
Abstract
This article reviews the clinical relevance of pharmacogenetics in cancer chemotherapy, with emphasis on drugs for which genetic differences in enzyme metabolism have been demonstrated to affect patient outcome.
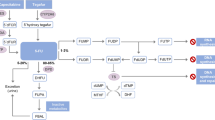
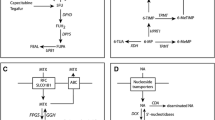
About 10% of children with leukaemia are intolerant to mercaptopurine (6-mercaptopurine) because of genetic defects in mercaptopurine inactivation by thiopurine S-methyltransferase. However, mercaptopurine dose intensity, a critical factor for outcome in patients deficient in thiopurine S-methyltransferase, can be maintained by means of thiopurine S-methyltransferase phenotyping or genotyping.
Patients with reduced fluorouracil (5-fluorouracil) catabolism are more likely to be exposed to severe toxicity. The measurement of dihydropyrimidine dehydrogenase activity in patients cannot be considered fully predictive, and the role of dihydropyrimidine dehydrogenase gene variants in this syndrome has yet to be clarified.
With regard to irinotecan, patients with Gilbert’s syndrome phenotype have reduced inactivation of the active topoisomerase I inhibitor 7-ethyl-10-hydroxy-camptothecin (SN-38) caused by a mutation in the UDP-glucuronosyltransferase 1A1 gene promoter. This subset of patients is more likely to be exposed to irinotecan toxicity and could be identified by genotyping for gene promoter variants.
Finally, the experience with amonafide represents a model for dose individualisation approaches that use simple phenotypic probes.



Similar content being viewed by others

References
Chabot GG. Factors involved in clinical pharmacology variability in oncology. Anticancer Res 1994; 14: 2269–72.
Iyer L, Ratain MJ. Pharmacogenetics and cancer chemotherapy. Eur J Cancer 1998; 34: 1493–9.
Iyer L. Inherited variations in drug-metabolizing enzymes: significance in clinical oncology. Mol Diagn 1999; 4: 327–33.
Shi MM, Bleavins MR, de la Iglesia FA. Technologies for detecting genetic polymorphisms in pharmacogenomics. Mol Diagn 1999; 4: 343–51.
Linder MW, Russell AP, Valdes RJ. Pharmacogenetics: a laboratory tool for optimizing therapeutic efficiency. Clin Chem 1997; 43: 254–66.
Tidd DM, Paterson AR. A biochemical mechanism for the delayed cytotoxic reaction of 6-mercaptopurine. Cancer Res 1974; 34: 738–46.
Deininger M, Szumlanski CL, Otterness DM, et al. Purine substrates for human thiopurine methyltransferase. Biochem Pharmacol 1994; 48: 2135–8.
Woodson LC, Ames MM, Selassie CD, et al. Thiopurine methyltransferase: aromatic thiol substrates and inhibition by benzoic acid derivatives. Mol Pharmacol 1983; 24: 471–8.
Lennard L, Lilleyman JS, Van Loon J, et al. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet 1990; 336: 225–9.
Lilleyman JS, Lennard L. Mercaptopurine metabolism and risk of relapse in childhood lymphoblastic leukaemia. Lancet 1994; 343: 1188–90.
Andersen JB, Szumlanski C, Weinshilboum RM, et al. Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr 1998; 87: 108–11.
McLeod HL, Coulthard S, Thomas AE, et al. Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br J Haematol 1999; 105: 696–700.
Lennard L, Gibson BE, Nicole T, et al. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69: 577–9.
Lennard L, Lewis IJ, Michelagnoli M, et al. Thiopurine methyltransferase deficiency in childhood lymphoblastic leukaemia: 6-mercaptopurine dosage strategies. Med Pediatr Oncol 1997; 29: 252–5.
Evans WE, Horner M, Chu YQ, et al. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119: 985–9.
Relling MV, Hancock ML, Boyett JM, et al. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93: 2817–23.
Dibenedetto SP, Guardabasso V, Ragusa R, et al. 6-Mercapto-purine cumulative dose: a critical factor of maintenance therapy in average risk childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol 1994; 11: 251–8.
