
Abstract
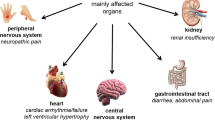
Fabry disease is an X-linked lysosomal storage disease caused by deficiency of the enzyme α-galactosidase A and results in pain, progressive renal impairment, cardiomyopathy, and cerebrovascular disease. The results of two major randomized, double-blind, placebo-controlled clinical trials and open-label extensions have shown that replacement of the deficient enzyme with either of two preparations of recombinant human α-galactosidase A, agalsidase-alfa, and agalsidase-beta is safe. Biweekly IV infusions of 0.2 mg/kg of agalsidase-alfa were associated with a significant decrease in pain and stabilization of renal function. Biweekly infusions of 1 mg/kg of agalsidase-beta were associated with virtually complete clearing of accumulated glycolipid substrate from renal and cutaneous capillary endothelial cells. Several smaller, open-label studies, along with observations made in the course of monitoring large numbers of patients on enzyme replacement therapy, indicated that treatment stabilizes renal function and produces significant improvements in myocardial mass and function. Treatment of Fabry disease by enzyme replacement has a significant impact on at least some serious complications of the disease.
Similar content being viewed by others


References
Desnick R. J., Ioannou Y. A., and Eng C. M. (2001) α-Galactosidase A deficiency: Fabry disease. In: The Metabolic and Molecular Bases of Inherited Disease, 8th ed, Scriver C. R., Beaudet A. L., Sly W. S., and Valle D., eds., McGraw-Hill, New York, NY, pp. 3733–3774.
Brady R. O. (1967). Enzymatic abnormalities in diseases of sphingolipid metabolism. Clin. Chem. 13, 565–577.
Clarke J. T. R, Wolfe L. S., and Perlin A. S. (1971). Evidence for a terminal α-d-galactopyranosyl residue in galactosylgalactosylglucosylceramide from human kidney. J. Biol. Chem. 246, 5563–5569.
Kint J. A. (1970). Fabry’s disease: alpha-galactosidase deficiency. Science 167, 1268,1269.
MacDermot K. D., Holmes A., and Miners A. H. (2001). Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J. Med. Genet. 38, 750–760.
MacDermot K. D., Holmes A., and Miners A. H. (2001). Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet. 38, 769–775.
Mehta A., Ricci R., Widmer U., et al. (2004). Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survery. Eur. J. Clin. Invest. 34, 236–242.
Cable W. J., Dvorak A. M., Osage J. E., and Kolodny E. H. (1982). Fabry disease: significance of ultrastructural localization of lipid inclusions in dermal nerves. Neurology 32, 347–353.
Clarke J. T. R, Knaack J., Crawhall J. C., and Wolfe L. S. (1971). Ceramide trihexosidosis (Fabry’s disease) without skin lesions. N. Engl. J. Med. 284, 233–235.
Mitsias P. and Levine S. R. (1996). Cerebrovascular complications of Fabry’s disease. Ann. Neurol. 40, 8–17.
Giacomini P. S., Shannon P. T., Clarke J. T. R., and Jaigobin C. (2004). Fabry’s disease presenting as stroke in a young female. Can. J. Neurol. Sci. 31, 112–114.
Alroy J., Sabnis S., and Kopp J. B. (2002). Renal pathology in Fabry disease. J. Am. Soc. Nephrol. 13, S134-S138.
Branton M., Schiffmann R., and Kopp J. B. (2002). Natural history and treatment of renal involvement in Fabry disease. J. Am. Soc. Nephrol. 13 (Suppl 2), S139-S143.
Nakao S., Kodama C., Takenaka T., et al. (2003). Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 64, 801–807.
Kotanko P., Kramar R., Devrnja D., et al. (2004). Results of nationwide screening for Anderson-Fabry disease among dialysis patients. J. Am. Soc. Nephrol. 15, 1323–1329.
Kampmann C., Wiethoff C. M., Perrot A., Beck M., Dietz R., and Osterziel K. J. (2002). The heart in Anderson Fabry disease. Z. Kardiol. 91, 786–795.
Linhart A., Lubanda J. C., Palecek T., et al. (2001). Cardiac manifestations in Fabry disease. J. Inherit. Metab. Dis. 24 (Suppl 2), 75–83.
Cantor W. J., Daly P., Iwanochko M., Clarke J. T. R., Cusimano R. J., and Butany J. (1998). Cardiac transplantation for Fabry’s disease. Can. J. Cardiol. 14, 81–84.
Sachdev B., Takenaka T., Teraguchi H., et al. (2002). Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 105, 1407–1411.
Chimenti C., Pieroni M., Morgante E., et al. (2004). Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation 110, 1047–1053.
Miners A. H., Holmes A., Sherr L., Jenkinson C., and MacDermot K. D. (2002). Assessment of healthy-related quality-of-life in males with Anderson Fabry disease before therapeutic intervention. Qual. Life Res. 11, 127–133.
Gold K. F., Pastores G. M., Botteman M. F., et al. (2002). Quality of life of patients with Fabry disease. Qual. Life Res. 11, 317–327.
Ries M., Ramaswami U., Parini R., et al. (2003). The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur. J. Pediatr. 162, 767–772.
Desnick R. J. and Brady R. O. (2004). Fabry disease in childhood. J. Pediatr. 144, S20-S26.
Clarke J. T. R., Guttmann R. D., Wolfe L. S., Beaudoin J. G., and Morehouse D. D. (1972). Enzyme replacement therapy by renal allotransplantation in Fabry’s disease. N. Engl. J. Med. 287, 1215–1218.
