
Abstract
Pure glyceryl mono-oleate (GMO) (lipid) and different batches of GMO commonly used for the preparation of GMO-chitosan nanoparticles were characterized by modulated differential scanning calorimetry (MDSC), cryo-microscopy, and cryo-X-ray powder diffraction techniques. GMO-chitosan nanoparticles containing poloxamer 407 as a stabilizer in the absence and presence of polymers as crystallization inhibitors were prepared by ultrasonication. The effect of polymers (polyvinyl pyrrolidone (PVP), Eudragits, hydroxyl propyl methyl cellulose (HPMC), polyethylene glycol (PEG)), surfactants (poloxamer), and oils (mineral oil and olive oil) on the crystallization of GMO was investigated. GMO showed an exothermic peak at around −10°C while cooling and another exothermic peak at around −12°C while heating. It was followed by two endothermic peaks between 15 and 30 C, indicative of GMO melting. The results are corroborated by cryo-microscopy and cryo-X-ray. Significant differences in exothermic and endothermic transition were observed between different grades of GMO and pure GMO. GMO-chitosan nanoparticles resulted in a significant increase in particle size after lyophilization. MDSC confirmed that nanoparticles showed similar exothermic crystallization behavior of lipid GMO. MDSC experiments showed that PVP inhibits GMO crystallization and addition of PVP showed no significant increase in particle size of solid lipid nanoparticle (SLN) during lyophilization. The research highlights the importance of extensive physical-chemical characterization for successful formulation of SLN.
Similar content being viewed by others


INTRODUCTION
Solid lipid nanoparticles (SLN) are nanoparticles, usually in the size range of 50–1000 nm, prepared by lipids that are solid at room temperatures. These lipids are usually purified triglycerides, complex glyceride mixtures, or even waxes, and are stabilized in solution by utilizing appropriate surfactants (1,2). SLN not only combines the advantages of other innovative carrier systems, including physical stability, protection of incorporated labile drugs from degradation, and controlled release, but also have excellent tolerability and site-specific targeting (2–6). Recent advancements in solid lipid nanoparticles include nanostructured lipid carriers (NLC), which are prepared by combining liquid lipids with solid lipids to control the properties of lipid matrix and drug release rate and enhance drug loading. These NLCs can be classified as three different types: consisting of (1) low oil, (2) high oil, and (3) amorphous matrix due to the combination of solid and liquid lipids (7,8).
SLNs are composed mostly of lipids, which dictate the stability, release, entrapment, and loading of drug in the formulation. One of the key challenges associated with the development of SLN preparation is the physicochemical characterization of lipids and the changes these lipids undergo during processing (9). Lipids of different purity and composition have resulted in significant variation in the physicochemical property and performance of formulations (10,11). SLNs are usually prepared using melt methods, via ultrasonication or high-pressure homogenization, which can result in the transformation or degradation of the lipids (12).
Another key challenge associated with SLN is to remove water to achieve maximum physical and chemical stability. Freeze-drying is commonly used to convert lipid emulsions into solids of sufficient physical stability for distribution and storage. The process involves the removal of water from the SLN by the aid of sublimation and desorption under vacuum. Freeze-drying offers many advantages, such as product elegance, long-term physical and chemical stability, and ease of reconstitution (13). In spite of these innumerable advantages offered by freeze-drying, the biggest shortcoming of the method is the increase in particle size due to aggregation of lipid nanoparticles (14).
Thus, there is a need to emphasize the selection of the appropriate grade of lipids utilized in the preparation of SLN at the early stages and its thorough physical-chemical characterization before and after lyophilization. The objective of this paper is to (1) prepare and characterize components of glyceryl mono-oleate-chitosan-based freeze-dried SLN; (2) prepare and characterize glyceryl mono-oleate (GMO) nanoparticles before and after freeze-drying; (3) understand particle size increase during freeze-drying; and (4) highlight the importance of physical-chemical characterization and formulation considerations for the preparation of SLN.
MATERIALS AND METHODS
Materials Used
Two batches of GMO, batch number G1017 (lot numbers ZG0467 and WA1499) were purchased from Spectrum Chemicals; pure GMO was purchased from Sigma-Aldrich. Poloxamer 407 was purchased from Spectrum Chemicals, whereas low molecular weight chitosan and citric acid were purchased from Sigma-Aldrich. Povidone K 90 (polyvinyl pyrrolidone) was purchased from BASF Chemicals. Polyethylene glycols (PEG), Eudragit E100, Eudragit L100, Eudragit S100, hydroxypropyl methyl cellulose (HPMC), mineral oil, and olive oil purchased from Fisher Scientific. Deionized water was used for all the experiments. Physical mixtures used for comparative analysis were prepared by geometrically mixing the lipids/excipients in the same amount as utilized in the preparation of solid lipid nanoparticles.