Szumlanski C, Otterness D, Her C, et al. Thiopurine methyltransferase pharmacogenetics: human gene cloning and characterization of a common polymorphism. DNA Cell Biol 1996; 15: 17–30.
Yates CR, Krynetski EY, Loennechen T, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med 1997; 126: 608–14.
Otterness D, Szumlanski C, Lennard L, et al. Human thiopurine methyltransferase pharmacogenetics: gene sequence polymorphisms. Clin Pharmacol Ther 1997; 62: 60–73.
Tai HL, Krynetski EY, Yates CR, et al. Thiopurine S-methyl-transferase deficiency: two nucleotide transitions define the most prevalent mutant allele associated with loss of catalytic activity in Caucasians. Am J Hum Genet 1996; 58: 694–702.
Otterness DM, Szumlanski CL, Wood TC, et al. Human thiopurine methyltransferase pharmacogenetics: kindred with a terminal exon splice junction mutation that results in loss of activity. J Clin Invest 1998; 101: 1036–44.
Hon YY, Fessing MY, Pui CH, et al. Polymorphism of the thiopurine S-methyltransferase gene in African-Americans. Hum Mol Genet 1999; 8: 371–6.
Collie-Duguid ES, Pritchard SC, Powrie RH, et al. The frequency and distribution of thiopurine methyltransferase alleles in Caucasian and Asian populations. Pharmacogenetics 1999; 9: 37–42.
McLeod HL, Pritchard SC, Githang’a J, et al. Ethnic differences in thiopurine methyltransferase pharmacogenetics: evidence for allele specificity in Caucasian and Kenyan individuals. Pharmacogenetics 1999; 9: 773–6.
Ameyaw MM, Collie-Duguid ES, Powrie RH, et al. Thiopurine methyltransferase alleles in British and Ghanaian populations. Hum Mol Genet 1999; 8: 367–70.
Hiratsuka M, Inoue T, Omori F, et al. Genetic analysis of thiopurine methyltransferase polymorphism in a Japanese population. Mutat Res 2000; 448: 91–5.
Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91: 2001–8.
McLeod HL, Krynetski EY, Relling MV, et al. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia 2000; 14: 567–72.
Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet 1980; 32: 651–62.
McLeod HL, Lin JS, Scott EP, et al. Thiopurine methyltransferase activity in American white subjects and black subjects. Clin Pharmacol Ther 1994; 55: 15–20.
McLeod HL, Relling MV, Liu Q, et al. Polymorphic thiopurine methyltransferase in erythrocytes is indicative of activity in leukemic blasts from children with acute lymphoblastic leukemia. Blood 1995; 85: 1897–902.
Lennard L, Van Loon JA, Lilleyman JS, et al. Thiopurine pharmacogenetics in leukemia: correlation of erythrocyte thiopurine methyltransferase activity and 6-thioguanine nucleotide concentrations. Clin Pharmacol Ther 1987; 41: 18–25.
Lennard L, Lilleyman JS. Variable mercaptopurine metabolism and treatment outcome in childhood lymphoblastic leukemia. J Clin Oncol 1989; 7: 1816–23.
Lennard L, Welch JC, Lilleyman JS. Thiopurine drugs in the treatment of childhood leukaemia: the influence of inherited thiopurine methyltransferase activity on drug metabolism and cytotoxicity. Br J Clin Pharmacol 1997; 44: 455–61.
Coulthard SA, Howell C, Robson J, et al. The relationship between thiopurine methyltransferase activity and genotype in blasts from patients with acute leukemia. Blood 1998; 92: 2856–62.
Welch JC, Lilleyman JS. 6-Mercaptopurine dose escalation and its effect on drug tolerance in childhood lymphoblastic leukaemia. Cancer Chemother Pharmacol 1996; 38: 113–6.
Ho DH, Townsend L, Luna MA, et al. Distribution and inhibition of dihydrouracil dehydrogenase activities in human tissues using 5-fluorouracil as a substrate. Anticancer Res 1986; 6: 781–4.
