm.[83]
It has been suggested that phenylbutyrate can produce p21-independent cytostasis, and enhance radiation sensitivity in p53 mutant human glioblastoma cells in vitro (D54, U87-MG, U251, and SKMG-3), suggesting the potential application of combined phenylbutyrate and radiotherapy in glioblastoma harboring mutant p53.[84]
An experimental study has shown that treatment of different metastatic melanoma cell lines (SK-Mel-19, SK-Mel-29, and SK-Mel-147 melanoma cells; 1.5 × 106) with 5-azacytidine-CdR, a potent inhibitor of cytosine methylation, significantly induces 14-3-3σ protein expressionFootnote 1 and additional treatment with phenylbutyrate further enhances 14-3-3σ expression. The induction of 14-3-3σ expression by 5-azacytidine-CdR/phenylbutyrate treatment leads to almost complete inhibition of cell proliferation, with cells predominantly arrested in G2-M. The antiproliferative effect of 5-azacytidine-CdR/phenylbutyrate is reversed in 14-3-3σ knockdown cells.[85]
A 6-hour pretreatment with phenylbutyrate and vitamin B3 on human promyelocytic leukemia cells HL-60, before the exposition to all-trans-retinoic acid (RA) alone or in combination with vitamin B3, has shown a significant acceleration and high level of granulocytic differentiation. The effects are associated with a rapid histone H4 acetylation and later histone H3 modifications.[86] It is important to underline that vitamin B3 inhibits sirtuins, NAD+-dependent protein deacetylases that are involved in transcriptional regulation, metabolism, apoptosis, differentiation, and aging.[87]
Phenylbutyrate, at different concentrations, has been used to treat hepatocellular carcinoma cells Bel-7402 and normal liver cell line L-02 showing: (i) a time-dependent growth inhibition in hepatocellular carcinoma cells Bel-7402; (ii) a significant decline in the fraction of S phase cells and significant increase in G0/G1 cells; (iii) an increase in the expression of P21WAF1/CIP1 and E-cadherin in Bel-7402 and a significant decrease in the level of HDAC4 in Bel-7402; (iv) a significant improvement in the level of acetylating histone H4; (v) no distinct changes in the normal liver cell line L-02 suggesting phenylbutyrate to be safe for clinical treatment; (vi) the inhibition in the growth of hepatocellular carcinoma cells Bel-7402 and induction of partial differentiation through the enhancement of the acetylating histones; and (vii) no significant effect on normal liver cells have been observed. These results underline the safety of phenylbutyrate, emphasizing that it may have great potential as a growth inhibitor for hepatocellular carcinoma.[88]
The clinical trials involving the use of phenylbutyrate for treatment of cancer are summarized in table SI (Supplemental Digital Content 1, http://links.adisonline.com/DRZ/A3).
4. Motor Neuron Disorders
Motor neuron diseases are a group of fatal neurologic progressive disorders, and the most common are spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS), both characterized by the loss of spinal motor neurons. No effective cure exists, but there have been major advances to understand the molecular and genetic mechanisms involved in these pathologies. Riluzole stops the advance of the disease by blocking the effects of the neurotransmitter glutamate and it is thought to extend the lifespan of ALS patients for few months.[89] In particular, the use of phenylbutyrate and its derivatives has been widely investigated both in vitro and in vivo. Important genes, playing a key role in motor neuron diseases, have been discovered during the past few years and several pathways have been recently implicated in these pathologies, i.e. pathways that affect RNA processing, axonal transport, and mitochondrial function.[90]
4.1 Phenylbutyrate and Spinal Muscular Atrophy
SMA is the term for a number of conditions characterized by the degeneration of α-motor neurons in the brainstem and spinal cord leading to hypotonia and muscle weakness.[91] SMA is an autosomal recessive disease and the leading cause of infant deaths caused by the gene survival motor neuron 1 (SMN1).[92] SMA has been subdivided into three clinical groups, based on the age of the onset of symptoms and the achievement of motor milestones, and that are due to homozygous loss of the functional motor neuron gene (SMN1) on chromosome 5q13. The age of the onset in patients with SMA I is less than 6 months and these patients never acquire the ability to sit unsupported. The onset of SMA II is usually between 6 and 18 months and the patients are able to sit unsupported, but are not able to walk. The onset of SMA III is usually >18 months and the patients are able to walk independently.[93] The fact that SMN genes have a unique organization has led to investigation of how to make SMN2, a gene existing in all patients, function like the missing SMN1 gene.[92] Histone acetylation leads to DNA relaxation, making it accessible to the transcriptional machinery, and its inhibition might enhance the expression in about 2% of human genes.[94] Phenylbutyrate, the first HDACI, found to enhance SMN2 expression, increases exon seven inclusion in lymphoid cell lines derived from patients with SMA. However, its half-life of about 6 minutes in human serum has made it impossible to develop a clinical use for this compound.[92] Andreassi et al.[95] reported that phenylbutyrate treatment caused an increase in full-length SMN2 transcripts in fibroblast cell cultures from 16 SMA patients and it is was also effective in enhancing SMN protein levels and the number of SMN-containing nuclear gems. Therefore, this compound could represent a valuable candidate for pharmacologic treatment of SMA.[95] It has been shown that the SMN2 gene is subject to gene silencing by DNA methylation, which might have major implications for SMA disease progression and upcoming pharmacologic interventions for epigenetic SMA therapy.[96]
It has been found that treatment of SMA-like transgenic mice with the hyperacetylating agent phenylbutyrate effectively increases the expression of SMN protein in motor neurons of the spinal cord,[97] and phenylbutyrate significantly increases SMN expression in fibroblast cultures from SMA patients.[95]
It has been shown that five pan-HDACIs, including SAHA, as well as the cyclic tetrapeptide romidepsin (FK-228), are able to bypass LT-SMN2 gene silencing in SMA fibroblasts and human hippocampal brain slice cultures (OHSCs), resulting in up to 5-fold inductions of total SMN2 transcript levels. In contrast, the HDAC isoenzyme selective inhibitors MS-275, VPA, and phenylbutyrate display only moderate effects, suggesting that pan-HDACIs possess superior functional capacities to activate SMN2. Among the pan-HDACIs, vorinostat appears to be promising, as this molecule has been shown to cross the blood-brain barrier. Moreover, ongoing clinical research in cancer patients has shown a good oral bioavailability and biologic activity.[96] The clinical trials involving the use of phenylbutyrate for treatment of SMA are summarized in table SII (see Supplemental Digital Content).
4.2 Phenylbutyrate and Amyotrophic Lateral Sclerosis
ALS is a fatal neurodegenerative disorder, characterized by a loss of upper and lower motor neurons in the brain and spinal cord, and leading to a progressive muscle wasting and limb paralysis, dysphagia, dysarthria, and respiratory failure.[98,99] About 10% of patients report a family history, and mutations in SOD1 (which encodes superoxide dismutase-1) are the cause in about 20% of these cases; 3% and 4% of familial cases, respectively, have been related to TARDBP (also known as TDP-43, which encodes TAR DNA-binding protein) and FUS (RNA-binding protein fusion), which have been linked to familial forms of the disease. Sporadic ALS has been associated with another gene, ELP3 (elongator protein 3), which encodes the catalytic subunit of the HAT complex elongator protein 3. ALS also features cytoplasmic deposits of misfolded proteins, consisting of SOD1, TARDBP, or FUS aggregates.[100] Riluzole (an FDA-approved ALS therapy) provides a 2- to 3-month prolongation of survival.[101,102] To date, no other drug therapies have been shown to slow or abrogate the disease process in ALS. It has been reported that the mean survival of patients from the onset of symptoms is 3–5 years.[103] The different types of ALS pathologies are studied in transgenic rodent models of ALS, expressing mutant forms of SOD1.[104] Mice, overexpressing a human SOD1 mutation, are characterized by a glycine, which is substituted by an alanine at amino acid position 93[105] since these transgenic models develop clinical and pathologic features that are similar to those in human diseases.[105–107] Apoptotic cell death is a common cellular event in both animal models and patients with ALS,[108] as suggested by the observations that increased expression of bcl-2, the dominant negative inhibition of caspase-1, and the administration of tetrapeptide caspase inhibitors delay the onset of the disease and prolong survival in ALS mice.[109–111] Neuroprotective therapies that target specific neurotoxic molecular mechanisms in ALS mice have the potential to delay the onset and slow the progression of the disease in ALS, and phenylbutyrate has been proposed as a good candidate for this kind of therapy.