Methods
Characterization by Modulated Differential Scanning Calorimetry
Thermal analysis of lipid (GMO) was conducted by modulated differential scanning calorimetry (MDSC) (TA instruments Q 2000 series, New Castle, DE) equipped with a cooling assembly. Samples of 5–10 mg were prepared in sealed pans. The nitrogen flow rate was kept at 40 ml/min. Initially, the temperature was increased from room temperature to 60°C at the ramp rate of 2°C/min with modulation at ±0.32 s every 60 s. This marked the end of cycle 1. Then, the system was brought down to −40°C at the ramp rate of 2°C/min, which marked the end of cycle 2. The temperature was again raised to 60°C to end cycle 3. Finally, samples were cooled down to 30°C at the ramp rate of 5°C/min to mark the end of the method. The data was analyzed by TA universal analysis (New Castle, DE) software. Detailed analysis of GMO was carried out using MDSC in various heat-cool-heat cycles to identify and correlate the exothermic and endothermic transitions. Experimental conditions utilized in these cycles are listed in Table I.
Characterization by Cryo-X-Ray Diffraction
X-ray diffractometry (CXD) was performed using a powder X-ray diffractometer (D8 ADVANCE, Bruker AXS, Madison, WI, USA) equipped with a variable temperature stage (TTK 450; Anton Paar, Graz-Straβgang, Austria) and Si strip one-dimensional detector (LynxEye; Bruker AXS). The temperature of the stage was controlled by a combination of liquid nitrogen and a resistive heater. A powder sample was placed on a specially designed sample holder made of copper. The temperature of the sample stage was monitored and controlled by platinum resistance temperature detectors (RTD). The environment of the sample chamber was controlled by flowing N2 gas. X-ray diffraction (XRD) data were taken after equilibrating the sample at the desired temperature. XRD measurements were made by exposing the sample to Cu Kα radiation (40 kV × 40 mA). The angular range was 5–400 2θ with a step size of 0.050 and a dwell time of 1 s.
Initial characterization of GMO by heat-cool-heat cycles of MDSC revealed broad melting of GMO (5–35°C) and two exothermic events at −10.50 and −11.94°C, respectively. The following procedure by Cryo-XRD method was designed to characterize and confirm these specific endothermic and exothermic transitions.
The procedure was as follows: (1) GMO sample was heated to 40°C and kept at 40°C for 5 min to ensure complete melting. XRD was taken at 40°C. (2) Sample was cooled at 2°C/min to −40°C. XRD was taken at 0, −12, −20, and −40°C during cooling of the sample to characterize changes observed as exothermic transition in MDSC. (3) Sample was heated at 2°C/min to 40°C. XRD was taken at −12, 0, 15, 25, and 40°C during heating cycle to characterize changes observed as exothermic and endothermic transition in MDSC.
Characterization by Cryo-microscopy
A Leica microscope was attached to a LINKAM TMS Hot stage LTS 350 with LINKAM LNP to control liquid nitrogen and assist in cooling the sample. A sample of pure GMO was removed from −20°C refrigeration and kept in the stage equilibrated to −20°C using liquid nitrogen. The sample was heated at 2°C/min to 40°C and equilibrated for 1 min. The sample was then again cooled to −40°C at 2°C/min and equilibrated for 1 min. The sample was then heated to 40°C at 2°C/min. Sample pics were taken every minute under plain polarized light (exposure 642.9; sat 1:30: gamma 0.68; gain 1.4×; 20×).
