
Abstract
Congenital lipoid adrenal hyperplasia (CLAH) is the most severe form of congenital adrenal hyperplasia, caused by defects in the steroidogenic acute regulatory protein (STAR). The STAR p.Q258* mutation is the most common mutation in China, Japan and Korea, suggesting a founder effect. This study aimed to investigate the phenotypic and mutation spectrum of STAR defects and identify the founder effect of the p.Q258* mutation in Korean patients with CLAH. For 45 patients from 42 independent pedigrees, haplotype analysis was performed in 10 unrelated trio families, including patients with the p.Q258* mutation whose DNA samples were available, using 1,972 single nucleotide polymorphism (SNP) and six short tandem repeat markers. An Illumina Infinium® Human Omni2.5-8 v1.3 performed the SNP genotyping. Among 84 alleles from 42 unrelated families, mutation p.Q258* was found in 74 alleles (88.1%) from 41 families. A shared haplotype was identified in 17 of 20 alleles from 10 patients (size, 198 kb). The age of the founder mutation was estimated as 4,875 years (95% credible set: 3,575–7,925 years), assuming an intergenerational time interval of 25 years. The STAR p.Q258* mutation is the most common in Korean patients with CLAH, suggesting a founder effect. The age of the mutation corresponds with the time when the Korean people settled in the Korean peninsula. The high prevalence of p.Q258* in Japan and China also suggests a founder effect in Asian countries.
Similar content being viewed by others


Introduction
Congenital lipoid adrenal hyperplasia (CLAH, OMIM #201710) is a rare autosomal recessive disorder caused by defects in the steroidogenic acute regulatory (STAR) protein (1,2). The defects in the synthesis of all adrenal steroid hormones caused by STAR mutations result in life-threatening adrenal insufficiency soon after birth. Most patients have female external genitalia regardless of genetic sex, because of a lack of testicular androgen production (1,3,4), except in nonclassical CLAH (5,6).
The STAR gene has seven exons, is located at chromosome 8p11.23, and encodes a 285-amino acid protein that regulates the flow of cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane enzyme, where the biosynthesis of steroid initiates (7). Glucose regulatory protein 78 (GRP78), a master endoplasmic reticulum chaperone, is necessary for STAR expression and activity by facilitating the intermediate folding of STAR for delivery to the outer mitochondrial membrane. In the absence of GRP78, appropriate folding of STAR is lost, resulting in ablation of STAR expression and complete shutdown of mitochondrial steroidogenesis (8).
Since the STAR gene was mapped in 1995 (7), 77 mutations have been identified in various ethnic groups to date (https://doi.org/www.hgmd.org/). Most mutations are private and nonrecurrent, whereas the recurrent mutations have been identified in specific ethnic groups in which CLAH is relatively common; e.g., the p.R182L and c.201_202delCT mutations among Palestinian Arabs (9,10), the p.R182H mutation in eastern Saudi Arabia (11), and the p.L260P mutation in the Swiss population (12). Among them, the c.201_202delCT mutation was documented as a founder mutation by haplotype analysis using intergenic short tandem repeat (STR) and several single nucleotide polymorphism (SNP) markers (10). In addition, the p.Q258* mutation has been reported in Chinese, Japanese and Korean patients with CLAH, with minor allele frequencies of 35%, 57.9% and 92%, respectively, which suggests a founder effect in Asian populations (13–15).
A recurrent mutation in different families can be a hot spot or a founder mutation. To distinguish a hot spot from a founder mutation, it is necessary to investigate genetic markers that are close to the mutation, including SNPs and STRs. A series of genetic markers that are inherited together through generations is called a haplotype, which has high linkage, resulting in little or no separation by meiotic recombination (16). A recurrent mutation that occurs in a single haplotype of a population can be considered a founder mutation, while a mutation that occurs in more than one haplotype is considered a hot-spot mutation (16).
Previously, our group reported the mutation spectrum of the STAR gene in patients with CLAH, suggesting the possibility of a founder effect of the p.Q258* mutation, which shows high minor allele frequency (92%) in Korea (9,13,15). However, this was not proven by haplotype analysis. Therefore, the present study was performed to investigate the phenotypic and mutation spectrum of STAR defects and to determine whether the p.Q258* mutation is descended from a common ancestor via the founder effect.
Materials and Methods
Subjects
This study included 45 patients with CLAH from 42 independent families. Diagnosis was based on clinical features, endocrine characteristics and molecular analysis of STAR. This study was approved by the Institutional Review Board at the Asan Medical Center, and written informed consent was obtained from all the patients or their parents.