Lu Z, Zhang R, Diasio RB. Dihydropyrimidine dehydrogenase activity in human peripheral blood mononuclear cells and liver: population characteristics, newly identified deficient patients, and clinical implication in 5-fluorouracil chemotherapy. Cancer Res 1993; 53: 5433–8.
Etienne MC, Lagrange JL, Dassonville O, et al. Population study of dihydropyrimidine dehydrogenase in cancer patients. J Clin Oncol 1994; 12: 2248–53.
Harris BE, Song R, Soong SJ, et al. Relationship between dihydropyrimidine dehydrogenase activity and plasma 5-flu-orouracil levels with evidence for circadian variation of enzyme activity and plasma drug levels in cancer patients receiving 5-fluorouracil by protracted continuous infusion. Cancer Res 1990; 50: 197–201.
Fleming RA, Milano G, Thyss A, et al. Correlation between dihydropyrimidine dehydrogenase activity in peripheral mononuclear cells and systemic clearance of fluorouracil in cancer patients. Cancer Res 1992; 52: 2899–902.
Johnson MR, Wang K, Tillmanns S, et al. Structural organization of the human dihydropyrimidine dehydrogenase gene. Cancer Res 1997; 57: 1660–3.
Wei X, Elizondo G, Sapone A, et al. Characterization of the human dihydropyrimidine dehydrogenase gene. Genomics 1998; 51: 391–400.
Meinsma R, Fernandez-Salguero P, Van Kuilenburg AB, et al. Human polymorphism in drug metabolism: mutation in the dihydropyrimidine dehydrogenase gene results in exon skipping and thymine uracilurea. DNA Cell Biol 1995; 14: 1–6.
Wei X, McLeod HL, McMurrough J, et al. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicity. J Clin Invest 1996; 98: 610–5.
McLeod HL, Collie-Duguid ES, Vreken P, et al. Nomenclature for human DPYD alleles. Pharmacogenetics 1998; 8: 455–9.
Collie-Duguid ES, Etienne MC, Milano G, et al. Known variant DPYD alleles do not explain DPD deficiency in cancer patients. Pharmacogenetics 2000; 10: 217–23.
Vreken P, Van Kuilenburg AB, Meinsma R, et al. A point mutation in an invariant splice donor site leads to exon skipping in two unrelated Dutch patients with dihydropyrimidine dehydrogenase deficiency. J Inherit Metab Dis 1996; 19: 645–54.
Ridge SA, Sludden J, Brown O, et al. Dihydropyrimidine dehydrogenase pharmacogenetics in Caucasian subjects. Br J Clin Pharmacol 1998; 46: 151–6.
Van Kuilenburg AB, Vreken P, Beex LV, et al. Severe 5-fluorouracil toxicity caused by reduced dihydropyrimidine dehydrogenase activity due to heterozygosity for a G→A point mutation. J Inherit Metab Dis 1998; 21: 280–4.
Johnson MR, Hageboutros A, Wang K, et al. Life-threatening toxicity in a dihydropyrimidine dehydrogenase-deficient patient after treatment with topical 5-fluorouracil. Clin Cancer Res 1999; 5: 2006–11.
Ridge SA, Sludden J, Wei X, et al. Dihydropyrimidine dehydrogenase pharmacogenetics in patients with colorectal cancer. Br J Cancer 1998; 77: 497–500.
Kouwaki M, Hamajima N, Sumi S, et al. Identification of novel mutations in the dihydropyrimidine dehydrogenase gene in a Japanese patient with 5-fluorouracil toxicity. Clin Cancer Res 1998; 4: 2999–3004.
Collie-Duguid ES, Johnston SJ, Powrie RH, et al. Cloning and initial characterization of the human DPYD gene promoter. Biochem Biophys Res Commun 2000; 271: 28–35.
Fleming RA, Milano GA, Gaspard MH, et al. Dihydropyrimidine dehydrogenase activity in cancer patients. Eur J Cancer 1993; 29A: 740–4.
McMurrough J, McLeod HL. Analysis of the dihydropyrimidine dehydrogenase polymorphism in a British population. Br J Clin Pharmacol 1996; 41: 425–7.