Characterization by Solid State Nuclear Magnetic Resonance
Solid state nuclear magnetic resonance (ss-NMR) spectra were acquired on a Bruker 400 MHz proton frequency wide bore spectrometer. Before obtaining carbon spectra, proton relaxation longitudinal relaxation times (1H T1) were determined by fitting proton saturation recovery data to an exponential function. These values were used to set an optimal recycle delay of carbon cross-polarization magic angle spinning experiment (13C CPMAS), which typically was set between 1.2 × 1H T1 and 1.5 × 1H T1. The carbon spectra were acquired with 2 ms contact time using a linear amplitude ramp on the proton channel (from 50 to 100%) and approximately 100 kHz SPINAL-64 decoupling. The typical magic angle spinning (MAS) speed was 12.5 kHz. To limit a frictional heating of the sample due to fast spinning, the probe head temperature was maintained at 275 K. Carbon spectra were referenced externally by setting the upfield resonance of solid phase sample of adamantane to 29.5 ppm. Using this procedure, carbon spectra were indirectly referenced to tetramethyl silane at 0 ppm.
Preparation of GMO-Chitosan Nanoparticles
The preparation method developed by Trickler et al. was used for the preparation of GMO-chitosan nanoparticles (15). In brief, 1.75 ml of GMO was melted and added to 12.5 ml of 1% w/v poloxamer 407 solution. This mixture was subjected to ultrasonication at 18 W for 2 min using a Misonix 3000 sonicator. This led to the formation of the primary emulsion. Thereafter, 12.5 ml of 2.4% w/v chitosan solution in 100 mM of citric acid solution was added to the primary emulsion. It was again ultrasonicated using the same parameters as used earlier to form the secondary emulsion.
Freeze-Drying of GMO-Chitosan Nanoparticles
The nanoemulsions prepared were freeze-dried in a Millrock Technologies freeze dryer to form dry, free-flowing powder. The freezing step involved cooling the sample from room temperature to −50°C, which was followed by primary drying. In this step, nanoemulsions were dried by increasing the temperature from −20 to 20°C. In the final secondary drying step, the samples were held at 20°C for 240 min until they were completely devoid of water.
Particle Size and Zeta Potential Measurement
The particle size of the nanoemulsion both before and after freeze-drying was determined by photon correlation spectroscopy (PCS) using ZetaPlus (Brookhaven Instruments Corporation) at 25°C. Each sample was diluted in the ratio of 1:1000 with deionized water before the measurement. Each measurement was performed in triplicate.
Crystallization Inhibitor Screening by MDSC
Excipients poloxamer 407, PEG, Eudragit E100, Eudragit L100, Eudragit S100, HPMC, mineral oil, olive oil, and PVP were screened for crystallization inhibition efficiency of GMO. Physical mixtures (70:30% w/w) of GMO and polymers were prepared by geometric dilution. A MDSC Q2000 Series from TA Instruments was used to analyze the changes in the exothermic crystallization events of GMO. A sample of 5–8 mg in a hermetically sealed aluminum pan with a nitrogen flow rate of 40 ml/min was used in the experiment. The temperature was increased from room temperature to 60°C at the heating rate of 2°C/min. The system then was cooled to −40°C at the cooling rate of 2°C/min. At the end, the temperature was raised to 60°C. Finally, samples were cooled to 30°C at the cooling rate of 5°C/min to mark the end of the method.
Preparation of Freeze-Dried Nanoparticles Containing PVP as Crystallization Inhibitor
GMO (1.75 ml) was melted at 40°C. PVP K90 1% (w/v) was added as a crystallization inhibitor to the molten GMO. Then, 12.5 ml of 2% w/v poloxamer 407 solution was added as stabilizer so the final poloxamer concentration was 1% w/v. This mixture was subjected to ultrasonication at 18 W for 2 min using a Misonix 3000 sonicator. This led to the formation of the primary emulsion. Thereafter, 12.5 ml of 2.4% w/v chitosan solution in 100 mM of citric acid solution was added to the primary emulsion. It was further ultrasonicated using the same parameters used earlier to form the secondary emulsion. The samples containing PVP as a crystallization inhibitor were freeze-dried in a Millrock Technologies Freeze dryer to form dry, free-flowing powder using a similar freeze-drying cycle to that described in “Freeze-Drying of GMO-Chitosan Nanoparticles” section.
RESULTS AND DISCUSSION
Physical-Chemical Characterization of Lipid GMO
MDSC was used extensively for characterization of GMO, since variations in its chemical composition can be reflected in its melting behavior. The processing and manufacturing of SLN involves melting and congealing of GMO; any degradation/transformation can be reflected in heat-cool-heat cycles of MDSC. Figure 1 shows the exothermic and endothermic transitions observed during cycles 2 and 3 of MDSC. Cycle 1 is the heating of GMO at a constant rate to completely convert it from semi-solid to melted liquid. Since GMO exists in a semi-solid state at room temperature, this cycle was not used to determine the melting behavior of GMO. Cycles 2 and 3 represent the cooling and heating of GMO at a constant rate; details can be found in the experimental section. The first exothermic event at −10.50°C was observed during cycle 2. This event can be assigned to the crystallization of GMO. Additionally, the second exothermic event at −11.94°C during cycle 3 (heating of GMO) was also observed. This exothermic event was difficult to assign. It might be due either to the crystallization of GMO (remaining) during first cycle or to the rearrangement of GMO.