Mutation Analysis of the STAR Gene
Genomic DNA was extracted from peripheral blood leukocytes using a Gentra Puregene blood kit (Qiagen, Hilden, Germany). The seven coding exons and exon-intron boundaries of STAR were amplified using polymerase chain reaction (PCR). The amplified PCR products were sequenced directly on both strands using a BigDye Terminator V3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) and an ABI3130xl genetic analyzer (Applied Biosystems), according to the manufacturer’s instructions.
Haplotype Analysis for STAR p.Q258* Mutation
Haplotype analysis was performed to determine whether the p.Q258* mutation represents a founder mutation. Ten unrelated patients and their parents, whose DNA samples were available and who had agreed to participate in this study, were included for the haplotype analysis. SNP and/or STR markers were selected within a genomic region encompassing at least three recombination hot spots on both sides of the mutation. This region covered 2,854 kb around the STAR gene (chromosome 8: 36,990,733–39,845,265). Five STR markers (D8S1722, D8S571, D8S1821, D8S2317 and D8S515) were selected from the UniSTS database (https://doi.org/www.ncbi.nlm.nih.gov/unists). PCR amplification was performed under standard conditions in a total volume of 50 µL, containing 50 ng of genomic DNA. Forward primers were labeled with fluorescein amidite. PCR products were sequenced using capillary electrophoresis in an ABI3130xl genetic analyzer (Applied Biosystems). Data were analyzed using GeneMarker software, version 2.4.1 (SoftGenetics, State College, PA, USA).
We performed genome-wide genotyping in the 10 unrelated probands and their parents who carried the STAR p.Q258* mutation. A total of 2,372,784 SNPs were genotyped using an Illumina Infinium® Human Omni2.5-8 v1.3 Bead-Chip (Illumina, San Diego, CA, USA). Of these, 1,972 SNP markers within the designated genomic region were selected for haplotype analysis.
To determine the familial segregation of polymorphic markers, DNA samples from patients and their parents were used for chromosomal phasing, which is the process of determining a particular allele is inherited from which of the two parental chromosomes. Haplotypes were established manually when phasing could be determined. The frequencies of the identified haplotypes were obtained from the International HapMap JPT population using Haploview 4.2 and the International HapMap database (https://doi.org/hapmap.ncbi.nlm.nih.gov/). A binomial probability formula was used to calculate the probability of whether a mutation occurred recurrently as a de novo event on the same haplotype, as previously described (17).
Estimating the Age of the Mutation
We also estimated the age of the mutation to further understand its history. The DMLE2.3 program (https://doi.org/www.dmle.org/) was used to estimate the age of the STAR p.Q258* mutation. The population growth rate was set as 0.025, with an intergenerational time interval of 25 years (18). The number of patients in the population was assumed using an estimated total carrier frequency of 1:135 and the Korean population of 50 million (19).
Results
Clinical and Endocrine Characteristics
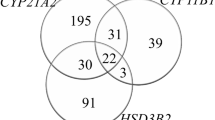
Among the 45 patients, 22 were genetic males and 23 had a 46,XX karyotype. Most patients (41/45, 91%) presented with adrenal insufficiency in the neonatal period. Four patients (one genetic male and three genetic females) presented with skin hyperpigmentation and chronic adrenal insufficiency, without adrenal crisis, after the infantile period and were diagnosed with late-onset nonclassic-type CLAH (Figure 1). Of these, one genetic male, who was compound heterozygous for p.Q258* and p.R272H, demonstrated perfect male external genitalia and have been raised as a boy. One genetic female, who had presented with chronic adrenal insufficiency without salt wasting after the age of 2.4 years, was also compound heterozygous for p.Q258* and p.R272H. The other two genetic females, who were both compound heterozygous for p.Q258* and p.V187M, showed skin hyperpigmentation and hyponatremia at the age of 1.2 years and 2.8 years, respectively.

Clinical outcomes of patients with congenital lipoid adrenal hyperplasia. Among the 45 patients, 22 were genetic males and 23 had a 46,XX karyotype. Most patients (41/45, 91.1%) presented with adrenal insufficiency in the neonatal period, while three late-onset patients showed skin hyperpigmentation after the infantile period. Two patients had neurodevelopmental deficits caused by adrenal insufficiency resulting from poor adherence to medication. Three genetic female patients experienced spontaneous puberty.
Two patients had neurodevelopmental deficits because of adrenal crisis. Three genetic female patients experienced spontaneous puberty. Ovarian cyst torsion occurred in one patient (20). She experienced thelarche at age 10.3 years and spontaneous menarche at 12.8 years. At the age of 14 years, she manifested acute pelvic pain and underwent laparoscopic left salpingo-oophorectomy. One patient died of adrenal crisis at the age of 2 months resulting from poor adherence to medication.