Pharmacologic treatment, using phenylbutyrate (the animals received either 200, 400, 600, or 800 mg/kg intraperitoneally or phosphate-buffered saline [PBS] injection, i.e vehicle; treatment was started at 21 and 70 days, before and after the occurrence of the symptoms), significantly extends survival and improves both the clinical and neuropathologic phenotypes in G93A transgenic ALS mice. Moreover, it ameliorates histone hypoacetylation and induces expression of nuclear factor-κB (NF-κB) p50, the phosphorylated inhibitory subunit of NF-κB (pIκB) and bcl-2, and it reduces cytochrome c and caspase expression. The same authors propose that the pharmacologic induction of NF-κB-dependent transcription and bcl-2 gene expression is neuroprotective in ALS mice by inhibiting programmed cell death, in particular: (i) phenylbutyrate acts to phosphorylate pIκB, translocating NF-κB p50 to the nucleus, or to directly acetylate NF-κB p50; (ii) NF-κB p50 transactivates bcl-2 gene expression; and (iii) upregulated bcl-2 blocks cytochrome c release and subsequent caspase activation, slowing motor neuron death. These transcriptional and post-translational pathways ultimately promote motor neuron survival and ameliorate disease progression in ALS mice.[38]
The catalytic antioxidant Mn (III) porphyrin AEOL 10150 (2.5 mg/kg/day; systemic administration), phenylbutyrate (400 mg/kg/day; intraperitoneal administration) and the combination of phenylbutyrate and AEOL 10 150 have been tested in G93A transgenic familial ALS mice (administered from the onset of the disease, i.e. after the appearance of motor dysfunction) observing that (i) AEOL 10 150 alone improved motor function and extended survival by 11%; (ii) phenylbutyrate alone significantly improved motor function and extended survival by 13%; and (iii) phenylbutyrate and AEOL 10 150 together increased survival by 19%. An increase in histone acetylation was confirmed by Western blot and quantitative real-time PCR analysis revealed the upregulation of the compounds capable of protecting cells against oxidative stress and apoptosis. Markers of oxidative damage were reduced in the lumbar spinal cord when compared with vehicle administration. This study suggests that agents inhibiting apoptosis and blocking oxidative stress show efficacy in treating mutant-SOD1-associated ALS and that a combination of agents, targeting different disease mechanisms, may exert additive therapeutic effects.[112]
The combined treatment of riluzole and phenylbutyrate significantly extends survival and improves both clinical and neuropathologic phenotypes in G93A transgenic ALS mice. In particular, the combination therapy increased survival by 21.5%, compared with the separate administration of riluzole (7.5%) and phenylbutyrate (12.8%), while improving both bodyweight loss and grip strength. Riluzole/phenylbutyrate treatment ameliorates gross lumbar and ventral horn atrophy, attenuates lumbar ventral horn neuronal cell death, and decreases reactive astrogliosis. In addition, riluzole/phenylbutyrate administration increases acetylation at H4 and increases NF-κB p50 translocation to the nucleus in G93A mice.[113]
An open-label study, involving 40 patients with ALS, has been performed to establish the safety and pharmacodynamics of escalating dosages of phenylbutyrate in 26 participants who completed a 20-week treatment phase. It showed that phenylbutyrate is safe and the majority of subjects tolerated higher dosages (21 g/day) of this drug, but the lowest dose (9 g/day) was therapeutically efficient in improving histone acetylation levels. No deaths or clinically relevant laboratory changes occurred with phenylbutyrate treatment. Histone acetylation decreased by approximately 50% in blood buffy-coat specimens at screening and significantly increased after phenylbutyrate administration. In addition, blood levels of phenylbutyrate and the primary metabolite (PAA) increased with dosage.[114]
4.3 Phenylbutyrate and Ischemia
Retinal ischemia is a common cause of visual impairment for humans and animals. Retinal ischemia has been typically observed as a consequence of the presence of diabetic retinopathy, glaucoma, and retinal-artery occlusion.[115] Ischemic retinal injury consists of a self-reinforcing cascade of destruction involving neuronal depolarization, calcium influx, and oxidative stress initiated because of energy failure, and increased glutamatergic stimulation.[116,117]
A study has investigated the neuroprotective effects of phenylbutyrate upon retinal ischemic injury using Sprague Dawley rats. Retinal ganglion cells (RGCs) were retrograde labeled with the fluorescent tracer fluorogold applied to the superior collicoli of test Sprague Dawley rats and, 7 days after that, high intraocular pressure and retinal ischemia were induced. The animals received either 100 or 400 mg/kg PBA intraperitoneally, 30 minutes prior to the induction of retinal ischemia and 1 hour subsequent to the cessation of the procedure inducing retinal ischemia. Histologic analysis has revealed that ischemic injury causes the loss of retinal RGCs and a net decrease in retinal thickness. Moreover, it has been observed that in the phenylbutyrate-treated groups, almost 100% of the RGCs have been preserved by a pre-ischemia treatment with phenylbutyrate (at a dose of either 100 or 400 mg/kg), while post-ischemia treatment of RGCs with phenylbutyrate has not led to the preservation of RGCs from ischemic injury by phenylbutyrate, as determined by the counting of whole-mount retinas. Pre-ischemia treatment of RGCs with phenylbutyrate (at a dose of either 100 or 400 mg/kg) has significantly reduced the level of ischemia-associated loss of thickness of the total retina, especially the inner retina and the inner plexiform layer of retina. Besides, phenylbutyrate treatment has significantly reduced the ischemia-induced loss of cells in the ganglion-cell layer of the retina.[118]
It has been observed that, if we target the ER by administration of intraperitoneal phenylbutyrate (1 hour before and 12 hours after reperfusion) the liver is protected against ischemia/reperfusion injury in male C57BL/6 mice subjected to warm ischemia (70% of the liver mass, 45 minutes), as evidenced by the decreased liver enzymes and reduction in the number of non-viable cells and PMN infiltrates into the liver. Moreover, the hepatoprotective properties of phenylbutyrate have further been demonstrated by a reduction in apoptosis after reperfusion. This study shows that phenylbutyrate reduces mortality in a lethal model of total ischemia/reperfusion injury to the liver (all vehicle-treated controls died within 3 days after reperfusion while 50% survival [>30 days] has been observed in animals given phenylbutyrate).[119]
4.4 Phenylbutyrate and Urea Cycle Disorders
The urea cycle is the final common pathway for the excretion of waste nitrogen in mammals, and the defects in this cycle, observed in the neonatal period, are usually associated with severe and rapidly worsening hyperammonemia which, if not treated, leads to cerebral and pulmonary hemorrage, leaving the baby severely handicapped or leading to death.[120] It has been reported that all the UCDs are associated with an accumulation of glutamine and alanine, and are characterized by a difficult diagnosis since they are associated with nonspecific symptoms such as poor feeding, vomiting, lethargy and/or irritability, and tachypnea. These babies can show a transient respiratory alkalosis, neuromuscular irritability and stridor, which are only transient, with the patients deteriorating rapidly, becoming unresponsive, and requiring intensive care. Other complications are changes of tone, with loss of normal reflexes, vasomotor instability and hypothermia, apnoea and fits, while a disordered liver function could be a secondary complication. Measurement of plasma ammonia concentration is the main diagnostic test for the UCD (plasma ammonia, in healthy neonates, is normally <65 μmol/L,[121] and reaches 180 μmol/L in sick neonates; it is >200 μmol/L in patients with inborn errors and achieves higher levels in patients with UCD).[120]
In cases of significant hyperammonemia, the following investigations could be useful: blood pH and gases, plasma urea and creatinine, electrolytes, glucose, liver function tests and clotting studies, plasma amino acids, urine organic acids, orotic acid and amino acids, and plasma free and acylcarnitines.[120] The therapy for this disease is directed at decreasing the requirement for urea biosynthesis by decreasing dietary nitrogen intake and increasing waste nitrogen excretion. It is achieved by prescribing phenylbutyrate, a precursor of PAA that conjugates with glutamine to yield phenylacetylglutamine, a waste nitrogen compound that is rapidly excreted in the urine.[122]
The use of phenylbutyrate to treat UCD is due to its ability to be oxidized in the liver to PAA, which is then conjugated with glutamine, resulting in phenylacetylglutamine, which is excreted in the urine and hence 2 mol of nitrogen is lost for each mole of the given phenylbutyrate.[123] An early diagnosis is fundamental because the prognosis often depends on urgent treatment. Family investigation and genetic counseling play a preventive key role. When elevated plasma ammonia levels are found, a low-protein diet, with supplemental arginine, and with both sodium benzoate and phenylbutyrate to remove excess nitrogen, has been suggested as treatment.[124]
Three cases of patients with hyperammonemia who received accidental inappropriate doses of intravenous sodium benzoate and PAA that have resulted in severe complications have been reported. All the patients showed alteration in mental status, Kussmaul respiration, and a partially compensated metabolic acidosis with an increased anion gap. Two patients developed cerebral edema and hypotension, and died; the third patient survived after hemodialysis, and plasma levels of benzoate and PAA were excessively high in all of them.[125] During pregnancy, benzoate might be the best choice because it is usually ingested with soft drinks and several types of food, while phenylbutyrate can affect many metabolic and other functions. Phenylbutyrate tablets are considered to be dangerous because they can cause serious tissue damage in the esophagus, underlining the importance of instructions to the patient to ensure correct swallowing of the drug.[126]
OTC deficiency is the most common urea cycle defect, usually affecting males in the neonatal period and causing acute hyperammonemic coma and leading to death if not treated.[124] OTC is an x-linked mitochondrial enzyme that catalyzes the synthesis of citrulline from carbamoyl phosphate and ornithine. A deficiency in this enzyme leads to hyperammonemia and hyperglutaminemia, hypoargininemia, hypocitrullinemia, and episodic encephalopathy that, if uncontrolled, results in brain injury and death. In boys, the disease is often fatal when its onset occurs during the neonatal period, but it is milder when the onset occurs later in childhood. Heterozygous girls may be normal or may have episodes of hyperammonemic encephalopathy and decline in cognitive function.[19] This condition has also been observed in the later childhood (between 15 months and 5 years) and has been often under-recognized.[127,128] This pathology is characterized, in the neonatal period, by feeding difficulties, lethargy, respiratory distress, impairment of consciousness, vomiting, convulsions, and coma. In later life the most important symptoms that might occur are headache, vomiting, lethargy, hyperventilation, episodes of abnormal behavior, and sometimes disorientation and confusion, ataxia, hypotonia, and focal neurologic signs such as hemiplegia[128] (proposed to be due to alterations in vascular endothelial wall integrity or alterations in cerebral perfusion and metabolism with an accumulation of toxic metabolites).[129] Cases of acrodermatitis and enteropathica-like rashes have been reported, probably associated with a deficiency of arginine, an amino acid that represents the 16% of the amino acid content of the human epidermal keratin.[130] The voluntary adoption of a vegetarian diet has been observed.[131] Acute pancreatitis has also been reported as a complication.[132] The clinical trials, involving the use of phenylbutyrate for treatment of UCD, are summarized in table SIII (see Supplemental Digital Content).