Exothermic and endothermic events observed in the MDSC of GMO. Cycle 2 represents the cooling cycle and cycle 3 represents the heating cycle. Cycle 1 is heating of GMO at constant rate to completely convert it from semi-solid to melted liquid and is not shown in the figure
Two broad endothermic events were observed during cycle 3 (heating of GMO) and both can be assigned to the melting of GMO. Lipids like GMO are known to exist in various polymorphic forms. Thus, the first endothermic event at 14.51°C can be due to the presence of a metastable polymorphic form of GMO. The second endothermic event 23.01°C can be due to the melting of GMO, which is similar to the reported values in the literature (16). This melting point corresponds to the most stable 1-monoolein, which is the beta form of GMO. GMO usually contains a mixture of monoglycerides, diglycerides, and triglycerides in varying proportion (17). The broad melting observed in GMO could be related to the presence of various glycerides. A drug delivery system containing GMO has been characterized by the thermal method; unfortunately, limited information is available on its crystallization behavior. This is the first attempt to extensively characterize GMO crystallization behavior, as the phenomenon is very important to developing stable SLN in the solid state. Table II summarizes the exothermic and endothermic events observed in cycle 2 and cycle 3.
Cryo-XRD experiments were used to identify and understand the nature of structural changes during heating and cooling of GMO. Cryo-XRD of melted GMO at 40°C showed an amorphous halo at a 2θ of 20° (Fig. 2). On cooling, X-ray diffraction at 0°C showed the crystallization of GMO. Further reduction in temperature to −20°C showed a large number of crystalline peaks (Fig. 2). During heating from −40 to 15°C, diffraction peaks appear to change from broad to sharp, possibly due to the rearrangement of GMO molecules. Sharp diffraction peaks disappears at 40°C, confirming the melting of crystallized GMO. Similarly, crystallization of GMO at around −12°C was observed in cryo-microscopy of melted GMO using polarized light microscopy. Two different types of crystals, individual and acicular, were formed (Fig. 3). No changes were observed during further cooling of GMO until −40°C. During heating, melting of acicular crystals started at around 6°C (Fig. 3). The melting of acicular crystals was followed by melting of individual crystals. Melting was completed between 25 and 30°C (Fig. 3). The observations from cryo-XRD and cryo-microscopy are in agreement with the observation in MDSC, except that no structural change is observed at −8 to −16°C (second exothermic event) during the heating cycle. Differential scanning calorimetry (DSC) has previously been successfully combined with other characterization techniques, such as microscopy, thermogravimetry, or XRD analysis for thorough characterization of SLN. A study of the drug olanzapine incorporated in glyceryl monostearate and glyceryl tristearate nanoparticles showed the absence of a melting endotherm using DSC (18). This corresponds to the fact that the drug is not in its crystalline form. The loss of crystalline form was confirmed using XRD, which showed the absence of sharp diffraction peaks.

Cryo-XRD of GMO. X-ray taken at different temperature during cooling of melted GMO. The temperatures are selected based on the endothermic and exothermic events observed in MDSC of GMO

Cryo-microscopy of GMO during cooling and heating of melted GMO. The figure represents the visual confirmation of crystallization and melting events of GMO with change in temperature
Correlation of Crystallization and Melting Transitions of GMO
The preparation, processing, and storage of SLN can change lipids present in these formulations into various polymorphic forms. These transitions can severely affect the physical stability and release profile of drugs from SLN (19). Slight variations in the heating and cooling of lipids during processing are unavoidable, so it is vital to understand the stability of lipids in these conditions. The relationship between exothermic and endothermic transition of GMO was explored by heating and cooling of GMO to various temperatures, as described in the experimental section. The exothermic peaks disappeared when GMO was cycled between 50 and 0–15°C. The appearance of new exothermic peak (5–10°C) while cooling was observed when GMO completed the first melting transition. The reappearance of old exothermic peaks (both cycles 1 and 2) was observed when the sample was allowed to undergo first complete and second partial melting transition. It appears that both the crystallization exothermic peaks are related to each other and to the second endothermic melting transition. These changes signify the complex thermal behavior of GMO during heat-cool-heat cycles, but confirm the absence of any irreversible polymorphic transition. In similar studies, C. Laura et al. performed thermal cycling (heat-cool-heat) on bulk lipids and identified α and β polymorphic forms present in SLN (20). A new exothermic peak during cooling disappeared when both melting transitions was completed. An exothermic transition similar to GMO (cycles 2 and 3, as shown in Fig. 1) was observed if the sample was melted above 35°C. The results are summarized in Fig. 4 and Table III.