Mutation Spectrum of the STAR Gene
Among the 45 patients from 42 independent pedigrees, eight different STAR mutations were identified (Supplementary Table S1). The most common mutation was c.772C > T (p.Q258*), which was identified in 74 of 84 alleles (88.1%) from 44 patients from 41 unrelated families. Two siblings from two unrelated pedigrees harbored a homozygous p.Q258* mutation. Six previously reported mutations were detected in nine alleles: c.545G > A (p.R182H) (1/84, 1.2%), c.543C > T (p.R182C) (1/84, 1.2%), c.559G > A (p.V187M) (2/84, 2.4%), c.815G > A (p.R272H) (2/84, 2.4%), c.653C > T (2/84, 2.4%) and c.745-6_810del (1/84, 1.2%) (5,13,15,21). The p.R182C mutation was found in twin siblings as a compound heterozygote with p.Q258* (22). The c.653C > T mutation, previously known as the p.A218V missense mutation, was reported to cause aberrant splicing, resulting in skipping of exon 6 or exons 5 and 6 (15). The c.745-6_810del mutation also causes aberrant splicing, leading to full or partial intron 6 retention (15). A novel frame-shift mutation was identified: c.407delA (p.E136Gfs*50), which was not found in the normative population databases, including the US National Heart, Lung and Blood Institute Exome Sequencing Project Exome Variant Server (https://doi.org/eversusgs.washington.edu/EVS/) and the Genome Aggregation Database (https://doi.org/gnomad.broadinstitute.org/).
Shared Haplotype and Estimated Age of the Mutation
Ten unrelated patients were analyzed using SNP and/or STR markers in a 2,854 kb region surrounding the p.Q258* mutation. Among the 10 patients, the p.Q258* mutation was identified in 17 of 20 alleles (85%). Seven patients were homozygous for the p.Q258* mutation, while the remaining three patients were compound heterozygotes for p.Q258* and other mutations (p.V187M, p.R272H and p.E136Gfs*50, respectively). The clinical and endocrinologic characteristics of the 10 patients are summarized in Table 1.
Their genotypes were analyzed manually to determine the most likely haplotypes carried by each of the 17 alleles with the p.Q258* mutation. The minimum size of the shared haplotype was approximately 198 kb (chromosome 8: 37,815,009–38,013,848) (Figure 2). All 17 alleles containing the p.Q258* mutation shared a common haplotype surrounding this mutation (Supplementary Table S2). The founder haplotype was present in 8.5% of the JPT population from the International HapMap database (https://doi.org/hapmap.ncbi.nlm.nih.gov/). The probability that the p.Q258* occurred recurrently as an independent de novo event, which would indicate a hot spot mutation, was extremely low (binomial probability of 6.311 × 10−19). These results suggest that the p.Q258* mutation was inherited from a common ancestor. The estimated age of the mutation was 195 generations (95% credible set; 143–317 generations); i.e., 4,875 years (95% credible set: 3,575–7,925 years), based on an intergeneration time of 25 years (18).

Haplotypes of the alleles with p.Q258* mutation in the STAR gene. The gray vertical line illustrates the location of the mutation, and the horizontal black bars indicate regions of the shared haplotype in each allele. The minimum size of the shared haplotype is approximately 198 kb. P, paternal allele; M, maternal allele.
Discussion
This study demonstrated that the p.Q258* mutation is a founder mutation that has been inherited from a common ancestor. All individuals carrying the p.Q258* mutation in the STAR gene shared a common haplotype encompassing the mutation. In this regard, CLAH is not a particularly rare disease in Asian countries, especially in Korea, where the estimated carrier frequency of STAR mutation is high. In the present study, among eight different STAR mutations, including one novel frameshift mutation, the p.Q258* mutation accounted for 88.1% of alleles, indicating a lower prevalence compared with our previous report (92%) (15). Most patients were classical CLAH, with severe adrenal insufficiency in the neonatal period, while nonclassical late-onset CLAH was identified rarely.
The genetic testing of STAR allows an accurate diagnosis of CLAH, which is necessary for timely life-saving treatment, prediction of clinical course and genetic counseling. Considering the 1 in 170–250 estimated carrier frequency of the p.Q258* mutation in the Korean population (19,23), the founder effect of this mutation has important clinical implications for the cost-effective and rapid genetic diagnosis of CLAH in Korea. The presence of a p.Q258* mutation in one allele in a symptomatic patient would increase the likelihood of the disease and suggest a second mutation in the other allele.