4.5 Sickle Cell Disease and Thalassemias
β-Thalassemia is an autosomal recessive genetic disease, caused by variations in the inactivation mechanism of the β-globin genes,[133] that shows clinical symptoms ranging from mild symptomless anemia to transfusion dependence. No adequate treatment for the various phenotypes has been established yet. The current management of this condition includes the use of regular red blood cell transfusions and iron chelation therapy. Bone marrow transplantation could be an important therapeutic choice, although it is not an option for the majority of the patients. The development of an effective therapy to increase hemoglobin levels in homozygous β-thalassemia, without the use of red blood cell transfusions, could allow normal growth and development while decreasing or eliminating transfusional iron overload, which is the leading life-threatening factor in patients with this disease.[134] Butyrate analogs are able to induce erythroid differentiation[135–137] and stimulate hemoglobin F production in human erythroid progenitors in vitro.[138–140] In vivo, they reactivate embryonic globin production in an avian model,[141] delay the switch from fetal to adult globin in ovine fetuses,[142] and increase fetal hemoglobin (HbF) production in adult primates.[140,143–145] In humans, phenylbutyrate is able to stimulate fetal hemoglobin production.[146,147] For the majority of children with β-hemoglobinopathies and β-thalassemias who do not have a transplant donor, survival is shortened and morbidity is high. Hydroxyurea, recombinant human erythropoietin preparations, phenylbutyrate, arginine butyrate, and 5-azacytidine/decitabine have shown efficacy in approximately 40–70% of sickle cell and β-thalassemia patients.[148]
The clinical trials involving the use of phenylbutyrate for treatment of sickle cell disease and thalassemias are summarized in table SIV (see Supplemental Digital Content).
4.6 Phenylbutyrate and Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the CF transmembrane conductance regulator (CFTR) gene.[149]
The ΔF508 mutation is caused by a three base pair deletion in the gene for CFTR, resulting in the loss of a phenylalanine residue at position 508 in the CFTR protein and this mutation fails to fold properly in the ER, which leads to ubiquitination and degradation of the protein in the proteasome.[150,151] The ΔF508-CFTR protein, which is retained in the ER and degraded rather than trafficked to the cell surface, forms a functional chloride channel in reconstituted bilayers, but with decreased mean open times, and therefore decreased conductance compared with wild-type CFTR. This fact underlines how treatments that would promote ΔF508-CFTR trafficking beyond the ER might restore partial CFTR chloride channel function at the cell surface.[152]
Two members of the 70 kDa heat shock protein family, HSP70 and HSC70, interact with CFTR, and the regulation of these heat shock protein-CFTR interactions can restore ΔF508-CFTR trafficking as shown in a study proving phenylbutyrate modulates heat shock protein function and restores ΔF508 maturation in vitro and in vivo.[15] Heat shock proteins are constitutive and stress-inducible proteins that can protect normal cells against protein damage by physical interaction during synthesis, folding, assembly, and degradation.[153]
In cultured cells, the deregulation of ΔF508-CFTR by butyrate has been observed.[154] Moreover, phenylbutyrate, at micromolar concentrations, induces CFTR channel function on the plasma membrane of ΔF508-expressing CF airway epithelial cells (in IB3-1 cells [an immortalized CF cell line containing the mutations ΔF508 and W1282X]) in vitro.[155] CFTR mutations have been categorized into five major classes: (i) the total absence of CFTR (class I); (ii) defects in CFTR folding and trafficking, leading to reduced amounts of mature CFTR (class II; the ΔF508 is the prototype class II trafficking mutation and has been found in >70% of patients with CF); (iii) abnormal regulation of chloride conduction in an otherwise mature CFTR (class III); (iv) decreased chloride conduction in a fully processed CFTR (class IV); and (v) decreased CFTR synthesis (class V).[156]
A study investigating the effects of genistein and phenylbutyrate on CFTR in three human airway epithelial cell lines expressing wild-type or ΔF508 CFTR (Calu-3, CFSMEo-, and CFBE41o- cells) loaded with the fluorescent dye N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide to study the chloride efflux, has shown that (i) forskolin and 3-isobutyl-1-methylxanthine (IBMX) induce chloride efflux in Calu-3 cells but not in CF lines; (ii) genistein (2.5–50 μmol/L) is able to induce chloride efflux in all cell lines but does not enhance the effect of forskolin and IBMX; and (iii) phenylbutyrate has little or no effect on genistein-induced chloride efflux.[157]
A study has investigated the ability of phenylbutyrate, genistein, and 8-cyclopentyl-1,3-dipropylxanthine (CPX) to activate the defective chloride channel in the CF bronchial epithelial cell line CFBE41o-, which expresses the ΔF508 mutation. It was investigated through x-ray microanalysis. Both genistein and CPX act by stimulating the chlorine flow end increasing the transmembrane conductance in CF. It was observed that 8-bromo-cyclic-adenosine monophosphate (cAMP) alone does not induce Cl- efflux in CFBE41o- cells, but, after incubation with phenylbutyrate, a significant efflux of Cl- occurs showing that phenylbutyrate allows the ΔF508 CF transmembrane conductance regulator to escape degradation and to be transported to the cell surface. The stimulation of cells, with a combination of genistein and cAMP, also induces Cl- efflux, whereas a combination of pretreatment with phenylbutyrate and a combined stimulation with genistein and cAMP induces an even larger Cl- efflux. Cl- efflux could also be stimulated by CPX, but this effect is not enhanced by phenylbutyrate pretreatment.[158]
It has been shown that arginine butyrate (AB) and two novel short-chain fatty acid derivatives, α-methylhydrocinnamic acid (ST7) and 2,2-dimethyl-butyrate (ST-20), functionally correct the ΔF508-CFTR defect in airway IB3-1 cells, which are heterozygotes for the CFTR mutations ΔF508 and W1282X.[159]
Singh et al.[160] have highlighted a specific subset of HSP70 system proteins that sense the rescue of ΔF508-CFTR from ER-associated degradation (ERAD). The phenylbutyrate-mediated rescue of ΔF508-CFTR is associated with changes in the network of ER-resident cytosolic chaperones and proinflammatory responses that mimic patterns of protein interaction observed with wild-type CFTR expression.