Endothermic and exothermic events of GMO as seen during different heat-cool-heat cycles of MDSC. The analysis showed that heating and cooling of GMO can result in changes in the exothermic and endothermic transitions
Difference in the Crystallization and Melting Transitions in Various Grades of GMO
Significant differences in exothermic and endothermic transitions were observed in a different batch of GMO from the same manufacturer (Fig. 5). The first batch of GMO was solid at room temperature, whereas the second batch was semi-liquid at room temperature. Clear separation of the second batch of GMO into supernatant and sediment was visually observed. Two exothermic events, possibly crystallization at −9.54 and −22.54°C during the cooling cycle, were observed in this batch of GMO. It showed a broad melting peak at around 5°C. To further understand the observed exothermic and endothermic transitions of GMO, it was separated into supernatant and sediment by ultracentrifugation. However, similar exothermic and endothermic transitions were observed in both supernatant and sediment (Fig. 5). These variations in lipids can have significant effect on the integrity of SLN. Previously, variations in the physical and chemical characteristics of glyceryl monostearate obtained from various sources were identified as the principal factors influencing the stability of the formulation (10). Glyceryl monostearate used in the study was found to have wide range of variability in its constituent composition, which affected the consistency and quality of a cream formulation (21). Typically, glyceryl monostearate is the mixture of the monoglycerides of stearic and palmitic acids with small quantities of diglycerides and triglycerides, but can also contain small quantities of free stearic, palmitic, or oleic acids, as well as glycerol and water. It was found that raw materials from different sources resulted in the soft consistency and syneresis during the scale-up phase development of oil-in-water cream product (10).

MDSC thermograms of different batch of GMO. Two samples of each GMO, its supernatant and its sediment were shown. (1, 2) represents cooling cycles and (3, 4) of heating cycle of GMO, (5, 6) represents cooling cycles and (7, 8) of heating cycle of supernatant, (9, 10) represents cooling cycles and (11, 12) of heating cycle of sediment
Difference in the Crystallization and Melting Transitions in Various Grades of GMO Versus Highly Pure GMO
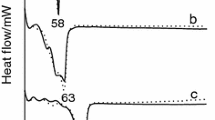
Most commercially available lipids used in SLN preparations are not pure and can result in significant differences in the properties of SLN. For example, commercial-grade stearates are permitted to contain a substantial amount of palmitate, although a relationship between the structure and function has not been extensively developed (22). Very few studies, including Sharpe et al., have developed and characterized the physical properties by preparing magnesium stearate and palmitate from chemically synthesized pure stearic and palmitic acids (23). Pure GMO showed sharp diffraction peaks at room temperature, confirming the crystalline nature of the lipid (Fig. 6a). A single exothermic transition at 9°C and a sharp endothermic melting peak at 35°C were observed in MDSC (Fig. 6b). Cryo-XRD confirmed the crystallization of pure GMO at around 10°C (during cooling) and melting at around 30°C (during heating) (Fig. 6c). The exothermic and endothermic transitions associated with pure GMO were significantly different than those associated with different grades of GMO. Cryo-microscopy also confirmed the crystallization and melting of pure GMO in a similar temperature range (data not shown). To explore the difference between pure GMO and GMO used in our studies, NMR was carried out; the results are presented in Fig. 7. Only slight a difference between pure GMO and lab-grade GMO was observed in NMR results. The one-dimensional spectra for 13°C when compared with lab-grade GMO showed that one glycerol peak is different; it is found to be a doublet in lab-grade GMO.