Grem JL, Yee LK, Venzon DJ, et al. Inter- and intraindividual variation in dihydropyrimidine dehydrogenase activity in peripheral blood mononuclear cells. Cancer Chemother Pharmacol 1997; 40: 117–25.
Van Kuilenburg AB, Van Lenthe H, Tromp A, et al. Pitfalls in the diagnosis of patients with a partial dihydropyrimidine dehydrogenase deficiency. Clin Chem 2000; 46: 9–17.
Van Kuilenburg AB, van Lenthe H, Blom MJ, et al. Profound variation in dihydropyrimidine dehydrogenase activity in human blood cells: major implications for the detection of partly deficient patients. Br J Cancer 1999; 79: 620–6.
Lu Z, Zhang R, Carpenter JT, et al. Decreased dihydropyrimidine dehydrogenase activity in a population of patients with breast cancer: implication for 5-fluorouracil-based chemotherapy. Clin Cancer Res 1998; 4: 325–9.
Chazal M, Etienne MC, Renee N, et al. Link between dihydropyrimidine dehydrogenase activity in peripheral blood mononuclear cells and liver. Clin Cancer Res 1996; 2: 507–10.
Stephan F, Etienne MC, Wallays C, et al. Depressed hepatic dihydropyrimidine dehydrogenase activity and fluorouracilrelated toxicities. Am JMed 1995; 99: 685–8.
Milano G, Etienne MC, Pierrefite V, et al. Dihydropyrimidine dehydrogenase deficiency and fluorouracil-related toxicity. Br J Cancer 1999; 79: 627–30.
Katona C, Kralovanszky J, Rosta A, et al. Putative role of dihydropyrimidine dehydrogenase in the toxic side effect of 5-fluorouracil in colorectal cancer patients. Oncology 1998; 55: 468–74.
Etienne MC, Cheradame S, Fischel JL, et al. Response to fluorouracil therapy in cancer patients: the role of tumoral dihydropyrimidine dehydrogenase activity. J Clin Oncol 1995; 13: 1663–70.
Salonga D, Danenberg KD, Johnson M, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res 2000; 6: 1322–7.
Huang CL, Yokomise H, Kobayashi S, et al. Intratumoral expression of thymidylate synthase and dihydropyrimidine dehydrogenase in non-small cell lung cancer patients treated with 5-FU-based chemotherapy. Int J Oncol 2000; 17: 47–54.
McLeod HL, Sludden J, Murray GI, et al. Characterization of dihydropyrimidine dehydrogenase in human colorectal tumours. Br J Cancer 1998; 77: 461–5.
Diasio RB, Beavers TL, Carpenter JT. Familial deficiency of dihydropyrimidine dehydrogenase: biochemical basis for familial pyrimidinemia and severe 5-fluorouracil-induced toxicity. J Clin Invest 1988; 81: 47–51.
Harris BE, Carpenter JT, Diasio RB. Severe 5-fluorouracil toxicity secondary to dihydropyrimidine dehydrogenase deficiency: a potentially more common pharmacogenetic syndrome. Cancer 1991; 68: 499–501.
Houyau P, Gay C, Chatelut E, et al. Severe fluorouracil toxicity in a patient with dihydropyrimidine dehydrogenase deficiency. J Natl Cancer Inst 1993; 85: 1602–3.
Lyss AP, Lilenbaum RC, Harris BE, et al. Severe 5-fluorouracil toxicity in a patient with decreased dihydropyrimidine dehydrogenase activity. Cancer Invest 1993; 11: 239–40.
Van Kuilenburg AB, Vreken P, Beex LV, et al. Heterozygosity for a point mutation in an invariant splice donor site of dihydropyrimidine dehydrogenase and severe 5-fluorouracil related toxicity. Eur J Cancer 1997; 33: 2258–64.
Takimoto CH, Lu ZH, Zhang R, et al. Severe neurotoxicity following 5-fluorouracil-based chemotherapy in a patient with dihydropyrimidine dehydrogenase deficiency. Clin Cancer Res 1996; 2: 477–81.