Brady R.O. (1967). Enzymatic abnormalities in diseases of sphingolipid metabolism. Clin. Chem. 13, 565–577.
Sly W.S. (2004). Enzyme replacement therapy for lysosomal storage disorders: successful transition from concept to clinical practice. Mo. Med. 101, 100–104.
Brady R. O., Tallman J. F., Johnson W. G., et al. (1973). Replacement therapy for inherited enzyme deficiency. Use of purified ceramidetrihexosidase in Fabry’s disease. N. Engl. J. Med. 289, 9–14.
Furbish F. S., Blair H. E., Shiloach J., Pentchev P. G., and Brady R. O. (1977) Enzyme replacement therapy in Gaucher’s disease: large-scale purification of glucocerebrosidased suitable for human administration. Proc. Natl. Acad. Sci. USA 74, 3560–3563.
Doebber T. W., Wu M. S., Bugianesi R. L., et al. (1982). Enhanced macrophage uptake of synthetically glycosylated human placental α-glucocerebrosidase. J. Biol. Chem. 257, 2193–2199.
Beutler E. (1997). Enzyme replacement therapy for Gaucher’s disease. Baillieres Clin. Haematol. 10, 751–763.
Grabowski G. A., Leslie N, and Wenstrup R. (1998). Enzyme therapy for Gaucher disease: the first 5 years. Blood Rev. 12, 115–133.
Grabowski G. A., Barton N. W., Pastores G., et al. (1995). Enzyme therapy in type I Gaucher disease. Comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann. Int. Med. 122, 33–39.
Weinreb N. J., Charrow J., Andersson H. C., et al. (2002). Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am. J. Med. 113, 112–119.
Eng C. M., Guffon N., Wilcox W. R., et al. (2001). Safety and efficacy of recombinant human a-b A replacement therapy in Fabry’s disease. N. Engl. J. Med. 345, 9–16.
Schiffmann R., Kopp J. B., Austin H. A., III et al. (2001). Enzyme replacement therapy in Fabry disease. A randomized controlled trial. JAMA 285, 2743–2749.
Moore D. F., Scott L. T., Gladwin M. T., et al. (2001). Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease: reversal by enzyme replacement therapy. Circulation 104, 1506–1512.
Moore D. F., Altarescu G., Herscovitch P., and Schiffmann R. (2002). Enzyme replacement reverses abnormal cerebrovascular responses in Fabry disease. BMC Neurology 2, 4.
Moore D. F., Altarescu G., Ling G. S., et al. (2002). Elevated cerebral blood flow velocities in Fabry disease with reversal after enzyme replacement. Stroke 33, 525–531.
Moore D. F., Ye F., Brennan M. -L., et al. (2004). Ascorbate decreases Fabry cerebral hyperperfusion suggesting a reactive oxygen species abnormality: an areterial spin tagging study. J. Magn. Reson. Imaging 20, 674–683.
Thurberg B. L., Rennke H., Colvin R. B., et al. (2002). Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 62, 1933–1946.
De Schoenmakere G., Chauveau D., and Grunfeld J. P. (2003). Enzyme replacement therapy in Anderson-Fabry’s disease: beneficial clinical effect on vital organ function. Nephrol. Dial. Transplant. 18, 33–35.
Dehout F., Schwarting A., Beck M., Mehta A., Ricci R., and Widmer U. (2003). Effects of enzyme replacement therapy with agalsidase alfa on glomerular filtration rate in patients with Fabry disease: preliminary data. Acta Paediatr. Suppl. 92, 14–15.
Weidemann F., Breunig F., Beer M., et al. (2003). Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease. A prospective strain rate imaging study. Circulation 108, 1299–1301.
Mignani R., Panichi V., Giudicissi A., et al. (2004). Enzyme replacement therapy with agalsidase beta in kidney transplant patients with Fabry disease: a pilot study. Kidney Int. 65, 1381–1385.
Baehner F., Kampmann C., Whybra C., Miebach E., Wiethoff C. M., and Beck M. (2003). Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J. Inherit. Metab. Dis. 26, 617–627.
Schiffmann R., Floeter M. K., Dambrosia J. M., et al. (2003). Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve 28, 703–710.
Hilz M. J., Brys M., Marthol H., Stemper B., and Dutsch M. (2004). Enzyme replacement therapy improves function of C-, Aδ- and Aβ-nerve fibers in Fabry neuropathy. Neurology 62, 1066–1072.
Hajioff D., Enever Y., Quiney R., Zuckerman J., MacDermot K., and Mehta A. (2003). Hearing loss in Fabry disease: the effect of agalsidase alfa replacement therapy. J. Inherit. Metab. Dis. 26, 787–794.
Hajioff D., Goodwin S., Quiney R., Zuckerman J., MacDermot K. D., and Mehta A. (2003). Hearing improvement in patients with Fabry disease treated with agalsidase alfa. Acta Paediatr. Suppl. 92, 28–30.
Palla A., Widmer U., and Straumann D. (2003). Head-impulse testing in Fabry disease—vestibular function in male and female patients. Acta Paediatr. Suppl. 92, 38–42.
Dehout F., Roland D., Treille de Granseigne S., Guillaume B., and Van Maldergem L. (2004). Relief of gastrointestinal symptoms under enzyme replacement therapy of patients with Fabry disease. J. Inherit. Metab. Dis. 27, 499–505.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clarke, J.T.R., Iwanochko, R.M. Enzyme replacement therapy of fabry disease. Mol Neurobiol 32, 43–50 (2005). https://doi.org/10.1385/MN:32:1:043
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1385/MN:32:1:043