a XRD of pure GMO at room temperature; b MDSC of pure GMO; and c cryo-XRD at various temperatures during cooling and heating

NMR of pure GMO and lab-grade GMO used in our study. The arrow indicates the difference observed between pure GMO and lab-grade GMO
Physical-Chemical Characterization of GMO Nanoparticles After Freeze-Drying
The particle size of prepared GMO-chitosan nanoparticle before lyophilization was found to be 183.5 ± 11.6 nm. However, a significant increase in particle size to 253.9 ± 24.2 nm was observed when they were analyzed after lyophilization. A similar increase in particle size was observed by Saez et al. after freeze-drying of polycaprolactone and poly (D, L-lactic-glycolic) nanoparticles (13). The main reason behind the increase in particle size is attributed to the fact that two or more individual nanoparticles come together to form a secondary nanoparticle of larger particle size. When the primary solid lipid nanoparticles are suspended in water, they get surrounded by an aqueous layer. During the freeze-drying process without any treatment of cryoprotectants, the hydrated layer disappears due to sublimation. This results in the amalgamation of lipid layers, leading to an increase in the effective particle size (24).
Another phenomena resulting in particle size increase is known as Ostwald ripening. It usually takes place in emulsions and suspensions. Ostwald ripening is the growth of one emulsion droplet or a particle at the expense of another smaller droplet or particle. This is due to the differences in the chemical potentials of the materials within the droplets. This arises from the difference in the radius of the curvature of the drops (25). Katayoun et al. have studied the effects of hydrophobic excipients such as Drakeol 7 and Miglyol 812 in reducing the rate of Ostwald ripening in case of local anesthetic emulsions (26). Cryoprotectants, such as trehalose, glucose, mannitol, sucrose, etc., have been successfully employed in many cases to deal with the issue of an increase in the effective particle size during freeze-drying (27,28). In our case, this mechanism was ruled out since adding a cryoprotectant like trehalose showed no effect on the particle size increase of the solid lipid nanoparticles. Another possible reason for the particle size could be due to the crystallization of the lipid present in the SLN, which was explored further.
MDSC of freeze-dried GMO nanoparticles and their comparison with physical mixtures and GMO are presented in Fig. 8. MDSC thermograms of freeze-dried GMO nanoparticles showed a similar exothermic crystallization peak at around −15°C (cycle 2), as observed in their physical mixtures and GMO. This was followed by a similar exothermic peak at −10°C, followed by a broad melting endothermic peak of GMO between 10 and 20°C in both physical mixtures and a freeze-dried formulation (Fig. 8). MDSC of other excipients, such as chitosan, trehalose, etc., present in the nanoparticle formulation showed no exothermic or endothermic transition during this temperature range. This indicates that the endothermic and exothermic events are representative events of GMO with no interference from other excipients used in the preparation of GMO nanoparticles. Further, annealing at −40°C for 24 h after cycle 2 did not show any changes in the thermal behavior of GMO during cycle 3. It appears that the crystallization of GMO during lyophilization results in particle size increase of GMO nanoparticles.

Exothermic and endothermic events observed in the MDSC of pure GMO, GMO-chitosan physical mixture, and freeze-dried GMO nanoparticles
Effects of Polymers on Crystallization of GMO
Chauhan et al. studied the crystallization inhibition efficiency of amorphous drugs by utilizing various polymers using MDSC (29,30). They successfully ranked ordered polymers based on their ability to inhibit crystallization and found a strong correlation in the polymers’ ability to inhibit precipitation in solution and amorphous stabilization in the solid state. Based on a similar principle, GMO was physically mixed with various polymers and analyzed by MDSC to study the effects of polymers/surfactants/oils on GMO crystallization. MDSC results are presented in Fig. 9. It was observed that the physical mixtures of GMO with excipients like Eudragit E-100, HPMC, and PEG showed similar exothermic crystallization peaks as GMO. Similar exothermic crystallization peaks were also observed in the case of physical mixtures of GMO with mineral oil and olive oil (Fig. 9). However, a GMO-PVP physical mixture showed no exothermic crystallization peak, indicating the possible inhibition of GMO crystallization. The simulation of lyophilization conditions using MDSC in this case helped in the selection of polymers/stabilizers that effectively inhibited the crystallization of GMO. The process has the potential to help in the optimization of lyophlization conditions for the preparation of SLN.

a MDSC of GMO physical mixtures with PVP, Eudragit, HPMC, and PEG; b MDSC of GMO physical mixtures with poloxamer, mineral oil, and olive oil
Preparation and Characterization of Freeze-Dried GMO Nanoparticles Containing PVP K90
The particle sizes of nanoparticles before and after lyophilization in the formulation containing PVP were found to be 192.4 ± 7.5 and 204.2 ± 7.2 nm, respectively. From these results, it was observed that the particle size of GMO-chitosan solid lipid nanoparticles containing PVP K90 remained almost the same after freeze-drying.