Shehata N, Pater A, Tang SC. Prolonged severe 5-fluorouracil-associated neurotoxicity in a patient with dihydropyrimidine dehydrogenase deficiency. Cancer Invest 1999; 17: 201–5.
Lu Z, Zhang R, Diasio RB. Population characteristics of hepatic dihydropyrimidine dehydrogenase activity, a key metabolic enzyme in 5-fluorouracil chemotherapy. Clin Pharmacol Ther 1995; 58: 512–22.
Tuchman M, Stoeckeler JS, Kiang DT, et al. Familial pyrimidinemia and pyrimidinuria associated with severe fluorouracil toxicity. N Engl J Med 1985; 313: 245–9.
Gamelin E, Boisdron-Celle M, Guerin-Meyer V, et al. Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5-FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: a potential interest for predicting 5-FU toxicity and determining optimal 5-FU dosage. J Clin Oncol 1999; 17: 1105–10.
Bocci G, Danesi R, Di Paolo A, et al. Comparative pharmacokinetic analysis of 5-fluorouracil and its major metabolite 5-fluoro-5,6-dihydrouracil after conventional and reduced test dose in cancer patients. Clin Cancer Res 2000; 6: 3032–7.
Humerickhouse R, Lohrbach K, Li L, et al. Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 andhCE-2. Cancer Res 2000; 60: 1189–92.
Kawato Y, Furuta T, Aonuma M, et al. Antitumor activity of a camptothecin derivative, CPT-11, against human tumor xenografts in nude mice. Cancer Chemother Pharmacol 1991; 28: 192–8.
Rivory LP, Riou JF, Haaz MC, et al. Identification and properties of a major plasma metabolite of irinotecan (CPT-11) isolated from the plasma of patients. Cancer Res 1996; 56: 3689–94.
Dodds HM, Haaz MC, Riou JF, et al. Identification of a new metabolite of CPT-11 (irinotecan): pharmacological properties and activation to SN-38. J Pharmacol Exp Ther 1998; 286: 578–83.
Gupta E, Lestingi TM, Mick R, et al. Metabolic fate of irinotecan inhumans: correlation of glucuronidation with diarrhea. Cancer Res 1994; 54: 3723–5.
Iyer L, King CD, Whitington PF, et al. Genetic predisposition to the metabolism of irinotecan (CPT-11): role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest 1998; 101: 847–54.
Sampietro M, Iolascon A. Molecular pathology of Crigler-Najjar type I and II and Gilbert’s syndromes. Haematologica 1999; 84: 150–7.
Bosma PJ, Chowdhury JR, Bakker C, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med 1995; 333: 1171–5.
Monaghan G, Ryan M, Seddon R, et al. Genetic variation in bilirubin UDP-glucuronyltransferase gene promoter and Gilbert’s syndrome. Lancet 1996; 347: 578–81.
Monaghan G, Foster B, Jurima-Romet M, et al. UGT1*1 genotyping in a Canadian Inuit population. Pharmacogenetics 1997; 7: 153–6.
Owens D, Evans L. Population studies on Gilbert’s syndrome. J Med Genet 1975; 12: 152–6.
Hall D, Ybazeta G, Destro-Bisol G, et al. Variability at the uridine diphosphate glucuronosyltransferase 1A1 promoter in human populations and primates. Pharmacogenetics 1999; 9: 591–9.
Ando Y, Chida M, Nakayama K, et al. The UGT1A1*28 allele is relatively rare in a Japanese population. Pharmacogenetics 1998; 8: 357–60.
Beutler E, Gelbart T, Demina A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA 1998; 95: 8170–4.
Lampe JW, Bigler J, Homer NK, et al. UDP-glucuronosyltransferase (UGT1A1*28 and UGT1A6*2) polymorphisms in Caucasians and Asians: relationships to serum bilirubin concentrations. Pharmacogenetics 1999; 9: 341–9.
DiRienzo A, Hall D, Iyer L, et al. Two new alleles in the promoter of the bilirubin UDP-glucuronosyl transferase 1 (UGT1A1) gene [abstract]. Clin Pharmacol Ther 1998; 63: 207.