The proteome profiling of IB3-1 CF bronchial epithelial cells, treated with phenylbutyrate, has been investigated to identify butyrate-responsive cellular chaperones, protein-processing enzymes, and cell-trafficking molecules associated with the amelioration of the chloride transport defect in these cells (analyzed by two-dimensional gel electrophoresis and mass spectrometry). The same study has shown that over a pI range of 4–7 and molecular weight 20–150 kDa, a total of 85 differentially expressed proteins have beem detected. They were mostly chaperones, catalytic enzymes, and proteins comprising structural elements, cellular defense, protein biosynthesis, trafficking activity, and ion transport.[161]
Phenylbutyrate decreases the expression of HSC70 mRNA and protein by inducing cellular adaptations that result in the decreased stability of HSC70 mRNA. Phenylbutyrate has been observed to stimulate the degradation of HSC70 mRNA in IB3-1 cells without significantly altering HSC70 mRNA synthesis, promoter activity, or protein turnover. The authors suggest that, since this response requires a preincubation with phenylbutyrate, it results from a cellular adaptation to phenylbutyrate treatment.[162]
In an in vitro experiment, IB3-1 cells (CF epithelial cells genotype; ΔF508/W1282X) have been treated with phenylbutyrate 0.05–5 mmol/L for 2 days in culture showing a dose-dependent reduction in Hsc70 protein immunoreactivity and mRNA levels. Immunoprecipitation with Hsc70-specific antiserum demonstrates that Hsc70 and CFTR are associated under control conditions. Levels of immunoreactive Hsp40, Hdj2, Hsp70, Hsp90, and calnexin were unaffected by phenylbutyrate treatment. This study suggests that phenylbutyrate may improve DF508-CFTR trafficking by allowing a greater proportion of mutant CFTR to escape association with Hsc70.[163]
Butyrate and phenylbutyrate, using patch clamp recording from CFTR-transfected mammalian cell lines, cause a voltage-dependent block of CFTR Cl- currents when applied to the cytoplasmic face of membrane patches, with apparent dissociation constants (Kds).[at 0 mV] of 29.6 mmol/L for butyrate and 6.6 mmol/L for phenylbutyrate. At the single channel level, both these fatty acids cause an apparent reduction in CFTR current amplitude, suggesting a kinetically fast blocking mechanism. The concentration-dependence of the block suggests that CFTR-mediated Cl- currents in vivo may be affected by both phenylbutyrate, used in the treatment of various diseases, including CF, and by butyrate produced endogenously within the colonic lumen.[164]
It has been proposed that a combination of chronic treatment with phenylbutyrate or selected flavonoids, followed by acute flavonoid exposure, may be beneficial in CF as proved by an increase in Cl- conductance measured by Cl- efflux in cells (IB3-1 cells [F508/W1282X]) that are treated for 24 hours with phenylbutyrate and then assayed with forskolin and genistein 1 μmol/L or 5 μmol/L, and also with cells treated for 24 hours with either phenylbutyrate, apigenin 5 μmol/L, or quercetin 1 μmol/L.[153]
Phenylbutyrate (5 μmol/L for 6 hours) increases the functional expression of epithelial sodium channels (ENaCs) in the apical membrane of non-CF and CF nasal epithelial cells by enhancing exocytosis of ENaC subunits likely via regulation of heat shock proteins, suggesting that (i) in non-CF patients, phenylbutyrate treatment may be useful to treat airway diseases in which ENaC trafficking may be disrupted, as it has been observed during hypoxia or endotoxemia; and (ii) in CF disease, although phenylbutyrate may be useful to restore functional mutant CFTR at the apical membrane, it is important to keep in mind that this effect may be counteracted by an increase in sodium hyperabsorption.[165]
The clinical trials involving the use of phenylbutyrate for treatment of cystic fibrosis are summarized in table SV (see Supplemental Digital Content).
4.7 Phenylbutyrate and Huntington Disease
The symptomatic Huntington disease (HD) onset occurs typically between 30 and 50 years of age, and mutant huntingtin has been shown to disrupt activator-dependent transcription in the early stages of HD pathogenesis, which has been related to transcriptional deregulation and functional loss of transcriptional co-activator proteins, leading to neuronal loss and gliosis, particularly in the cortex and basal ganglia regions of the HD patient’s brain.[166] HD is an inherited autosomal dominant neurodegenerative disorder caused by the expansion of the polyglutamine tract near the amino terminus of the huntington protein (HTT; CAG trinucleotide repeat located in the Huntington gene), leading to polyglutamine-dependent inclusions, which have been seen in some HD brains, and some mouse models that express a small fragment of mutant huntingtin.[167] This disease leads to the loss of medium spiny neurons affecting mainly the striatum (caudate nucleus, putamen, and globus pallidus), and also the frontal and temporal cortex causing chorea, psychiatric disturbances, and cognitive impairments.[168] There is evidence that mutant HTT interacts with the transcription factors, leading to reduced histone acetylation.[169] In particular phenylbutyrate improves survival and attenuates striatal atrophy in the R6/2 transgenic mouse HD model, when administered presymptomatically, starting at 21 days of age.[170]
Administration of the histone deacetylase inhibitor phenylbutyrate (intraperitoneal injections of phenylbutyrate [100 mL/kg/day, volume 3.33 mL/kg] or vehicle [PBS, 3.33 mL/kg], 6 days per week from 75 days of age) after the onset of symptoms in a transgenic mouse HD model (transgenic N171-82Q mice maintained on a B6C3F1 background) has shown that (i) a significant attenuation of gross brain atrophy and ventricular enlargement, as well as a neuronal atrophy after phenylbutyrate treatment (significant at 120 days) has been observed; (ii) phenylbutyrate treatment significantly reduces striatal neuron atrophy in N171-82Q mice; (iii) the administration of phenylbutyrate for 2 weeks increases immunostaining for both acetylated histone H3 and histone H4 in striatal neurons, and the administration of phenylbutyrate at 100 mg/kg increases histone acetylation in the spleen and brain at 2 hours post-administration; (iv) the immunocytochemistry shows a marked increase in methylation of histone 3 in the striatum at 120 days of age, which is markedly attenuated by phenylbutyrate treatment; (v) the expression of selected genes (Gfer, Gstm3, and Psma3) is significantly upregulated after phenylbutyrate treatment compared with controls, whereas the other genes (Casp9, Cflar, and Prkce), after phenylbutyrate treatment, have shown a significantly lower expression; and (vi) the caspase 3 immunoactivity (caspase 9 is involved in activation of caspase 3, and has increased in HD patients) at 120 days of age is markedly increased in the striatum of the N171-82Q mice compared with controls, and the increase is markedly attenuated by phenylbutyrate treatment.[169]
Affymetrix GeneChip U133A and Amersham Biosciences CodeLink (Amersham Biosciences) oligonucleotide microarrays have been used to analyze global gene expression changes in blood samples from 17 HD subjects (12 symptomatic and five late presymptomatic carriers of the HD mutation) and 14 healthy, age- and gender-matched control subjects. 322 mRNAs, showing significantly altered expression in HD blood samples, have been identified in this study. Some changes in blood mRNAs that clearly distinguish HD patients from controls, and that correlate with disease progression and response to experimental treatment, have also been identified as follows: (i) a subset of upregulated mRNAs, selected from this group, is able to distinguish controls, presymptomatic individuals carrying the HD mutation, and symptomatic HD patients; (ii) early presymptomatic subjects have shown gene expression profiles similar to those of controls, whereas late presymptomatic subjects have shown altered expression that resembles that of symptomatic HD patients; (iii) these elevated mRNAs were significantly reduced in HD patients involved in a dose-finding study of phenylbutyrate; and (iv) the expression of the marker genes is significantly upregulated in post mortem HD caudate, suggesting that alterations in blood mRNAs may reflect disease mechanisms observed in HD brain.[171]