Physicochemical Characterization and Formulation Consideration for Lipid Nanoparticles
GMO physical-chemical characterization revealed differences within batches of GMO; it is a significant possibility that SLN prepared from them will behave differently. This compromises the reproducibility and quality of SLN, and could be attributed to difficulty in appropriately characterizing lipids due to their complex nature, composition, and modification/transformation during processing. Variations in the physical-chemical properties of lipids can lead to changes in particle size and zeta potential, degree of crystallinity, and coexistence of additional colloidal structures and can significantly impact the stability and release kinetics of SLN. For GMO, it has been found that its crystallization resulted in the particle size aggregation during lyophilization. Conventionally, formulation approaches, such as the addition of a cryoprotectant, have been found to inhibit particle size increase during lyophilization; however, in the case of GMO, addition of PVP resulted in the desired SLN preparation. Figure 10 summarizes the physical-chemical characterization and formulation considerations for the preparation of SLN. It can be noted that the key to successful formulation of SLN for optimum drug delivery system is to understand the physicochemical properties of lipids, along with drugs and surfactants. Further, it is vital to characterize any changes during the preparation of primary and secondary SLN. For the stability of SLN, it is important to consider the composition, processing parameters, and storage conditions.

Physicochemical characterization and formulation consideration for preparation of SLN
CONCLUSIONS
The crystallization and melting behavior of GMO were successfully characterized using MDSC. Results were corroborated by cryo-microscopy and cryo-XRD measurements, which provided detailed understanding of the crystallization phenomena in GMO. Significant differences between various grades of GMO were observed, highlighting the importance of thoroughly characterizing lipids used in the preparation of SLN and optimizing their formulation process. The observed crystallization of GMO was found to be associated with the increase in particle size during lyophilization. Successful inhibition of GMO crystallization using PVP was achieved, resulting in the formation of similar particle size SLN before and after lyophilization.
Abbreviations
- SLN:
-
Solid lipid nanoparticles
- GMO:
-
Glyceryl mono-oleate
- MDSC:
-
Modulated differential scanning calorimetry
- PVP:
-
Polyvinyl pyrrolidone
- HPMC:
-
Hydroxyl propyl methyl cellulose
- PEG:
-
Polyethylene glycol
- NLC:
-
Nanostructured lipid carriers
- TA:
-
Thermal analysis
- XRD:
-
X-ray diffraction
- ss-NMR:
-
Solid state nuclear magnetic resonance
REFERENCES
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery—a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–77.
Wissing S, Kayser O, Müller R. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56:1257–72.
Joshi MD, Müller RH. Lipid nanoparticles for parenteral delivery of actives. Eur J Pharm Biopharm. 2009;71:161–72.
Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–96.
Muchow M, Maincent P, Müller RH. Lipid nanoparticles with a solid matrix (SLN®, NLC®, LDC®) for oral drug delivery. Drug Dev Ind Pharm. 2008;34:1394–405.
Schwarz C, Mehnert W, Lucks J, Müller R. Solid lipid nanoparticles (SLN) for controlled drug delivery. I. Production, characterization and sterilization. J Control Release. 1994;30:83–96.
Hu L, Tang X, Cui F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J Pharm Pharmacol. 2004;56:1527–35.
Zur Mühlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery–drug release and release mechanism. Eur J Pharm Biopharm. 1998;45:149–55.
Kathe N, Henriksen B, Chauhan H. Physicochemical characterization techniques for solid lipid nanoparticles: principles and limitations. Drug Dev Ind Pharm. 2014;40:1–11.
O’Laughlin R, Sachs C, Brittain H, Cohen E. Effects of variations in physicochemical properties of glyceryl monostearate on the stability of an oil-in. J Soc Cosmet Chem. 1989;40:215–29.
Prasad D, Chauhan H, Atef E. Studying the effect of lipid chain length on the precipitation of a poorly water soluble drug from self‐emulsifying drug delivery system on dispersion into aqueous medium. J Pharm Pharmacol. 2013;65:1134–44.
Radomska-Soukharev A. Stability of lipid excipients in solid lipid nanoparticles. Adv Drug Deliv Rev. 2007;59:411–8.