The frequency of disease-associated mutations in specific human populations can be influenced by multiple factors, including natural selection, genetic drift and migration (16,24). Although disease-causing mutations are typically deleterious, certain mutations could survive because of a selective advantage for a specific environment, a heterozygote advantage or environmental exposure (25,26). Genetic drift is another force that results in different frequencies of disease-causing mutations in human populations, and this effect is greater in small and isolated populations (26).
Establishing a founder effect aids cost-effective population-based screening of disease susceptibility genes. Targeted screening for three founder mutations (BRCA1 c.68_69delAG, c.5266dupC and BRCA2 c.5946delT) in the Ashkenazi Jewish population could be cost-effective and technically simple and would allow a clear interpretation to diagnose and predict the disease risk (27–29).
Studying the founder effect can provide a detailed analysis of a population’s evolution and its migration pathways. The Korean population has been settled in the Korean peninsula as a single nation for a long time, and several recurrent mutations have been identified as founder mutations. For example, the type 3 familial hemophagocytic lymphohistiocytosis (FLH3) c.118-308C > T and c.754-1G > C mutations, which account for 38% and 26%, respectively, of all mutant alleles, were shown to be founder mutations by haplotype analysis (30). The CDH23 p.P240L mutation in autosomal recessive sensorineural hearing loss, which is predominant in Japanese and Korean populations, was shown to be a founder mutation by genotyping using STR markers (31). Recently, newborn screening of inherited metabolic diseases based on next-generation sequencing identified several founder mutations, such as p.Y138* in DUOXA2, p.R885Q in DUOX2, p.Y439C in PCCB, p.R285Pfs*2 in SLC25A13 and p.R224Q in GALT (32).
Some founder mutations identified in the Korean population are shared with other Asian countries, especially Japan and China. The p.Q258* mutation in STAR has been reported only in Korean, Japanese and Chinese populations, and appears to have resulted from a founder effect (13–15). These countries are geographically adjacent and are influenced culturally by the movement of people. The estimated age of the p.Q258* mutation is about 5,000 years, which corresponds to the Neolithic age, the period during which agriculture began in settled communities. The distribution patterns of such founder mutations might help determine the estimated migration pathways of Asian ancestry populations. However, the time of origin of most founder mutations is unknown in Korea. Thus, further studies on the prevalence of shared mutations in Asian countries, and haplotype analyses in other ethnic groups, would be helpful to trace the common ancestor.
There are limitations to this study. The frequency of shared haplotypes in the general population was compared with that in the JPT population because of a lack of information in the Korean population. In addition, the prevalence of CLAH in the Korean population was based on the estimated carrier frequency (19), which could influence the estimation of the age of mutation.
Conclusion
In conclusion, haplotype analysis confirmed that the c.772C > T (p.Q258*) mutation in STAR is a Korean founder mutation. The mutation spectrum of the STAR gene in Korean patients is homogeneous because of the founder effect. Identification of a founder effect enables easier identification of at-risk groups.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Lin D, et al. (1995) Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 267:1828–31.
Miller WL. (2017) Disorders in the initial steps of steroid hormone synthesis. J. Steroid Biochem. Mol. Biol. 165:18–37.
Bhangoo A, Anhalt H, Ten S, King SR. (2006) Phenotypic variations in lipoid congenital adrenal hyperplasia. Pediatr. Endocrinol. Rev. 3:258–71.
Miller WL, Bose HS. (2011) Early steps in steroidogenesis: intracellular cholesterol trafficking. J. Lipid Res. 52:2111–35.
Baker BY, et al. (2006) Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J. Clin. Endocrinol. Metab. 91:4781–5.
Metherell LA, et al. (2009) Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. J. Clin. Endocrinol. Metab. 94:3865–71.
Sugawara T, et al. (1995) Structure of the human steroidogenic acute regulatory protein (StAR) gene: StAR stimulates mitochondrial cholesterol 27-hydroxylase activity. Biochemistry. 34:12506–12.
Prasad M, et al. (2017) Mitochondrial metabolic regulation by GRP78. Sci. Adv. 3:e1602038.
Bose HS, Sugawara T, Strauss JF 3rd, Miller WL, Intl. Congenital Lipoid Adrenal Hyperplasia Consortium. (1996) The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N. Engl. J. Med. 335:1870–8.
Abdulhadi-Atwan M, et al. (2007) Role of a founder c.201_202delCT mutation and new phenotypic features of congenital lipoid adrenal hyperplasia in Palestinians. J. Clin. Endocrinol. Metab. 92:4000–8.