A phenylbutyrate dose-finding study in 21 subjects with HD found that phenylbutyrate was safe and well tolerated up to 15 g/day (maximum tolerated dose), while at 18 g/day, dose-limiting toxicity emerged in 5 of 11 subjects with gait instability, which was the most commonly reported adverse event (observed in one-third of the subjects) at all doses tested, and it was dose-limiting in two subjects. At higher doses, other reported toxicities were vomiting, lightheadedness, and confusion. A gene expression assay was able to detect a biologic response to the drug in peripheral lymphocytes. These results provide safety and dose-ranging data to support a phase II clinical trial with phenylbutyrate in HD and related neurodegenerative disorders.[172]
5. Conclusions
HDACIs have been widely studied since they seem to give the oppurtunity to manipulate gene expression and, through it, they seem to be promising to treat several pathologic conditions. Apart from the previously described conditions, recently, phenylbutyrate has also been investigated in Wilson disease, an autosomal recessive disorder of copper homeostasis. Wilson disease is caused by mutations in the gene encoding the copper transporting P1B-type adenosine triphosphatase (ATPase) ATP7B, and, currently, more than 300 different mutations have been described. Treatment with phenylbutyrate and curcumin has partially restored protein expression of most ATP7B mutants (p.G85V, p.R778L, p.H1069Q, p.C1104F, p.V1262F, p.G1343V, and p.S1363F).[173] Phenylbutyrate has also been investigated recently in deficiency in P-type ATP8B1 (caused by autosomal recessive mutations in the gene encoding ATP8B1), a severe and clinically highly variable hereditary disorder characterized by intrahepatic cholestasis. This condition is clinically divided into progressive familial intrahepatic cholestasis type 1 or intermittent, i.e. benign recurrent intrahepatic cholestasis type 1 disease. In this study, it has been observed that a large proportion of ATP8B1 mutations have resulted in aberrant folding and decreased expression at the plasma membrane, and, interestingly, these effects have been partially restored by treatment with phenylbutyrate. Therefore, according to the two previously described studies, it has been pointed out that pharmacologic chaperone may be an important strategy in the future management of hereditary liver disease in general.[174] A study reveals that phenylbutyrate possesses chemical chaperone activity in vitro, which prevents the aggregation of denatured α-lactalbumin and bovine serum albumin. The same authors investigated the effects of phenylbutyrate on the accumulation of Parkin-associated endothelin receptor-like receptor (Pael-R), pathologically relevant to the loss of dopaminergic neurons in autosomal recessive juvenile parkinsonism, showing that (i) phenylbutyrate restores the normal expression of Pael-R protein and suppresses ER stress induced by the overexpression of Pael-R; (ii) phenylbutyrate attenuates the activation of ER stress-induced signal transduction pathways and subsequent neuronal cell death; and (iii) phenylbutyrate restores the viability of yeasts that fail to induce an ER stress response under ER stress conditions. These findings lead the author to conclude that phenylbutyrate suppresses ER stress by directly reducing the amount of misfolded protein, including Pael-R accumulated in the ER.[175]
A recent study has shown that 5-week administration of phenylbutyrate reverses spatial learning and memory deficits in an established mouse model of Alzheimer disease, i.e. Tg2576 mice, without altering β-amyloid burden. In addition, the same authors have observed that transgenic mice have showed a significant decrease in phosphorylated tau in the hippocampus, after phenylbutyrate treatment, which may be due to an increase in the inactive form of the glycogen synthase kinase 3b (GSK3b). A consistent decrease in brain histone acetylation in the transgenic mice, which may reflect an indirect transcriptional repression underlying memory impairment, has also been observed. The administration of phenylbutyrate has restored brain histone acetylation levels and activated the transcription of synaptic plasticity markers, such as the selective glutamate receptor (GluR)-1 subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor, PSD95, and microtubule-associated protein-2. According to these results, phenylbutyrate may represent a new therapeutic agent to restore memory function in AD.[176]
A recent experimental study showed using both an in vivo and in vitro approach that phenylbutyrate treatment may be successfully used to reduce the plasma levels of neurotoxic branched-chain amino acids and their corresponding α-keto acids in a subset of maple syrup urine disease patients encouraging further studies of its long-term efficacy.[177]
A recent study has also analyzed the efficacy of phenylbutyrate on lipid-induced insulin resistance and β-cell dysfunction in eight overweight or obese nondiabetic men who were randomized to undergo four studies each 4–6 weeks apart. Two studies were preceded by 2 weeks of phenylbutyrate (7.5 g/day) orally, followed by a 48-hour intravenous infusion of intralipid/heparin or saline, and two studies were preceded by placebo treatment, followed by similar infusions. It was observed that lipid infusion reduced insulin sensitivity (S[I]), as assessed by hyperinsulinemic-euglycemic clamps, while it was significantly ameliorated by pretreatment with phenylbutyrate. Absolute insulin secretion rate (ISR), assessed by hyperglycemic clamps, was not affected by any treatment. Phenylbutyrate partially ameliorated the lipid-induced reduction in the disposition index (DI = ISR × S[I]) pointing out how phenylbutyrate prevented lipid-induced β-cell dysfunction.[178]
Particularly in this review we have reported the different clinical uses of phenylbutyrate, both in vitro and in vivo, in lethal illnesses that do not have any effective treatment. In our opinion, this drug should be more widely and deeply investigated in the clinical area but, unfortunately, the absence of viable international patents to protect commercial use of the molecule reduces the chances of financing new studies. Despite this frustrating position, there is a wide speculative attention to further investigate phenylbutyrate, even on an anedoctical basis, and we definitely support this challenge.
Notes
14-3-3 proteins play an important role in cancer biology by interfering with intracellular signaling pathways and cell cycle checkpoints,and in particular the 14-3-3σ isoform acts as a tumor suppressor and is often inactivated during the tumor development.
References
Waddington CH. The epigenotype. Endeavour 1942; 1: 18
Waddington CH. Towards a theoretical biology: the basic ideas of biology. Edinburgh: Edinburgh University Press, 1968: 1–32
Suzuki T, Miyata N. Epigenetic control using natural products and synthetic molecules. Curr Med Chem 2006; 13 (8): 935–58
Monneret C. Histone deacetylase inhibitors. Eur J Med Chem 2005 Jan; 40 (1): 1–13
Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta 2009 Jan; 1789 (1): 45–57
Gray SG, Ekström TJ. The human histone deacetylase family. Exp Cell Res 2001 Jan 15; 262 (2): 75–83
Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000 Jan 6; 403 (6765): 41–5
Chang KT, Min KT. Regulation of lifespan by histone deacetylase. Ageing Res Rev 2002 Jun; 1 (3): 313–26
Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol 2010 Feb; 77 (2): 126–35
Egler V, Korur S, Failly M, et al. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin Cancer Res 2008 May 15; 14 (10): 3132–40
Spira AI, Carducci MA. Differentiation therapy. Curr Opin Pharmacol 2003; 3: 338–43
Brusilow SW. Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res 1991; 29: 147–50
Feillet F, Leonard JV. Alternative pathway therapy for urea cycle disorders. J Inherit Metab Dis 1998; 21 Suppl. 1: 101–11
Carducci MA, Nelson JB, Chan-Tack KM, et al. Phenylbutyrate induces apoptosis in human prostate cancer and is more potent than phenylacetate. Clin Cancer Res 1996; 2: 379–87
Zeitlin PL, Diener-West M, Rubenstein RC, et al. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol Ther 2002 Jul; 6 (1): 119–26
Gilbert J, Baker SD, Bowling MK, et al. A phase I dose escalation and bioavailability study of oral sodium phenylbutyrate in patients with refractory solid tumor malignancies. Clin Cancer Res 2001; 7: 2292–300
Kemp S, Wei H-M, Lu J-F, et al. Gene redundancy of pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med 1998; 4: 1261–8