Saez A, Guzman M, Molpeceres J, Aberturas M. Freeze-drying of polycaprolactone and poly (D, L-lactic-glycolic) nanoparticles induce minor particle size changes affecting the oral pharmacokinetics of loaded drugs. Eur J Pharm Biopharm. 2000;50:379–87.
Hou D, Xie C, Huang K, Zhu C. The production and characteristics of solid lipid nanoparticles (SLNs). Biomaterials. 2003;24:1781–5.
Trickler WJ, Khurana J, Nagvekar AA, Dash AK. Chitosan and glyceryl monooleate nanostructures containing gemcitabine: potential delivery system for pancreatic cancer treatment. AAPS PharmSciTech. 2010;11:392–401.
Rowe RC, Sheskey PJ, Owen SC, Association AP. Handbook of pharmaceutical excipients. London: Pharmaceutical Press; 2006.
Revision, U.S.P.C.C.o., United States Pharmacopeia, The National Formulary. United States Pharmacopeial Convention, Inc., 2004.
Vivek K, Reddy H, Murthy RS. Investigations of the effect of the lipid matrix on drug entrapment, in vitro release, and physical stability of olanzapine-loaded solid lipid nanoparticles. AAPS PharmSciTech. 2007;8:16–24.
Freitas C, Müller R. Correlation between long-term stability of solid lipid nanoparticles (SLN™) and crystallinity of the lipid phase. Eur J Pharm Biopharm. 1999;47:125–32.
Laura C, Milena S, Giovanna B, Cristina BM, Giuseppina S, Giampiero B. Characterization of silver sulfadiazine-loaded solid lipid nanoparticles by thermal analysis. J Therm Anal Calorim. 2013;111:2149–55.
Harry RG, Myddleton WW. Polyhydric alcohol esters of fatty acids: the principles and practice of modern cosmetics. New York: Chemical Publishing Co; 1963.
Chariot M, Ser J. Optimisation de la granulation en lit fluidisr. Labo-Pharma Prob Tech. 1983;31:456–60.
Sharpe SA, Celik M, Newman AW, Brittain HG. Physical characterization of the polymorphic variations of magnesium stearate and magnesium palmitate hydrate species. J Struct Chem. 1997;8:73–84.
Zhang L, Liu L, Qian Y, Chen Y. The effects of cryoprotectants on the freeze-drying of ibuprofen-loaded solid lipid microparticles (SLM). Eur J Pharm Biopharm. 2008;69:750–9.
Taylor P. Ostwald ripening in emulsions. Adv Colloid Interf Sci. 1998;75:107–63.
Welin-Berger K, Bergenståhl B. Inhibition of Ostwald ripening in local anesthetic emulsions by using hydrophobic excipients in the disperse phase. Int J Pharm. 2000;200:249–60.
Ausborn M, Nuhn P, Schreier H. Stabilization of liposomes by freeze-thaw- and lyophilization techniques: problems and opportunities. Eur J Pharm Biopharm. 1992;38:133–9.
Schwarz C, Mehnert W. Freeze-drying of drug-free and drug-loaded solid lipid nanoparticles (SLN). Int J Pharm. 1997;157:171–9.
Chauhan H, Hui‐Gu C, Atef E. Correlating the behavior of polymers in solution as precipitation inhibitor to its amorphous stabilization ability in solid dispersions. J Pharm Sci. 2013;102:1924–35.
Chauhan H, Kuldipkumar A, Barder T, Medek A, Gu C-H, Atef E. Correlation of inhibitory effects of polymers on indomethacin precipitation in solution and amorphous solid crystallization based on molecular interaction. Pharm Res. 2014;31:500–15.
ACKNOWLEDGMENTS
The author wishes to acknowledge Creighton University, Omaha, NE, for funding and the use of instruments in completing this project. Authors would also like to acknowledge Dr. Raj Suryanarayanan, Professor, College of Pharmacy, University of Minnesota, for carrying out cryo-X-ray experiments and Dr. Sorin Luca, Assistant Professor, University of Nebraska Medical Center, Omaha, NE, for carrying out ss-NMR analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chauhan, H., Mohapatra, S., Munt, D.J. et al. Physical-Chemical Characterization and Formulation Considerations for Solid Lipid Nanoparticles. AAPS PharmSciTech 17, 640–651 (2016). https://doi.org/10.1208/s12249-015-0394-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0394-x