Chen X, Baker BY, Abduljabbar MA, Miller WL. (2005) A genetic isolate of congenital lipoid adrenal hyperplasia with atypical clinical findings. J. Clin. Endocrinol. Metab. 90:835–40.
Fluck CE, et al. (2005) A novel mutation L260P of the steroidogenic acute regulatory protein gene in three unrelated patients of Swiss ancestry with congenital lipoid adrenal hyperplasia. J. Clin. Endocrinol. Metab. 90:5304–8.
Nakae J, et al. (1997) Analysis of the steroidogenic acute regulatory protein (StAR) gene in Japanese patients with congenital lipoid adrenal hyperplasia. Hum. Mol. Genet. 6:571–6.
Huang Z, et al. (2016) Identification of five novel STAR variants in ten Chinese patients with congenital lipoid adrenal hyperplasia. Steroids. 108:85–91.
Kim JM, et al. (2011) High allele frequency of the p.Q258X mutation and identification of a novel mis-splicing mutation in the STAR gene in Korean patients with congenital lipoid adrenal hyperplasia. Eur. J. Endocrinol. 165:771–8.
Cavalli-Sforza LL, Feldman MW. (2003) The application of molecular genetic approaches to the study of human evolution. Nat. Genet. 33 Suppl:266–75.
Avbelj Stefanija M, et al. (2012) An ancient founder mutation in PROKR2 impairs human reproduction. Hum. Mol. Genet. 21:4314–24.
Slatkin M, Rannala B. (1997) Estimating the age of alleles by use of intraallelic variability. Am. J. Hum. Genet. 60:447–58.
Song MJ, et al. (2012) Estimation of carrier frequencies of six autosomal-recessive Mendelian disorders in the Korean population. J. Hum. Genet. 57:139–44.
Jin HY, et al. (2011) Ovarian cyst torsion in a patient with congenital lipoid adrenal hyperplasia. Eur. J. Pediatr. 170:535–8.
Achermann JC, et al. (2001) Molecular and structural analysis of two novel StAR mutations in patients with lipoid congenital adrenal hyperplasia. Mol. Genet. Metab. 73:354–7.
Park HW, Kwak BO, Kim GH, Yoo HW, Chung S. (2013) p.R182C mutation in Korean twin with congenital lipoid adrenal hyperplasia. Ann. Pediatr. Endocrinol. Metab. 18:40–3.
Yoo HW, Kim GH. (1998) Molecular and clinical characterization of Korean patients with congenital lipoid adrenal hyperplasia. J. Pediatr. Endocrinol. Metab. 11:707–11.
Gamelon M, et al. (2017) Interactions between demography and environmental effects are important determinants of population dynamics. Sci. Adv. 3:e1602298.
Williams TN, et al. (2005) Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J. Infect. Dis. 192:178–86.
Lynch M, et al. (2016) Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 17:704–14.
Foulkes WD, Knoppers BM, Turnbull C. (2016) Population genetic testing for cancer susceptibility: founder mutations to genomes. Nat. Rev. Clin. Oncol. 13:41–54.
Roa BB, Boyd AA, Volcik K, Richards CS. (1996) Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat. Genet. 14:185–7.
Hartge P, Struewing JP, Wacholder S, Brody LC, Tucker MA. (1999) The prevalence of common BRCA1 and BRCA2 mutations among Ashkenazi Jews. Am. J. Hum. Genet. 64:963–70.
Seo JY, et al. (2013) Founder effects in two predominant intronic mutations of UNC13D, c.118-308C>T and c.754-1G>C underlie the unusual predominance of type 3 familial hemophagocytic lymphohistiocytosis (FHL3) in Korea. Ann. Hematol. 92:357–64.
Kim SY, et al. (2015) Strong founder effect of p.P240L in CDH23 in Koreans and its significant contribution to severe-to-profound nonsyndromic hearing loss in a Korean pediatric population. J. Transl. Med. 13:263.
Park KJ, et al. (2016) A Population-Based Genomic Study of Inherited Metabolic Diseases Detected Through Newborn Screening. Ann. Lab. Med. 36:561–72.
Acknowledgments
This work was supported by a grant (2015-01) from the Korean Society of Pediatric Endocrinology.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Kang, E., Kim, YM., Kim, GH. et al. Mutation Spectrum of STAR and the Founder Effect of p.Q258* in Korean Patients with Congenital Lipoid Adrenal Hyperplasia. Mol Med 23, 149–154 (2017). https://doi.org/10.2119/molmed.2017.00023
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2017.00023