Berg S, Serabe B, Aleksic A, et al. Pharmacokinetics and cerebrospinal fluid penetration of phenylacetate and phenylbutyrate in the nonhuman primate. Cancer Chemother Pharmacol 2001; 47: 385–90
Maestri NE, Brusilow SW, Clissold DB, et al. Longterm treatment of a girl with ornithine transcarbamilase deficiency. N Engl J Med 1996; 335: 855–9
Redonnet-Vernhet I, Rouanet F, Pedespan JM, et al. A successful pregnancy in a heterozygote for OTC deficiency treated with sodium phenylbutyrate. Neurology 2000; 54: 1008
Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst 2000; 92: 1210–6
Svechnikova I, Almqvist PM, Ekström TJ. HDAC inhibitors effectively induce cell type-specific differentiation in human glioblastoma cell lines of different origin. Int J Oncol 2008 Apr; 32 (4): 821–7
Dasgupta S, Zhou Y, Jana M, et al. Sodium phenylacetate inhibits adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. J Immunol 2003 Apr 1; 170 (7): 3874–82
Qi X, Hosoi T, Okuma Y, et al. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol 2004 Oct; 66 (4): 899–908
Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov 2008 Oct; 7 (10): 854–68
Chiurazzi P, Neri G. Pharmacological reactivation of inactive genes: the fragile X experience. Brain Res Bull 2001 Oct–Nov 1; 56 (3–4): 383–7
Chiurazzi P, Pomponi MG, Willemsen R, et al. In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum Mol Genet 1998; 7: 109–13
Yoshida M, Furumai R, Nishiyama M, et al. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother Pharmacol 2001; 48 Suppl. 1: S20–6
Nakano K, Mizuno T, Sowa Y, et al. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. J Biol Chem 1997; 272: 22199–206
Huang L, Sowa Y, Sakai T, et al. Activation of the p21WAF1/ CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene 2000; 19: 5712–9
Kim YH, Park JW, Lee JY, et al. Sodium butyrate sensitizes TRAIL-mediated apoptosis by induction of transcription from the DR5 gene promoter through Sp1 sites in colon cancer cells. Carcinogenesis 2004; 25: 1813–20
Feinman R, Clarke KO, Harrison LE. Phenylbutyrate-induced apoptosis is associated with inactivation of NF-kappaB IN HT-29 colon cancer cells. Cancer Chemother Pharmacol 2002; 49: 27–34
Svechnikova I, Gray SG, Kundrotiene J, et al. Apoptosis and tumor remission in liver tumor xenografts by 4-phenylbutyrate. Int J Oncol 2003; 22: 579–88
Yokota N, Mainprize TG, Taylor MD, et al. Identification of differentially expressed and developmentally regulated genes in medulloblastoma using suppression subtraction hybridization. Oncogene 2004; 23: 3444–53
Drummond DC, Noble CO, Kirpotin DB, et al. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol 2005; 45: 495–528
Bhalla K, List A. Histone deacetylase inhibitors in myelodysplastic syndrome. Best Pract Res Clin Haematol 2004; 17: 595–611
Phuphanich S, Baker SD, Grossman SA, et al. Oral sodium phenylbutyrate in patients with recurrent malignant gliomas: a dose escalation and pharmacologic study. Neurooncol 2005; 7: 177–82
Ryu H, Smith K, Camelo SI, et al. Sodium phenylbutyrate prolongs survival and regulates expression of antiapoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem 2005; 93: 1087–98
Qi X, Hosoi T, Okuma Y, et al. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol 2004; 66: 899–908
Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006; 313: 1137–40
Liu XL, Done SC, Yan K, et al. Defective trafficking of nephrin missense mutants rescued by a chemical chaperone. J Am Soc Nephrol 2004; 15: 1731–8
Scaglia F, Carter S, O’Brien WE, et al. Effect of alternative pathway therapy on branched chain amino acid metabolism in urea cycle disorder patients. Mol Genet Metab 2004; 81: S79–85
Maestri NE, Brusilow SW, Clissold DB, et al. Long-term treatment of girls with ornithine, transcarbamylase deficiency. N Engl J Med 1996; 335: 855–9
Peart MJ, Tainton KM, Ruefli AA, et al. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res 2003; 63: 4460–71
Blagosklonny MV, Robey R, Sackett DL, et al. Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther 2002; 1: 937–41
Ishiguro K, Sartorelli AC. Activation of transiently transfected reporter genes in 3T3 Swiss cells by the inducers of differentiation/apoptosis-dimethylsulfoxide, hexamethylene bisacetamide and trichostatin A. Eur J Biochem 2004; 271: 2379–90
Piekarz R, Bates S. A review of depsipeptide and other histone deacetylase inhibitors in clinical trials. Curr Pharm Des 2004; 10: 2289–98
Yamano T, Ura K, Morishita R, et al. Amplification of transgene expression in vitro and in vivo using a novel inhibitor of histone deacetylase. Mol Ther 2000; 1: 574–80
Takimoto R, Kato J, Terui T, et al. Augmentation of antitumor effects of p53 gene therapy by combination with HDAC inhibitor. Cancer Biol Ther 2005; 4: 421–8
Lin J, Gilbert J, Rudek MA, et al. A phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clin Cancer Res 2009 Oct 1; 15 (19): 6241–9
Piscitelli SC, Thibault A, Figg WD, et al. Disposition of phenylbutyrate and its metabolites, phenylacetate and phenylacetylglutamine. J Clin Pharmacol 1995; 35: 368–73
Liu L, Hudgins WR, Miller AC, et al. Transcriptional upregulation of TGF-alpha by phenylactate and phenylbutyrate is associated with differentiation of human cells. Cytokine 1995; 7: 449–56
Zhang X, Wei L, Yang Y, et al. Sodium 4-phenylbutyrate induces apoptosis of human lung carcinoma cells through activating JNK pathway. J Cell Biochem 2004; 93 (4): 819–29
Carducci MA, Gilbert J, Bowling MK, et al. A phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin Cancer Res 2001 Oct; 7: 3047–55
Camacho LH, Olson J, Tong WP, et al. Phase I dose escalation clinical trial of phenylbutyrate sodium administered twice daily to patients with advanced solid tumors. Invest New Drugs 2007; 25: 131–8
Gore SD, Weng LJ, Figg WD, et al. Impact of prolonged infusions of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin Cancer Res 2002 Apr; 8 (4): 963–70
Christov K, Grubbs CJ, Shilkaitis A, et al. Short-term modulation of cell proliferation and apoptosis and preventive/ therapeutic efficacy of various agents in a mammary cancer model. Clin Cancer Res 2007 Sep 15; 13 (18 Pt 1): 5488–96
Daosukho C, Chen Y, Noel T, et al. Phenylbutyrate, a histone deacetylase inhibitor, protects against adriamycin-induced cardiac injury. Free Radic Biol Med 2007 Jun 15; 42 (12): 1818–25
DiGiuseppe JA, Weng L-J, Yu KH, et al. Phenylbutyrateinduced G1 arrest and apoptosis in myeloid leukemia cells: structure-function analysis. Leukemia (Baltimore) 1999; 13: 1243–53
Gore SD, Samid D, Weng L-J. Impact of the putative differentiating agents sodium phenylbutyrate and sodium phenylacetate on proliferation, differentiation, and apoptosis of primary neoplastic myeloid cells. Clin Canc Res 1997; 3: 1755–62
Yu KH, Weng L-J, Fu S, et al. Augmentation of phenylbutyrate- induced differentiation of myeloid leukemia cells using all trans-retinoic acid. Leukemia (Baltimore) 1999; 13: 1258–65
Redner RL, Wang J, Liu JM. Chromatin remodeling and leukemia: new therapeutic paradigms. Blood 1999; 94: 417–28
Ryningen A, Stapnes C, Bruserud Ø. Clonogenic acute myelogenous leukemia cells are heterogeneous with regard to regulation of differentiation and effect of epigenetic pharmacological targeting. Leukemia Res 2007; 31: 1303–13
Burkitt K, Ljungman M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol Cancer 2008; 7: 24
Bandres E, Agirre X, Bitarte N, et al. Epigenetic regulation of microRNA expression in colorectal cancer. Int J Cancer 2009; 125: 2737–43
Hurtubise A, Bernstein ML, Momparler RL. Preclinical evaluation of the antineoplastic action of 5-aza-2’-deoxycytidine and different histone deacetylase inhibitors on human Ewing’s sarcoma cells. Cancer Cell Int 2008 Nov 17; 8: 16
Vine AL, Bertram JS. Cancer chemoprevention by connexins. Cancer Metastasis Rev 2002; 21: 199–216
Seo MS, Park JS, Yang SR, et al. Expression ofMAP kinases and connexins in the differentiation of rat mammary epithelial cells. J Vet Med Sci 2006 Jun; 68 (6): 567–71
Yamasaki H, Naus CC. Role of connexin genes in growth control. Carcinogenesis 1996 Jun; 17 (6): 1199–213
Lee SW, Tomasetto C, Paul D, et al. Transcriptional downregulation of gap-junction proteins blocks junctional communication in human mammary tumor cell lines. J Cell Biol 1992; 118: 1213–21
Pelin K, Hirvonen A, Linnainmaa K. Expression of cell adhesion molecules and connexins in gap junctional intercellular communication deficient human mesothelioma tumour cell lines and communication competent primary mesothelial cells. Carcinogenesis 1994; 15: 2673–5