Iolascon A, Faienza MF, Centra M, et al. (TA)8 allele in the UGT1A1 gene promoter of a Caucasian with Gilbert’s syndrome. Haematologica 1999; 84: 106–9.
Akaba K, Kimura T, Sasaki A, et al. Neonatal hyperbilirubinemia and a common mutation of the bilirubin uridine diphosphate-glucuronosyltransferase gene in Japanese. J Hum Genet 1999; 44: 22–5.
Akaba K, Kimura T, Sasaki A, et al. Neonatal hyperbilirubinemia and mutation of the bilirubin uridine diphosphate-glucuronosyltransferase gene: a common missense mutation among Japanese, Koreans and Chinese. Biochem Mol Biol Int 1998; 46: 21–6.
Huang C-S, Luo G-A, Huang M-J, et al. Variations of the bilirubin uridine-diphosphoglucuronosyl transferase 1A1 gene in healthy Taiwanese. Pharmacogenetics 2000; 10: 539–44.
Koiwai O, Yasui Y, Hasada K, et al. Three Japanese patients with Crigler-Najjar syndrome type I carry an identical nonsense mutation in the gene for UDP-glucuronosyltransferase. Jpn J Hum Genet 1995; 40: 253–7.
Sato H, Adachi Y, Koiwai O. The genetic basis of Gilbert’s syndrome. Lancet 1996; 347: 557–8.
Iyer L, Hall D, Das S, et al. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther 1999; 65: 576–82.
Iyer L, Bergseth AC, Ybazeta G, et al. High variability in SN-38 glucuronidation in liver samples from African population as predicted by UGT1A1 genotypes [abstract]. Clin Pharmacol Ther 1999; 65: 197.
Ando Y, Saka H, Asai G, et al. UGT1A1 genotypes and glucuronidation of SN-38, the active metabolite of irinotecan. Ann Oncol 1998; 9: 845–7.
Wasserman E, Myara A, Lokiec F, et al. Severe CPT-11 toxicity in patients with Gilbert’s syndrome: two case report. Ann Oncol 1997; 8: 1049–51.
Iyer L, Janisch L, Das S, et al. UGT1A1 promoter genotype correlates with pharmacokinetics of irinotecan (CPT-11) [abstract]. Proc Am Soc Clin Oncol 2000; 19: 178a.
De Isabella P, Zunino F, Capranico G. Base sequence determinants of amonafide stimulation of topoisomerase II DNA cleavage. Nucleic Acids Res 1995; 23: 223–9.
Costanza ME, Berry D, Henderson IC, et al. Amonafide: an active agent in the treatment of previously untreated advanced breast cancer — a cancer and leukemia group B study (CALGB 8642). Clin Cancer Res 1995; 1: 699–704.
Felder TB, McLean MA, Vestal ML, et al. Pharmacokinetics and metabolism of the antitumor drug amonafide (NSC-308847) in humans. Drug Metab Dispos 1987; 15: 773–7.
Price Evans DA, Manley KA, McKusick VA. Genetic control of isoniazid metabolism in man. BMJ 1960; 2: 485–91.
Ratain MJ, Mick R, Berezin F, et al. Paradoxical relationship between acetylator phenotype and amonafide toxicity. Clin Pharmacol Ther 1991; 50: 573–9.
Ratain MJ, Mick R, Berezin F, et al. Phase I study of amonafide dosing based on acetylator phenotype. Cancer Res 1993; 53: 2304–8.
Ratain MJ, Mick R, Janish L, et al. Individualized dosing of amonafide based on a pharmacodynamic model incorporating acetylator phenotype and gender. Pharmacogenetics 1996; 6: 93–101.
Acknowledgements
We wish to thank Jackie Ramírez, Carla Buterman and Larry House for their assistance in literature searches.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Innocenti, F., Iyer, L. & Ratain, M.J. Pharmacogenetics. Clin Pharmacokinet 39, 315–325 (2000). https://doi.org/10.2165/00003088-200039050-00001
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003088-200039050-00001