Zhang ZQ, Zhang W, Wang NQ, et al. Suppression of tumorigenicity of human lung carcinoma cells after transfection with connexin43. Carcinogenesis 1998; 19: 1889–94
Mehta PP, Perez-Stable C, Nadji M, et al. Suppression of human prostate cancer cell growth by forced expression of connexin genes. Dev Genet 1999; 24: 91–110
Hattori Y, Fukushima M, Maitani Y. Non-viral delivery of the connexin 43 gene with histone deacetylase inhibitor to human nasopharyngeal tumor cells enhances gene expression and inhibits in vivo tumor growth. Int J Oncol 2007; 30: 1427–39
Bueno MJ, Pérez de Castro I, Gómez de Cedrón M, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell 2008 Jun; 13 (6): 496–506
Ammerpohl O, Trauzold A, Schniewind B, et al. Complementary effects of HDAC inhibitor 4-PB on gap junction communication and cellular export mechanisms support restoration of chemosensitivity of PDAC cells. Br J Cancer 2007; 96: 73–81
Phillips JA, Griffin BE. Pilot study of sodium phenylbutyrate as adjuvant in cyclophosphamide-resistant endemic Burkitt’s lymphoma. Trans R Soc Trop Med Hyg 2007 Dec; 101 (12): 1265–9
Hanada S, Harada M, Kumemura H, et al. Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J Hepatol 2007 Jul; 47 (1): 93–102
Egger G, Aparicio AM, Escobar SG, et al. Inhibition of histone deacetylation does not block resilencing of p16 after 5-aza-2’-deoxycytidine treatment. Cancer Res 2007 Jan 1; 67 (1): 346–53
Gray SG, Al-Sarraf N, Baird AM, et al. Regulation of EP receptors in non-small cell lung cancer by epigenetic modifications. Eur J Cancer 2009 Nov; 45 (17): 3087–97
Hauswald S, Duque-Afonso J, Wagner MM, et al. Histone deacetylase inhibitors induce a very broad, pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by modulation of multiple ABC transporter genes. Clin Cancer Res 2009 Jun 1; 15 (11): 3705–15
Svechnikova I, Ammerpohl O, Ekström TJ. p21waf1/Cip1 partially mediates apoptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun 2007 Mar 9; 354 (2): 466–71
Verheul HM, Qian DZ, Carducci MA, et al. Sequencedependent antitumor effects of differentiation agents in combination with cell cycle-dependent cytotoxic drugs. Cancer Chemother Pharmacol 2007 Aug; 60 (3): 329–39
Lopez CA, Feng FY, Herman JM, et al. Phenylbutyrate sensitizes human glioblastoma cells lacking wild-type p53 function to ionizing radiation. Int J Radiat Oncol Biol Phys 2007 Sep 1; 69 (1): 214–20
Schultz J, Ibrahim SM, Vera J, et al. 14-3-3sigma gene silencing during melanoma progression and its role in cell cycle control and cellular senescence. Mol Cancer 2009 Jul 30; 8: 53
Merzvinskyte R, Treigyte G, Savickiene J, et al. Effects of histone deacetylase inhibitors, sodium phenyl butyrate and vitaminB3, incombinationwith retinoic acidongranulocytic differentiation of human promyelocytic leukemia HL-60 cells. Ann N Y Acad Sci 2006 Dec; 1091: 356–67
Denu JM. Vitamin B3 and sirtuin function. Trends Biochem Sci 2005 Sep; 30 (9): 479–83
Wang CT, Meng M, Zhang JC, et al. Growth inhibition and gene induction in human hepatocellular carcinoma cell exposed to sodium 4-phenylbutanoate. Chin Med J (Engl) 2008 Sep 5; 121 (17): 1707–11
Miller RG, Mitchell JD, Lyon M, et al. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev 2007 Jan; 24 (1): CD001447
Dion PA, Daoud H, Rouleau GA. Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet 2009 Nov; 10 (11): 769–82
Darras BT, Kang PB. Spinal muscular atrophy. Curr Opin Ped 2007; 19: 675–9
Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008 Jun 21; 371 (9630): 2120–33
Zerres K, Rudnick-Schoneborn S. Natural history in proximal spinal muscular atrophy. Arch Neurol 1995; 52: 518–23
Pazin MJ, Kadonaga JT. What’s up and down with histone deacetylation and transcription? Cell 1997; 89: 325–8
Andreassi C, Angelozzi C, Tiziano FD, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet 2004; 12: 59–65
Hauke J, Riessland M, Lunke S, et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet 2009 Jan 15; 18 (2): 304–17
Chang JG, Hsieh-Li HM, Jong YJ, et al. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA 2001; 98: 9808–13
Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med 2001 May 31; 344 (22): 1688–700
Chiò A, Logroscino G, Hardiman O, et al., Eurals Consortium. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler 2009 Oct–Dec; 10 (5–6): 310–23
Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol 2009 Nov; 8 (11): 1056–72
Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994 Mar 3; 330 (9): 585–91
Miller RG, Mitchell JD, Lyon M, et al. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph Lateral Scler Other Motor Neuron Disord 2003 Sep; 4 (3): 191–206
Armon C. Motor neuron disease. In: Gorelick PB, Alter M, editors. Handbook of neuroepidemiology. New York: Marcel Dekker, 1994: 407–54
Brown Jr RH, Robberecht W. Amyotrophic lateral sclerosis: pathogenesis. Semin Neurol 2001 Jun; 21 (2): 131–9
Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994 Jun 17; 264 (5166): 1772–5. Erratum in: Science 1995 Jul 14; 269 (5221): 149
Bruijn LI, Beal MF, Becher MW, et al. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci U S A 1997 Jul 8; 94 (14): 7606–11
Bruijn LI, Becher MW, Lee MK, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997 Feb; 18 (2): 327–38
Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2001 Nov; 2 (11): 806–19
Kostic V, Jackson-Lewis V, de Bilbao F, et al. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science 1997 Jul 25; 277 (5325): 559–62
Wakabayashi K, Saito H, Ebinuma H, et al. Bcl-2 related proteins are dramatically induced at the early stage of differentiation in human liver cancer cells by a histone deacetylase inhibitor projecting an anti-apoptotic role during this period. Oncol Rep 2000 Mar–Apr; 7 (2): 285–8
Vukosavic S, Stefanis L, Jackson-Lewis V, et al. Delaying caspase activation by Bcl-2: a clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 2000 Dec 15; 20 (24): 9119–25
Petri S, Kiaei M, Kipiani K, et al. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 2006 Apr; 22 (1): 40–9
Del Signore SJ, Amante DJ, Kim J, et al. Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph Lateral Scler 2009 Apr; 10 (2): 85–94
Cudkowicz ME, Andres PL, Macdonald SA, et al. Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph Lateral Scler 2009 Apr; 10 (2): 99–106
Osborne NN, Casson RJ, Wood JP, et al. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res 2004; 23: 91e147
Lagrèze WA, Knörle R, Bach M, et al. Memantine is neuroprotective in a ratmodel of pressure-induced retinal ischemia. Invest Ophthalmol Vis Sci 1998 May; 39 (6): 1063–6
Lagrèze WA, Knörle R, Bach M, et al. Retinal ganglion cell dysfunction induced by hypoxia and glutamate: potential neuroprotective effects of beta-blockers. Surv Ophthalmol 1999; 43: 162e170
Jeng YY, Lin NT, Chang PH, et al. Retinal ischemic injury rescued by sodium 4-phenylbutyrate in a rat model. Exp Eye Res 2007 Mar; 84 (3): 486–92
Vilatoba M, Eckstein C, Bilbao G, et al. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery 2005 Aug; 138 (2): 342–51
Leonard JV, Morris AAM. Urea cycle disorders. Semin Neonatol 2002; 7: 27–35
Batshaw ML, Berry GT. Use of citrulline as a diagnostic marker in the prospective treatment of urea cycle disorders. J Pediatr 1991; 118: 914–7
Brusilow SW, Finkelstein JE. Restoration of nitrogen homeostasis in a man with ornithine transcarbamylase deficiency. Metabolism 1993; 42: 1336–9
Leonard JV. Disorders of the urea cycle and related enzymes. 4th rev ed. Berlin: Springer, 2006
Gordon N. Ornithine transcarbamylase deficiency: a urea cycle defect. Eur J Paed Neurol 2003; 7: 115–21
Praphanphoj V, Boyadjiev SA, Waber LJ, et al. Three cases of intravenous sodium benzoate and sodium phenylbutyrate toxicity occurring in the treatment of acute hyperammonaemia. J Inherit Metab Dis 2000; 23: 129–36
Wilcken B. Problems in the management of urea cycle disorders. Mol Genet Metab 2004 Apr; 81 Suppl. 1: S86–91
Schultz REH, Salo MK. Under recognition of late onset ornithine transcarbamylase deficiency. Arch Dis Child 2000; 82: 390–1
Nicolaides P, Liebsch D, Dale N, et al. Neurological outcome of patients with ornithine carbamoyltransferase deficiency. Arch Dis Child 2002; 86: 54–6
Schwab S, Scharz S, Mayatepek E, et al. Recurrent brain edema in ornithine transcarbamylase deficiency. J Neurol 1999; 246: 609–11
Lee J-Y, Chang S-E, Sub C-W, et al. A case of acrodermatitis enteropathica-like dermatosis caused by ornithine transcarbamylase deficiency. J Am Acad Dermatol 2002; 46: 965–7
Legras A, Labarthe F, Maillot F, et al. Late diagnosis of ornithine transcarbamylase defect in three related female patients: polymorphic presentations. Crit Care Med 2002; 30: 241–4
Anadiotis G, Ierardi-Curto L, Kaplan PD, et al. Ornithine transcarbamylase deficiency and pancreatitis. J Pediatr 2001; 138: 123–4
Winichagoon P, Fucharoen S, Chen P, et al. Genetic factors affecting clinical severity in β-thalassemia syndromes. J Ped Hematol Oncol 2000 Nov/Dec; 22 (6): 573–80
Dover GJ. Hemoglobin switching protocols in thalassemia: experience with sodium phenylbutyrate and hydroxyurea. Ann N Y Acad Sci 1998 Jun 30; 850: 80–6
Orkin SH, Swan D, Leder P. Differential expression of α- and β-globin genes during differentiation of cultured erythroleukemic cells. J Biol Chem 1975; 250: 8753
Anderson LC, Jokinen M, Gahmberg CG. Induction of erythroid differentiation in the human leukemia cell line K562. Nature 1979; 278: 364
Samid D, Shack S, Sherman LT. Phenylacetate: a novel nontoxic inducer of tumor cell differentiation. Cancer Res 1992; 52: 1988
Perrine SP, Miller BA, Faller DV, et al. Sodium butyrate enhances fetal globin expression in erythroid progenitors of patients with HbSS and β-thalassemia. Blood 1989; 74: 454
Fibach E, Prasanna P, Rodgers GP, et al. Enhanced fetal hemoglobin production by phenylacetate and 4-phenylbutyrate in erythroid precursors derived from normal blood donors and patients with sickle cell anemia and β-thalassemia. Blood 1993; 82: 2203
Stamatoyannopoulos G, Nakamoto B, Josephson B, et al. Acetate, a product of butyrate catabolism, stimulates β-globin expression in adult cells in vivo and in culture [abstract]. Blood 1993; 82: 313a
Ginder GD, Whitters MJ, Pohlman JK. Activation of a chicken embryonic globin gene in adult erythroid cells by 5-azacytidine and sodium butyrate. Proc Natl Acad Sci USA 1984; 81: 3954
Perrine SP, Rudolph A, Faller DV, et al. Butyrate infusions in the ovine fetus delay the biologic clock for globin gene switching. Proc Natl Acad Sci U S A 1988 Nov; 85 (22): 8540–2
Constantoulakis P, Papayannopoulou TH, Stamatoyannopoulos G. Alpha-amino-N-butyric acid stimulates fetal hemoglobin in the adult. Blood 1988; 72: 1961. 21
Constantoulakis P, Knitter G, Stamatoyannopoulos G. On the induction of fetal hemoglobin by butyrates: in vivo and in vitro studies with sodium butyrate and comparison of combination treatment with 5-azaC and Ara C. Blood 1989; 74: 1963. 22
Blau CA, Constantoulakis P, Shaw CM, et al. Fetal hemoglobin induction with butyric acid: efficacy and toxicity. Blood 1993; 81: 529
Dover GJ, Brusilow SW, Samid D. Increased fetal hemoglobin in patients receiving sodium 4-phenylbutyrate [letter 29]. N Engl J Med 1992; 327: 569
Dover GJ, Brusilow SW, Charache S. Induction of HbF production in subjects with sickle cell anemia by oral sodium phenylbutyrate. Blood 1994; 84: 339
Perrine SP. Fetal globin stimulant therapies in the betahemoglobinopathies: principles and current potential. Pediatr Ann 2008 May; 37 (5): 339–46
Tsui LC. Mutations and sequence variations detected in the cystic fibrosis transmembrane conductance regulator (CFTR) gene: a report from the Cystic Fibrosis Genetic Analysis Consortium. Hum Mutat 1992; 1: 197–203
Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066–73
Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995; 83: 121–7
Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in delta F508- homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med 1998 Feb; 157 (2): 484–90
Lim M, McKenzie K, Floyd AD, et al. Modulation of deltaF508 cystic fibrosis transmembrane regulator trafficking and function with 4-phenylbutyrate and flavonoids. Am J Respir Cell Mol Biol 2004 Sep; 31 (3): 351–7
Cheng SH, Fang SL, Zabner J, et al. Functional activation of the cystic fibrosis trafficking mutant delta F508-CFTR by overexpression. Am J Physiol 1995; 268: L615–24
Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest 1997 Nov 15; 100 (10): 2457–65
Lim M, Zeitlin PL. Therapeutic strategies to correct malfunction of CFTR. Paediatr Respir Rev 2001 Jun; 2 (2): 159–64
Andersson C, Servetnyk Z, Roomans GM. Activation of CFTR by genistein in human airway epithelial cell lines. Biochem Biophys Res Commun 2003 Aug 29; 308 (3): 518–22
Andersson C, Roomans GM. Activation of deltaF508 CFTR in a cystic fibrosis respiratory epithelial cell line by 4-phenylbutyrate, genistein and CPX. Eur Respir J 2000 May; 15 (5): 937–41
Nguyen TD, Kim US, Perrine SP. Novel short chain fatty acids restore chloride secretion in cystic fibrosis. Biochem Biophys Res Commun 2006 Mar 31; 342 (1): 245–52
Singh OV, Pollard HB, Zeitlin PL. Chemical rescue of deltaF508-CFTR mimics genetic repair in cystic fibrosis bronchial epithelial cells. Mol Cell Proteomics 2008 Jun; 7 (6): 1099–110
Singh OV, Vij N, Mogayzel Jr PJ, et al. Pharmacoproteomics of 4-phenylbutyrate-treated IB3-1 cystic fibrosis bronchial epithelial cells. J Proteome Res 2006 Mar; 5 (3): 562–71
Rubenstein RC, Lyons BM. Sodium 4-phenylbutyrate downregulates HSC70 expression by facilitating mRNA degradation. Am J Physiol Lung Cell Mol Physiol 2001 Jul; 281 (1): L43–51
Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am J Physiol Cell Physiol 2000 Feb; 278 (2): C259–67
Linsdell P. Direct block of the cystic fibrosis transmembrane conductance regulator Cl(−) channel by butyrate and phenylbutyrate. Eur J Pharmacol 2001 Jan 12; 411 (3): 255–60
Prulière-Escabasse V, Planès C, Escudier E, et al. Modulation of epithelial sodium channel trafficking and function by sodium 4-phenylbutyrate in human nasal epithelial cells. J Biol Chem 2007 Nov 23; 282 (47): 34048–57
Ebbel EN, Leymarie N, Schiavo S, et al. Identification of phenylbutyrate-generated metabolites in Huntington disease patients using parallel liquid chromatography/ electrochemical array/mass spectrometry and off-line tandem mass spectrometry. Anal Biochem 2010 Apr 15; 399 (2): 152–61
Atwal RS, Truant R. A stress sensitive ER membraneassociation domain in Huntington protein defines a potential role for Huntington in the regulation of autophagy. Autophagy 2008 Jan 1; 4 (1): 91–3
Marambaud P, Dreses-Werringloer U, Vingtdeux V. Calcium signaling in neurodegeneration. Mol Neurodegener 2009 May 6; 4: 20
Gardian G, Browne SE, Choi DK, et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem 2005 Jan 7; 280 (1): 556–63
Ferrante RJ, Kubilus JK, Lee J, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci 2003 Oct 15; 23 (28): 9418–27
Borovecki F, Lovrecic L, Zhou J, et al. Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc Natl Acad Sci U S A 2005 Aug 2; 102 (31): 11023–8
Hogarth P, Lovrecic L, Krainc D. Sodium phenylbutyrate in Huntington’s disease: a dose-finding study. Mov Disord 2007 Oct 15; 22 (13): 1962–4
Van den Berghe PV, Stapelbroek JM, Krieger E, et al. Reduced expression of ATP7B affected by Wilson diseasecausing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology 2009 Dec; 50 (6): 1783–95
Van der Velden LM, Stapelbroek JM, Krieger E, et al. Folding defects in P-type ATP 8B1 associated with hereditary cholestasis are ameliorated by 4-phenylbutyrate. Hepatology 2010 Jan; 51 (1): 286–96
Kubota K, Niinuma Y, Kaneko M, et al. Suppressive effects of 4-phenylbutyrate on the aggregation of Pael receptors and endoplasmic reticulum stress. J Neurochem 2006 Jun; 97 (5): 1259–68
Ricobaraza A, Cuadrado-Tejedor M, Pérez-Mediavilla A, et al. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 2009 Jun; 34 (7): 1721–32. Epub 2009 Jan 14
Brunetti-Pierri N, Lanpher B, Erez A, et al. Phenylbutyrate therapy for maple syrup urine disease. Hum Mol Genet 2011 Feb 15; 20 (4): 631–40
Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 2011 Mar; 60 (3): 918–24
Acknowledgements
The authors have contributed equally to this work. This review has not been supported by grants.
Competing interests: the authors certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
Authors’ contribution: the authors hereby certify that all work contained in this review is original work of Tommaso Iannitti and Beniamino Palmieri. All the information taken from other articles, including tables and illustrations, have been referenced in the ‘References’ section. The authors claim full responsibility for the contents of the article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Iannitti, T., Palmieri, B. Clinical and Experimental Applications of Sodium Phenylbutyrate. Drugs R D 11, 227–249 (2011). https://doi.org/10.2165/11591280-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11591280-000000000-00000